
Abstract
It has been shown that Caspy2, a zebrafish active caspase, can efficiently suppress the growth of malignant tumor. The present study was designed to test whether combined gene therapy with IP-10, a potent antitumor chemokine, and Caspy2 would improve therapy efficacy. Recombinant plasmid expressing both Caspy2 and IP-10 genes was mixed with DOTAP–cholesterol nanoparticles. Immunocompetent mice bearing CT26 colon carcinoma, B16-F10 melanoma, and 4T1 breast carcinoma were treated with the complex. We found that the combined gene therapy more efficiently inhibited tumor growth, while efficiently prolonging the survival of tumor-bearing animals, compared with monotherapy. Moreover, a significant reduction in spontaneous lung metastasis could be observed in the 4T1 breast carcinoma model. Infiltration of CD8+ T lymphocytes was also observed. In addition, apoptotic cells were widely detected by TUNEL assay and caspase-3 immunostaining in coadministered tumor tissues. The combination treatment also successfully inhibited angiogenesis and tumor cell proliferation as assessed by CD31 and Ki-67 immunostaining, respectively. Furthermore, depletion of CD8+ T lymphocytes could significantly abrogate the antitumor activity, whereas the depletion of CD4+ cells or natural killer cells showed partial abrogation. Rechallenged CT26 tumors were rejected in all of the surviving mice treated by combination therapy. Our results suggest that combined therapy with Caspy2 and IP-10 can significantly enhance antitumor activity by acting as an immune response initiator, apoptosis inducer, and angiogenesis inhibitor, which may be important for further applications in clinical cancer therapy.
Introduction
Caspy2, a zebrafish protease containing N-terminal pyrin domains and C-terminal caspase catalytic domains, shares highest homology with human caspase-5. Biochemical analysis has revealed that Caspy2 is an active caspase in apoptosis of mammalian cells, which is inhibited by general caspase inhibitors (Masumoto et al., 2003). It has been reported that expression of Caspy2 induced apoptosis in tumor cells and enhanced specific antitumor immunity in tumor cells (Liu et al., 2009).
Interferon γ-inducible protein of 10 kDa (IP-10/CXCL10), a member of the non-ELR-CXC chemokine superfamily, is known to be a highly inducible chemoattractant binding to the CXCR3 chemokine receptor (Farber, 1997; Loetscher et al., 1998). Current research has demonstrated IP-10 to be a pleiotropic molecule eliciting potent biological effects, including stimulation of monocytes, natural killer (NK) cells, and T cells; regulation of T cells and bone marrow progenitor maturation; modulation of adhesion molecule expression; as well as inhibition of angiogenesis (Neville et al., 1997). At present, much interest is focused on the antineoplastic effects of IP-10. IP-10 has been shown to display antitumor and antimetastatic properties by immunological, antiangiogenic, and antineoplastic mechanisms (Maru et al., 2008; Fujita et al., 2009).
In this study, we have investigated the effects of combined Caspy2 and IP-10 on murine malignant neoplasms. We wondered whether the addition of IP-10 could enhance the antitumor immune response of Caspy2 through immunocyte recruitment and improve tumor growth inhibition through antiangiogenesis; bring the superiority of Caspy2 to induce apoptosis simultaneously. We coexpressed Caspy2 and IP-10 by DOTAP–cholesterol nanoparticle-mediated gene transfer in murine malignant tumors and explored the possible mechanisms underlying the action of Caspy2 and IP-10. We present here the first evidence that enhanced expression of Caspy2 and IP-10 dramatically inhibits tumor cell growth and metastasis.
Materials and Methods
Cell lines
Murine colon carcinoma cell line CT26, murine breast carcinoma cell line 4T1, and murine melanoma cell line B16-F10 were purchased from the American Type Culture Collection (ATCC, Manassas, VA). CT26 and 4T1 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), and B16-F10 cells was grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS.
Plasmid construction
A dual-promoter plasmid, pVITRO2 (Invitrogen, San Diego, CA), was used to express both Caspy2 and IP-10 genes. Total RNA was isolated from zebrafish and the open reading frame of the Caspy2 gene was cloned by RT-PCR. pBLAST49-mIP-10 was purchased from InvivoGen (San Diego, CA). The following PCR primers were used: Caspy2-forward, 5′-CG ACG CGT AAG ATG GAG GAT ATT ACC CAG CTG CT-3′; Caspy2-reverse, 5′-ACGC GTC GAC TCA CAG TCC AGG AAA CAG GTA GAAC-3′; IP-10-forward, 5′-TTA CAC CGG TG AAG ATG AAC CCA AGT GCT GCC GT-3′; IP-10-reverse, 5′-CTA GCT AGC TTA AGG AGC CCT TTT AGA CCTT-3′. The amplified products were subcloned into the pVITRO2 expression vector to generate IP-10 (pIP-10)/Caspy2 (pCaspy2)/IP-10 plasmid and Caspy2(pIP-10-Caspy2) plasmid. All the sequences were determined by DNA sequencing. An empty pVITRO2 was used as an empty vector control. Plasmids were purified with an Endo-free Giga kit (Qiagen, Hilden, Germany), in accordance with the instructions of the manufacturer.
