
Abstract
Oncolytic virotherapy poses unique challenges to the evaluation of tumor response. We hypothesized that the addition of [18F]fluorodeoxyglucose (FDG) positron emission tomography (PET) to standard computed tomography (CT) evaluation would improve diagnostic and prognostic power of the measurement of tumor response to oncolytic virotherapy. A phase I/II trial was conducted to investigate treatment of hepatic metastases from colorectal carcinoma using intra-arterial administration of the oncolytic herpes virus NV1020. Both contrast-enhanced CT and FDG PET were obtained on each patient at each time point. Quantitative FDG PET and CT responses were correlated with each other and with clinical outcome metrics. A majority of patients showed initial post–viral infusion increases in tumor size (69%) or in standardized uptake value (SUV) (80%) large enough to qualify as progressive disease. Most showed subsequent decreases in tumor size (64%) or SUV (83%) enough to be reclassified as partial response or stable disease. Late PET and CT imaging results correlated well with each other and with clinical outcomes, but results from early in the treatment scheme did not correlate with each other, with later results, or with clinical outcomes. The addition of FDG PET to the evaluation of tumor response to the oncolytic virus NV1020 did not provide useful diagnostic or prognostic data. More sophisticated molecular imaging will need to be developed to monitor the effects of this novel class of antineoplastic agents.
Introduction
Oncolytic virotherapy is an investigational locoregional treatment that has posed uniquely challenging obstacles to accurate evaluation of tumor response (Chang et al., 2009). Oncolytic viruses create radiographic patterns of response different from those of traditional chemotherapeutic and targeted agents, where not only the magnitude of observed changes is poorly understood, but even the timing and desired direction of change are not understood. In a previous trial evaluating the safety and efficacy of the first-generation oncolytic adenovirus ONYX-015, a peculiar but reproducible pattern of radiographic response was observed where targeted lesions enlarged and showed changes in vascularity, as demonstrated by changes in contrast enhancement (Sze et al., 2003). This enlargement led to withdrawal of some enrolled patients for presumed disease progression. However, a large proportion of patients with early enlargement who were not withdrawn went on to show tumor regression, casting doubt on whether the patients withdrawn for progression were appropriately evaluated.
The advent of functional and molecular imaging has greatly improved the sensitivity and specificity of imaging of malignancy. For many locoregional tumor therapies, 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography (FDG PET) has become the standard of care to evaluate response to treatment, because it allows depiction of metabolic activity of a tumor rather than just its size (Pysz et al., 2010). We hypothesized that the addition of FDG PET evaluation to the standard computerized tomography (CT) evaluation could improve diagnostic and prognostic power in the evaluation of tumor response to oncolytic virotherapy. We analyzed the FDG PET and CT results of a multicenter, prospective, open-label phase I/II trial evaluating the safety and efficacy of the oncolytic herpes virus NV1020, administered to patients with hepatic metastases from colorectal carcinoma.
Materials and Methods
NV1020 is a replication-selective, highly attenuated derivative of wild-type herpes simplex virus 1 (HSV-1) that has shown antitumor properties both in vitro and in vivo, with additive and/or synergistic effects when administered concurrently or sequentially with conventional chemotherapeutics (Kelly et al., 2008). Multiple genetic deletions greatly weaken its ability to infect normal cells and subjects, and the insertion of a functional HSV-1 thymidine kinase gene confers sensitivity to acyclovir, an additional safety provision. The safety and tolerability of single intra-arterial infusion of NV1020 into the hepatic artery were previously demonstrated in a phase I study (Kemeny et al., 2006).
The design and clinical outcomes of this clinical trial were recently reported (Geevarghese et al., 2010). In brief, an initial phase I dose escalation was performed to determine dose-limiting toxicity and to identify an optimal biological dose (OBD) for repeated administrations of NV1020. Sequential cohorts of three patients each received half-log increments of NV1020, starting at 3×106 pfu and increasing to 1×108 pfu for the last cohort. This was followed by a phase II dose expansion at the OBD.