Transfection
Twenty-four hours before transfection, tumor cells were trypsinized and seeded in 6-well culture plates at 1×105 or 2×105 cells per well. DNA transfection was performed with FuGENE HD transfection reagent (Roche, Indianapolis, IN) according to the manufacturer's protocol. In brief, DNA–FuGENE HD transfection reagent complexes, which contained 2 μg of DNA and 5 μl of transfection reagent, were prepared in 100 μl of serum-free medium for 15 min at room temperature. The transfection complexes were then added to cells in a drop-wise manner and incubated for 48 or 72 hr for further studies.
Apoptosis analysis by flow cytometry
Apoptotic cells were visualized with an annexin V–fluorescein isothiocyanate (FITC) apoptosis detection kit (BD Biosciences, San Jose, CA) according to the manufacturer's protocol. Briefly, cells were harvested 48 hr after transfection, washed twice with phosphate-buffered saline (PBS), and resuspended in 500 μl of annexin V binding buffer. Two microliters of FITC-conjugated annexin V was added to the cell solutions, followed by the addition of 5 μl of propidium iodide (PI). After incubation for 5 min at room temperature in the dark, samples were immediately analyzed with a FACSCalibur flow cytometer (BD Biosciences). Data from approximately 1×104 cells were analyzed with CellQuest software (BD Biosciences).
Preparation of cationic liposome and liposome–DNA complex
Biodegradable cationic liposomes consisting of 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP)–cholesterol (1:1, molar ratio) were prepared by hydration in 5% dextrose solution and sequentially extruded through polycarbonate membranes (400, 200, 100, and 50 nm) to generate small unilaminar vesicles, and then the recovered liposome reagent was stored at 4°C. The preparation of plasmid and liposome was done as described previously (Templeton et al., 1997; Shi et al., 2009). Briefly, DNA and stock liposome were diluted in 5% glucose solution and then mixed at a DNA-to-liposome ratio of 1:5 (w/w). The particle size of the complex was between 100 and 120 nm. The resulting complexes were incubated at room temperature for 30 min before administration to mice.
Murine tumor models and treatment of established tumors
Animal studies were performed in accordance with institutional guidelines concerning animal use and care. All studies involving animals included female C57BL/6 mice and female BALB/c mice aged 6–8 weeks, which were purchased from the Laboratory Animal Center of Sichuan University (Chengdu, Sichuan, China) and allowed to acclimate for 1 week before use.
About 2×105 CT26 cells and 1×105 B16-F10 cells were inoculated subcutaneously into the right flanks of BALB/c mice and C57BL/6 mice, respectively. When the size of tumors reached approximately 100 mm3 (5 days after tumor cell inoculation), mice were randomly divided into the following five groups: pIP-10-Caspy2, pIP-10, pCaspy2, empty pVITRO2 (e-p), and 5% glucose (5% GS) (10 mice each group), and treatment was initiated. Tumor-bearing mice were injected intratumorally with a 100-μl total volume containing plasmid–liposome complex (10 μg of plasmid DNA and 50 μg of liposome). Injections, done 12 times, were distributed equally into each of four tumor quadrants at 1-day intervals. Tumors were measured at 2-day intervals with Vernier calipers. Tumor volume was determined by the following formula: tumor volume (mm3)=[length (mm)×width (mm)2]/2. Three days after the last injection, animals were killed and tumor tissues were excised. The tumor tissues were paraffin embedded or quick-frozen and sectioned for further pathological analysis. After observing a definite curative effect in the first animal experiment, we repeated the experiment with these two tumor models to investigate whether the treatment could prolong the survival of tumor-bearing mice, before proceeding to further studies.
To establish a spontaneous metastasis model, 2×105 4T1 breast carcinoma cells were injected into the right flank of BALB/c mice; this model can spontaneously form pulmonary metastasis after 3 weeks. Seven days after the implantation, mice were randomly allocated to five groups as described previously. Mice were treated via the caudal vein with DNA–liposome complex, 12 times with a 1-day interval. Three days after the last treatment, the animals were killed, and their lung tissues were excised. Tumor colonies on lung surfaces were counted under a dissecting microscope, and lung tissues were paraffin embedded and sectioned for further pathological analysis.
Clone formation assay
Twenty-four hours after transfection, 4T1 cells were plated in triplicate at 500 cells per well in 6-well plates, and cultured with RPMI 1640 complete culture medium. After 10 days, cells were stained with crystal violet, and aggregates of more than 100 cells were scored as colonies. Clone formation efficiency was determined as follows: (number of clones/number of cells inoculated)×100%. All experiments were repeated three times and average values are reported.
Tube formation assay
Antiangiogenic effects of pIP-10-Caspy2/pIP-10/pCaspy2 were analyzed in an in vitro tube formation assay as described previously (Danussi et al., 2008). Human umbilical vein endothelial cells (HUVECs) were cultured in complete M199 medium before being plated in 96-well plates (2×104 per well), which were previously coated with 50 μl of growth factor-reduced Matrigel (BD Biosciences). We collected the supernatant of transfected CT26 or B16-F10 cells after incubation for 48 hr as condition medium with which to culture HUVECs. Test condition medium and control supernatant of tumor cells were added in triplicate, and untreated HUVECs were used as blank control. After 6 hr, the appearance of capillary-like structures formed by HUVECs was visualized with an inverted Axioskop microscope (Carl Zeiss, Jena, Germany) and photographed with a digital camera (Olympus, Lake Success, NY). Tube formation was analyzed with ImagePro Plus software (Media Cybernetics, Silver Spring, MD) as previously described (Qian et al., 2004).
T lymphocyte cytotoxicity assay
To detect cytotoxic T lymphocyte (CTL) activity, lymphocytes were isolated from the spleen of each treated mouse as described previously (Zhao et al., 2008). Briefly, splenocytes obtained from treated and control mice were depleted of erythrocytes with ammonium chloride–Tris buffer. Specific CTL activity was measured by 51Cr release assay, using CT26 cells as target cells, as described elsewhere (Takeda et al., 2004). The percentage of cytotoxicity was calculated by the following formula: cytotoxicity (%)=[(experimental release – effector spontaneous release)/(target maximum release – target spontaneous release)]×100%. In the cytotoxicity inhibition assays, effector cells or CT26 cells were pretreated with monoclonal antibody (mAb) at room temperature for 30 min, washed, and tested. The mAbs used included anti-CD4 (10 μg/ml), anti-CD8 (10 μg/ml), and anti-NK (10 μg/ml) (BD Biosciences). The above-cited concentrations of mAb were effective in mediating their activity in preliminary experiments. A control cytotoxicity assay was performed in the presence of isotype IgG mAb (BD Biosciences).
Tumor cell rechallenge
We repeated the CT26 model therapy experiment to observe the life span of treated mice, and then, to determine whether surviving mice in the CT26 model had developed a long-term immunological memory, the treated animals were divided into two groups (three mice each group) and rechallenged subcutaneously via the other flank with 2×105 parental CT26 cells or irrelevant 4T1 cells.
In situ TUNEL assay
A terminal deoxynucleotidyltransferase-mediated nick end labeling (TUNEL) assay was performed to detect fluorescence of apoptotic cells in situ, using a TUNEL kit (Promega, Madison, WI) in accordance with the manufacturer's protocol. Apoptotic cells were counted under a microscope (magnification, ×200) in randomly selected fields.
Histological analysis
Immunohistochemical staining or immunofluorescence of Ki-67, caspase-3, CD8, and CD31 was performed to detect proliferation, apoptosis, immunoreaction, and neovascularization in tumors, which was done as described previously (Zhang et al., 2009). Primary antibodies included rabbit anti-mouse Ki-67 antibody (Abcam, Cambridge, UK), active caspase-3 antibody (Abcam), anti-mouse CD8 antibody (Cy5–PE conjugate; eBioscience, San Diego, CA), and rat anti-mouse CD31 antibody (BD Biosciences).
For visualization of pulmonary metastasis, lung tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Sections of 5 μm were stained with hematoxylin and eosin (H&E).
In addition, to evaluate potential side effects and toxicity of the treatment, relevant indexes such as weight loss, diarrhea, anorexia, cachexia, skin ulcerations, and toxic deaths were monitored. Heart, liver, spleen, lung, and kidney tissues were harvested and fixed in 4% paraformaldehyde solution and embedded in paraffin. Sections of 5 μm were stained with H&E and observed by two pathologists in a blinded manner.