Patients were required to have histologically proven colorectal carcinoma with liver-dominant metastasis and radiologic evidence of intrahepatic disease progression. They were heavily pretreated patients (≥2 prior regimens of conventional systemic chemotherapy), with Karnofsky performance status of ≥70%. Mild hematological and hepatic serum laboratory abnormalities were permitted. All patients were required to be seropositive for HSV-1 as a safety precaution.
Patients underwent hepatic arteriography and whole-liver administration of NV1020 weekly for 4 weeks. Approximately 1 week after the final viral infusion, patients underwent at least two cycles of systemic chemotherapy with or without targeted agents. Choice of individual agents was left to the discretion of the medical oncologists, based on each patient's prior exposures and responses. Clinical follow-up included physical examination, and hematology, coagulation, chemistry, and tumor marker tests. In addition, a limited cytokine panel, neutralizing antibody titer, and virus-shedding assays were performed. Toxicity was graded by National Cancer Institute Common Toxicity Criteria version 3 (NCI-CTCv.3).
The protocol was approved by each local Institutional Review Board and was overseen by an independent Data Monitoring Committee. All data were handled in compliance with the Health Insurance Portability and Accountability Act (HIPAA). Each patient provided written, informed consent at the time of enrollment.
Imaging evaluation and follow-up schema
Contrast-enhanced CT and FDG PET were obtained for each patient at the time of screening, after completion of four viral infusions, after completion of two cycles of systemic chemotherapy, and every 3 months thereafter. Independent blinded readers reviewed all images and recorded lesion-specific responses. Quantitative PET responses were graded according to European Organisation for Research and Treatment of Cancer (EORTC) criteria (Young et al., 1999), and quantitative CT responses were graded according to RECIST criteria (Therasse et al., 2000).
Statistical correlations, tests
CT and FDG PET responses at every time point were correlated with each other, with earlier and later CT and FDG PET responses, and with best CT and FDG PET responses. In addition, CT and FDG PET responses at each time point were correlated with clinical metrics, including overall survival (OS), time to progression (TTP), tumor-marker response, baseline tumor size, baseline maximum standardized uptake value (SUVmax), viral antibody titer, and time interval since original diagnosis. Pearson product-moment coefficients were calculated for each correlation, and p values were calculated using the Fisher transformation.
Results
The clinical outcomes of this trial have been reported elsewhere (Geevarghese et al., 2010). In brief, 13 patients were enrolled in the dose-escalation portion of the protocol, where grade 3/4 toxicity was limited to transient, asymptomatic lymphopenia in two patients. Mild grade 1 perturbation of the prothrombin time and pronounced increases in circulating cytokines were deemed potentially dose-limiting at 1×108 pfu, and this dose was designated the OBD. An additional 19 patients were treated at the OBD.
One patient in the dose-escalation phase and two patients in the OBD expansion phase underwent only two of the four viral infusions, with completion of the protocol aborted because of rapidly progressive disease (PD) and declining performance status. Two additional patients in the OBD expansion phase refused chemotherapy for personal reasons. Although these patients were included in the safety and early response evaluations, their longer-term follow-up data were censored in the response evaluations.
All but one of the patients treated with lower doses in the dose-escalation phase demonstrated PD throughout the follow-up protocol, with one showing stable disease (SD). At the OBD, 22 patients were evaluable for early response. Twenty-one of the 22 (95%) showed initial enlargement of tumor size and/or increase in SUVmax after viral infusions alone. In 11 patients (50.0%), the increase in size was sufficiently large to be categorized as PD by RECIST criteria. Likewise, in 12 patients (54.6%), the increase in SUVmax was sufficiently pronounced to be categorized as PD by EORTC criteria.