Statistical analysis
Data are expressed as means±SD. Statistical significance was determined by one-way analysis of variance (ANOVA). Animal survival data was analyzed by Kaplan–Meier survival analysis. All p values were two sided and p<0.05 was considered statistically significant. The Statistical Package for the Social Sciences, version 18.0 (SPSS, Chicago, IL) was used for all statistical analyses.
Results
In vitro coexpression of Caspy2 and IP-10 inhibits tumor cell growth
Plasmids were generated as described in Materials and Methods. Tumor cells were transfected with the following expression vectors: pIP-10, pCaspy2, pIP-10-Caspy2, or empty pVITRO2 (e-p). Expression was confirmed by RT-PCR (Supplementary Fig. 1; supplementary data are available online at

Effects of combination treatment in vitro.
In vitro inhibition of angiogenesis
We tested the ability of Caspy2 and IP-10 to inhibit endothelial cell activity in vitro, using the tube formation assay. Untreated HUVECs display healthy tubular network formation. In the case of HUVECs treated with e-p supernatant, there was tubular network formation almost similar to that of the control groups. In the case of HUVECs treated with pIP-10 or pCaspy2 supernatant, we found obvious inhibition of tubular network formation, whereas the combination treatment resulted in complete inhibition (Fig. 1C).
In vivo growth inhibition of established tumors in mice
Consistent with the results in vitro, combined treatment with Caspy2 and IP-10 nanoparticles significantly inhibited tumor formation and growth in the CT26 and B16-F10 subcutaneous tumor models (Fig. 2). The results showed that both pIP-10 and pCaspy2 resulted in effective suppression of tumor growth. However, combined treatment with Caspy2 and IP-10 had a superior antitumor effect (Fig. 2A and B).
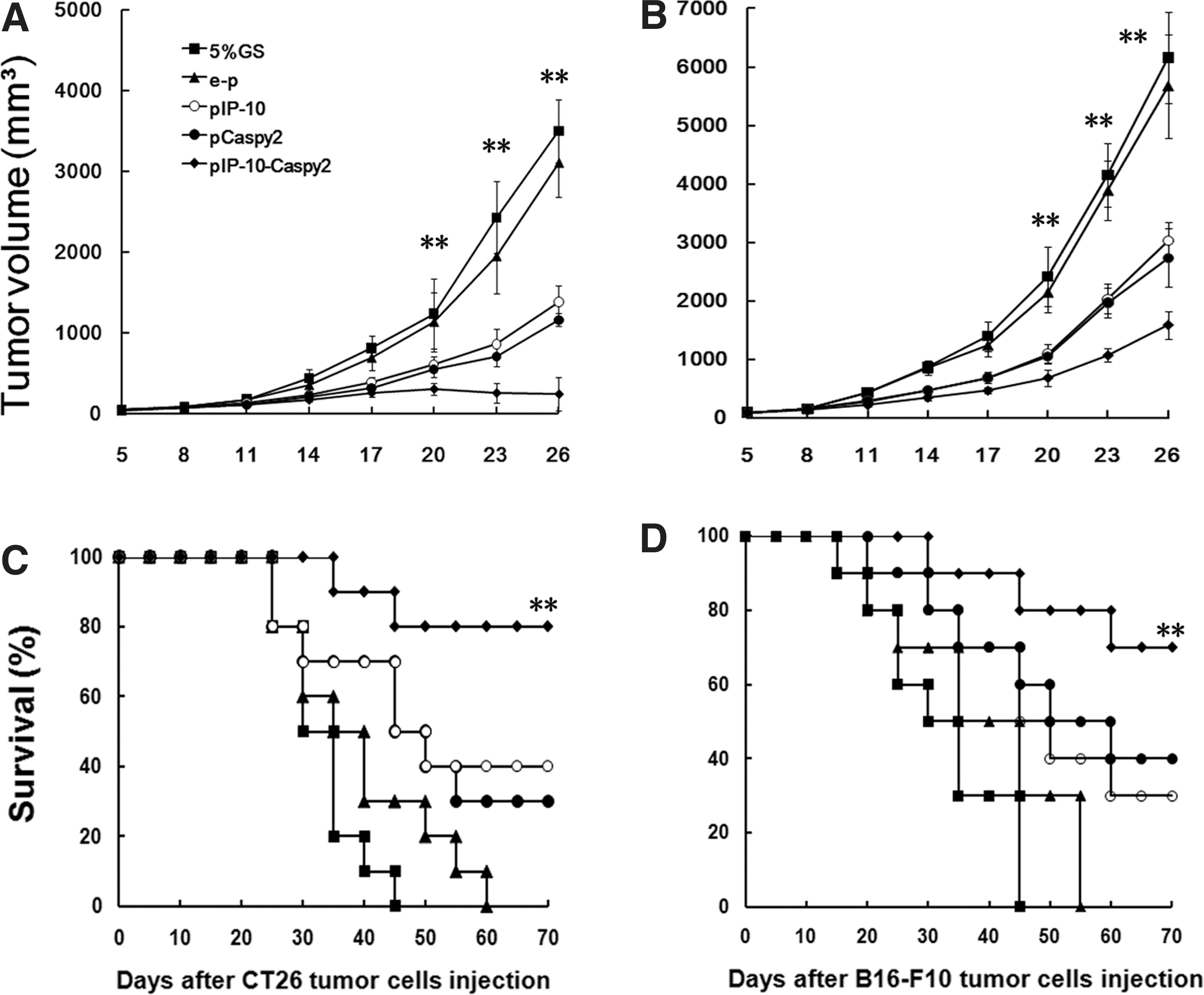
Tumor suppression and survival advantage in mice. Tumor-bearing mice (10 mice per group) were treated as described in Materials and Methods.
In the second animal experiment, the long-term therapeutic effect of the combined treatment was examined by measuring animal survival in each treatment group. In the CT26 and B16-F10 tumor models, combined treatment with Caspy2 and IP-10 resulted in a significant increase in life span compared with the pIP-10 monotherapy group, pCaspy2 monotherapy group, and controls (Fig. 2C and D). Furthermore, in the CT26 tumor model, 60% of the mice in the pIP-10-Caspy2 group were cured, showing no histological evidence of tumor. Forty percent of mice treated with pIP-10 or pCaspy2 alone showed no evidence of tumor until the end of observation (70 days). These findings demonstrate that combined injection with Caspy2 and IP-10 markedly improved antitumor outcomes in vivo.
Systemic treatment suppresses lung metastasis in 4T1 mouse model
Metastasis to the lung is the major cause of death in 4T1-bearing mice. In both the pIP-10 and pCaspy2 groups, the number of lung metastases was clearly lower than in the 5% GS and e-p groups. In addition, in the pIP-10-Caspy2 group, the number of lung metastases was significantly reduced compared with either monotherapy group (Fig. 3). This result showed that coadministration of Caspy2 and IP-10 nanoparticles resulted in effective therapeutic efficacy, suppressing metastasis in the 4T1 breast tumor model.
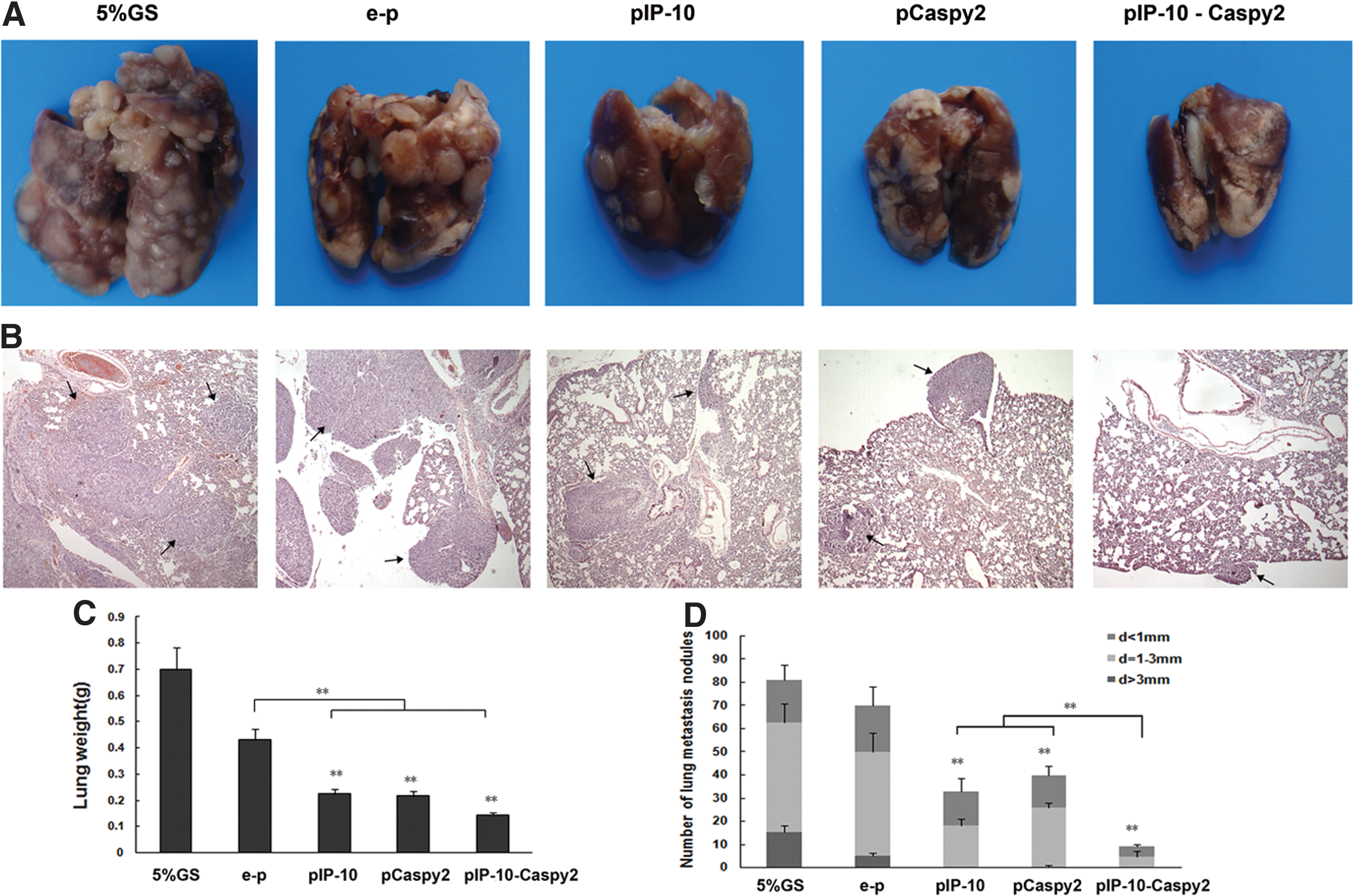
Suppression of lung metastasis in the 4T1 mouse model. A BALB/c mouse lung metastasis model was established by subcutaneous inoculation with 2×105 4T1 cells. Mice were killed 34 days after tumor cell inoculation. Lung weight and the number of lung metastases were assessed.
In vivo inhibition of angiogenesis
Angiogenesis within tumor tissue was assessed by CD31 immunohistochemical staining. The most highly vascularized area of each tumor was identified on low power, and five high-power fields were counted in this area of greatest vessel density. pIP-10-Caspy2 therapy resulted in an apparent inhibition of tumor angiogenesis compared with the control groups, and the pIP-10 and pCaspy2 monotherapy groups (Fig. 4A). Our results suggested that the antitumor effects of the combined treatment acted partly via an antiangiogenic mechanism to promote tumor regression.