At the OBD, 18 patients completed the protocol, adequate CT imaging for response evaluation was available for 16, and adequate FDG PET imaging was available for 15. A large majority of the patients showed regression of tumor size and SUVmax on later follow-up after administration of systemic chemotherapy. The overall rates of partial response (PR) and SD by RECIST criteria were 6.3% and 50.0%, respectively. The overall rates of PR and SD by EORTC criteria were 26.7% and 26.7%, respectively. Of the 11 patients that initially showed tumor size increases large enough to qualify as PD after viral infusions alone, seven (63.6%) regressed enough to be reclassified as PR or SD. Likewise, of the 12 patients that initially showed increase in SUVmax large enough to qualify as PD, 10 (83.3%) regressed enough to be reclassified as PR or SD (Fig. 1).

A 40-year-old man who had progressive hepatic metastases after undergoing resection of the primary colonic adenocarcinoma and receiving 8 months of 5-fluorouracil/leucovorin–based systemic chemotherapy regimens. He was treated with four intra-arterial administrations of NV1020, each accompanied by fever, chills, and fatigue, followed by 7 months of FOLFOX, followed by 2 months of cetuximab. Survival from initial NV1020 administration was 51 weeks.
Although trends in changes of tumor size and SUVmax were relatively consistent, quantitative evaluation of changes showed only weak correlations. Early response after viral infusions alone as measured by CT and by FDG PET correlated only moderately with each other, with a Pearson coefficient of 0.39 (Table 1). These early imaging measures of response did not correlate well with later imaging responses or best imaging responses (Figs. 2 and 3). Most importantly, these early measures of response did not correlate closely with clinical metrics such as OS and TTP. Later CT and FDG PET measurements of response after systemic chemotherapy correlated more closely with each other, and also with clinical metrics. The only correlations that reached statistical significance were between CT and FDG PET responses after two cycles of chemotherapy, and between best CT and best FDG PET responses with OS and best carcinoembryonic antigen (CEA) responses (Table 1).
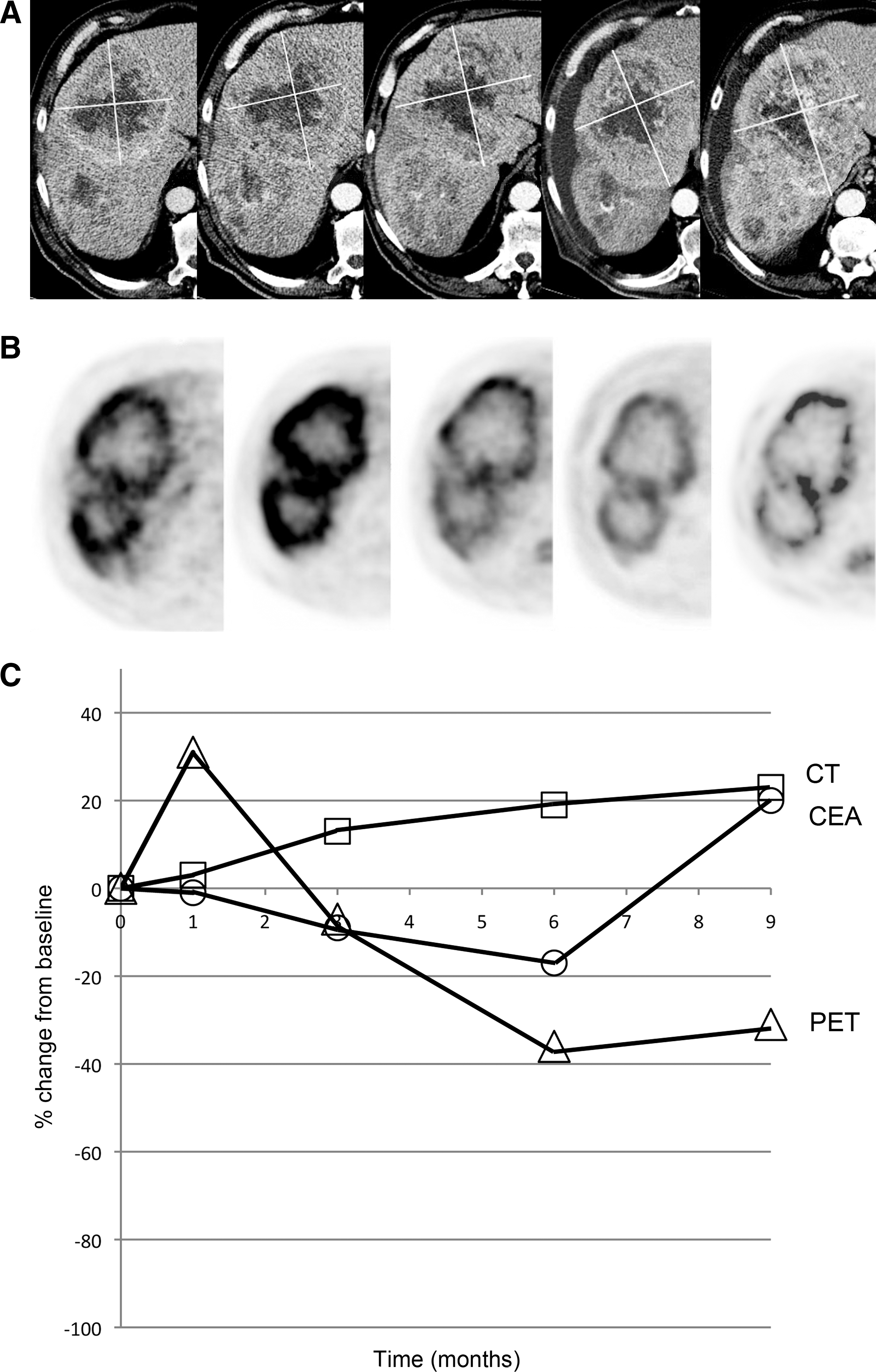
A 72-year-old man with metastatic colon cancer had progressed on FOLFOX after 5 months of therapy. After four intra-arterial infusions of NV1020, he was treated with FOLFIRI. Overall survival was 50 weeks.
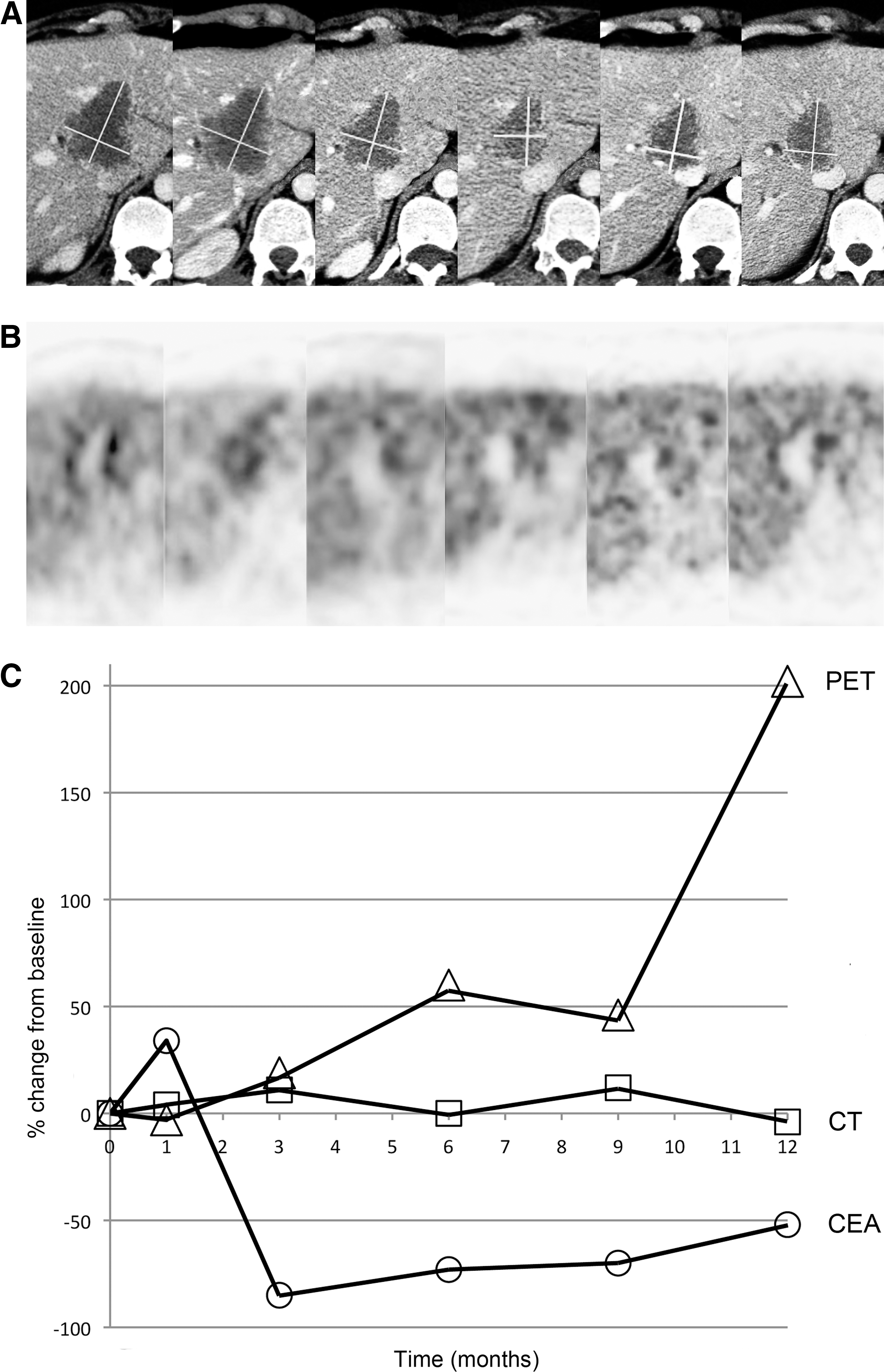
A 39-year-old woman with sigmoid cancer metastatic to the liver, who had intrahepatic progression after undergoing primary resection and 5 months of FOLFOX/bevacizumab systemic therapy.
CEA, carcinoembryonic antigen; CT, computed tomography; OS, overall survival; PET, positron emission tomography; SUVmax, maximum standardized uptake value.
Statistically significant correlation.
Although this was a blinded evaluation of data drawn from a U.S. Food and Drug Administration–approved, prospective trial with strict enrollment and follow-up requirements, limitations were evident. Its multicenter nature introduced considerable variability in the quality, timing, and reproducibility of the imaging. Fortunately, imaging protocols were standardized and enforced with more rigor as the trial progressed, resulting in a higher quality data set for the patients treated at the OBD. Additional variables contributing to response were also introduced by allowing oncologists to prescribe systemic chemotherapy at their own discretion, and the prompt