Histochemical analysis of CT26 tumors.
In vivo inhibition of tumor cell proliferation
Ki-67 staining was performed to detect cell proliferation. As shown in Fig. 4B, fewer proliferation cells (brown) in tumor tissues were observed in mice treated with pIP-10 or pCaspy2 than in the control groups. In the pIP-10-Caspy2 group, the Ki-67-labeling index was the lowest.
Fluorescence analysis of intratumoral T cell infiltration
To determine whether the therapy facilitated the infiltration of CD8+ lymphocytes into the tumors, anti-CD8 mAb was used in immunofluorescence staining. The results showed that pIP-10 or pCaspy2 alone significantly increased the infiltration of CD8+ CTLs, and that the combined therapy resulted in the most infiltration of CD8+ CTLs (Fig. 4C).
In vivo apoptosis induction of combined treatment
To investigate whether apoptosis was indeed the mechanism that enhanced the antitumor effects of combined treatment in vivo, the TUNEL assay was carried out to detect early DNA fragmentation associated with apoptosis. More apoptotic cells were found in pCaspy2-treated tumor tissue versus e-p-treated or control group tissue samples, whereas an apparent increase in apoptotic cells was observed within the tumors of the combined treatment group (Fig. 4D).
Furthermore, we performed immunostaining associated with apoptosis, using anti-caspase-3 antibody. Analysis of the numbers of caspase-3-labeling cells (brown) showed that combined treatment with Caspy2 and IP-10 was clearly more potent in eliciting tumor cell apoptosis than was monotherapy with Caspy2 or IP-10 (Fig. 4E). These results showed that IP-10 could enhance the apoptosis effect induced by Caspy2 and related to caspase-3.
Effect of combination therapy on CTL activity
Determination of CTL activity provided evidence for the involvement of an immunological mechanism in the antitumor effects of the treatments. Spleen T lymphocytes isolated from mice treated with pIP-10-Caspy2 showed higher cytotoxicity against parental CT26 cells than did those from the other groups (Fig. 5A). Next, we observed that this cytotoxicity could be almost completely blocked by anti-CD8 mAb, but fewer by anti-CD4 mAb or anti-NK mAb in vitro (Fig. 5B).