instigation of systemic therapy after viral infusions did not allow for longer-term evaluation of the effects of viral infusions alone. No adjunctive investigations were performed to differentiate between hypermetabolic uptake from viral infection and that due to neoplasm.
Discussion
Although the treatment protocol of this trial studying the biological activity of NV1020 was complicated by sequential administration of systemic chemotherapies, this study supported prior observations that oncolytic viral activity manifests initially as paradoxical increase in tumor size (Sze et al., 2003). The mechanism of this morphologic change is likely one of inflammation and edema, as supported by the febrile response, pronounced increases in circulating cytokines and C-reactive protein, and decrease in circulating lymphocytes. The systemic symptoms and early CT response were nearly identical to those observed after intra-arterial administration of other oncolytic viruses (Reid et al., 2002).
Unfortunately, FDG PET is sensitive to not only hypermetabolic states seen in malignancy, but also those seen in infection and inflammation (Gotthardt et al., 2010). Thus, the initial increases in SUVmax seen in this trial confirmed that metabolic activity in tumors increased after administration of oncolytic virus, but did not distinguish tumor progression from favorable biological activity. Despite the functional and physiological nature of PET evaluation, FDG as a substrate is of insufficient biological specificity to be of clinical utility in this application. Specifically, we found no advantages between CT and FDG PET for prognostic value in imaging early response. There was no superiority proven for CT or FDG PET when imaging responses were correlated with clinical metrics such as OS and TTP.
The limitations of using WHO and RECIST criteria for evaluation of response to locoregional treatments are becoming well known (Forner et al., 2009; Lencioni and Llovet, 2010; Riaz et al., 2011). In situ ablative technologies, such as radiofrequency ablation, microwave ablation, and cryoablation, aim to ablate a normal margin surrounding each tumor, thereby giving the appearance of enlarging a lesion. In addition, these ablative technologies [as well as embolization technologies including chemoembolization, radioembolization, drug-eluting bead embolization, and bland embolization, and possibly oncolytic virotherapy (Breitbach et al., 2011)] disrupt the vascular infrastructure in and around a tumor, compromising the subject's ability to resorb necrotic tissue. Thus, modifications such as EASL modification of WHO, mRECIST, and Choi criteria aim to factor in the viability of the target lesions, as measured by enhancement by intravenously administered contrast media (Benjamin et al., 2007; Lencioni and Llovet, 2010). Estimates of viability, though, are unavoidably subject to reader error and to inconsistencies of imaging protocols, including uncontrollable variables such as patient intravascular volume status and cardiac output, gauge and site of intravenous access, and venous and arterial occlusive disease. Standard tumor response criteria may be even more inadequate when evaluating response to oncolytic viruses, because there is currently no accurate way of estimating viability of a tumor undergoing infection, inflammation, and edema.
Although FDG PET was not shown to be useful in this application, the concept of molecular or functional imaging warrants additional investigation. A multitude of new PET radiopharmaceuticals are currently being developed to complement FDG for use in cancer imaging. Promising agents include the thymidine analogue 3′-deoxy-3′-[18F]fluorothymidine (FLT), which provides a measure of DNA replication (Barwick et al., 2009). Potential advantages of FLT over FDG include fewer false-positive lesions due to inflammation, increased sensitivity of detecting brain lesions due to decreased neuronal uptake, and theoretically greater reliability in monitoring response to selective chemotherapies. PET radiopharmaceuticals that exploit the high vascularity of tumors include radiopharmaceuticals that measure angiogenesis, such as [18F]galacto-RGD or [18F]FPPRGD2 (Mittra et al., 2011). These potential agents target the expression of αvβ3-integrins, proteins selectively expressed on activated endothelial cells.
Intrinsic viral proteins may function as markers, or may function as enzymes to produce markers. In addition, exogenous genes may be inserted as therapeutic armament or as reporters to allow virus-specific measurement and localization of infection (Chang et al., 2009; Stanford et al., 2010). Specifically for HSV-1, viral thymidine kinase may be exploited as a reporter gene by using 9-(4-[18F]fluoro-3-hydroxymethylbutyl)guanine (FHBG) as the radiopharmaceutical for PET imaging (Kuruppu et al., 2007). This substrate has the advantage that it is not converted to a cytotoxic metabolite and thus should not alter the biological activity of the oncolytic virus. Temporal specificity of HSV-1 gene expression may also allow imaging of products of immediate-early versus strict-late promoter activity, which could allow the differentiation of infection from replication (Yamamoto et al., 2006).
In conclusion, the addition of FDG PET imaging to the evaluation of tumor response to intra-arterial administration of the oncolytic virus NV1020 did not provide data useful for detecting biological activity or for prognostication. More sophisticated molecular imaging will need to be developed to monitor the effects of this novel class of antineoplastic agents.
Footnotes
Author Disclosure Statement
This clinical trial and a portion of the subset analysis presented herein were supported by MediGene, Inc. This trial was registered as ClinicalTrials.gov #NCT00149396. D.Y. Sze, A.H. Iagaru, and S.S. Gambhir were consultants to MediGene, Inc. from 2006 to 2008. H.A. De Haan is an employee of MediGene, Inc.