CTL-mediated antitumor activity.
Combination treatment leads to systemic and persistent antitumor immunity
To examine whether long-term tumor-specific protective immunity was established by the combined treatment, 80 days after the first injection of CT26 cells long-term-surviving mice were rechallenged subcutaneously with parental CT26 cells or irrelevant syngeneic 4T1 cells. The results showed that detectable flank tumors were not observed with inoculation of CT26 tumor cells into previously treated mice, and that all the mice survived in apparent good health. However, when these mice were inoculated with 4T1 cells, the tumors grew vigorously and all had died by 45 days after rechallenge (Fig. 6). In the CT26 model, animals treated with pIP-10 or pCaspy2, and especially those treated with pIP-10 and Caspy2, were all nearly cured and exhibited long-term protection against parental tumor rechallenge. These results suggested that persistent, systemic, and CT26-specific immunity was induced either by the combined treatment, or by monotreatment (data not shown).
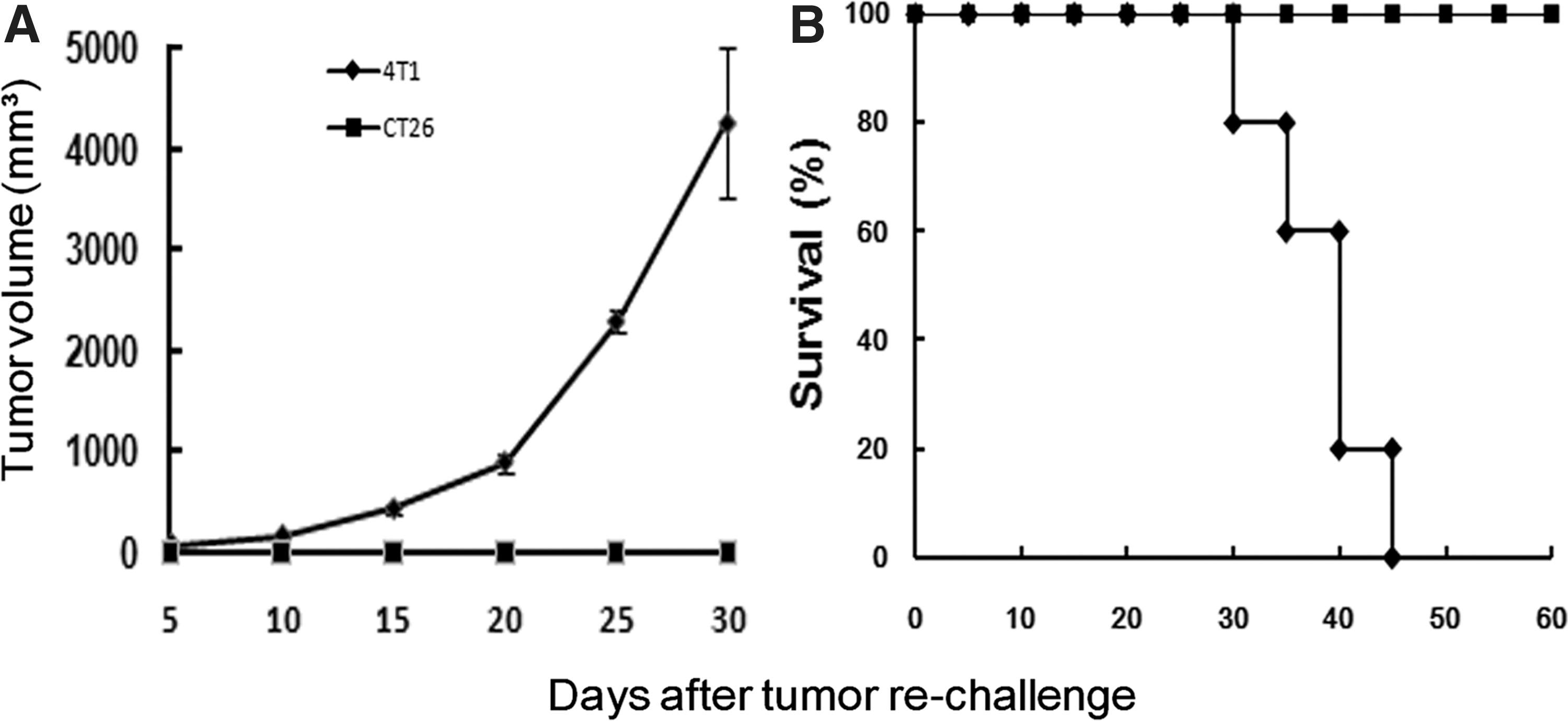
Development of specific and long-term antitumor immune responses. Recovered mice from the pIP-10-Caspy2-treated group in the CT26 model were rechallenged with 2×105 parental CT26 cells or irrelevant syngeneic 4T1 cells in the other flank. Mice rechallenged with 4T1 cells developed large flank tumors
Discussion
Evidence indicates that the antitumor strategy based on immunological approaches has been particularly important. It has been reported that introduction of new antigens into tumor cells would stimulate immune responses against autologous malignant cells (Schirrmacher et al., 1999; Rosenberg, 2001). Caspy2 is an active zebrafish caspase in the apoptosis of mammalian cells and tumor cells. Our previous studies have demonstrated that the expression of Caspy2 as a foreign antigen in tumor cells could improve antitumor immunity through the acquisition of tumor-associated antigens from apoptotic tumor cells. In the present study, in order to further enhance the antitumor activity of Caspy2, we hypothesized that coexpression of the multifunctional murine chemokine IP-10 could generate a more effective immune response and boost antitumor efficacy in part by its antiangiogenic activity. Our data showed that the combined therapy was sufficiently effective against primary tumors in the CT26 colon carcinoma model and the B16-F10 melanoma model, and suppressed spontaneous pulmonary metastases in the 4T1 breast carcinoma model. In fact, we have shown here that plasmid DNA coencoding Caspy2 and IP-10 can indeed induce an antitumor immune response against established tumors, by mechanisms involving both induction of tumor cell apoptosis and suppression of tumor angiogenesis.
In the present study, apoptotic cells observed in treated tumor tissues were in accordance with the results in vitro. Apoptosis is carried out by the caspases, and one of the family members, caspase-3, is involved extensively in the execution of apoptosis and the proteolytic cleavage of subcellular substrates (Tan and Nasirudeen, 2005). Caspase-3-like activity associated with apoptosis has been detected in zebrafish (Negron and Lockshin, 2004), and the relevant genes are caspy and caspy2. Immunohistochemical analysis showed that the apoptosis induced by pCaspy2 or pIP-10-Caspy2 was proceeding by the caspase-3 associate pathway. This result was consistent with our previous study, which detected the activity of caspase with a caspase-3/7 activity detection kit (Liu et al., 2009).
In this study, we found that delivery of pIP-10-Caspy2 could significantly increase the infiltration of CD8+ T lymphocytes into tumor tissues and induce tumor-specific CTL activity in vivo. This indicates that the expression of caspy2 in tumor cells enhances the presentation ability of embellished apoptotic tumor cells to antigen-presenting cells (APCs) and induces a T cell-mediated immune response against unembellished tumor cells (Plautz et al., 1993). IP-10, acting as a chemokine to activate T cells and to attract monocytes and T cells (including neutrophils and NK cells) to tumor tissues, while upregulating MHC class I antigen expression, may enhance the antitumor immunity effect. In addition, the effective generation and maturation of CD8+ CTLs, assisting the differentiation of CD4+ T cells, should result in an effective helper T cell type 1 (Th1) immune response. Furthermore, when pIP-10-Caspy2 cured mice were rechallenged with CT26 cells, the mice exhibited immune memory, preventing tumor recurrence. It is indeed relevant that some of the CD8+ T cells were subsequently found as long-term basis memory T cells that were ready to respond to stimulation with the same tumor antigen (Luo et al., 2005). Furthermore, IFN-γ, which is found with IP-10, is also capable of promoting the turnover of memory phenotype CD8+ T cells (Tough et al., 2001).
Immunohistochemical analysis of tumor samples from the pIP-10-Caspy2 group demonstrated cooperative inhibition of cancer cell proliferation and neovascularization. Previous studies indicated that antiangiogenic factor might produce a morphologically and functionally “normalized” vascular network (Tong et al., 2004; Hamzah et al., 2008). Abnormal angiogenesis prevents migration of immune cells into established tumors, thereby representing a major hindrance to successful immune therapy. IP-10 may induce vascular normalization and enhance the movement of immune effector cells into the tumor parenchyma, markedly increasing the immune response activated by Caspy2 treatment.
In summary, we have shown that the antitumor efficacy of a novel strategy based on the coexpression of Caspy2 and IP-10 could induce a CTL response sufficiently effective to attack tumor cells as well as trigger apoptosis of tumor cells and suppress angiogenesis in the tumor neovasculature, resulting in the inhibition of tumor progression and metastasis. It may prove a promising candidate for further clinical application.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (30973451) and the National Key Basic Research Program (973 Program) of China (2010CB529900).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.