
Abstract
Small antisense RNAs targeted to the HIV-1 promoter have been shown to remodel the surrounding chromatin to a state unfavorable for transcriptional activation, yet transcriptional gene silencing (TGS) of HIV-1 has, to date, not been shown in primary human cells. We demonstrate here that TGS can reduce viral transcription in primary human CD4+ T cells; however, increasing viral burden results in the loss of this antiviral effect. This observation suggests a critical level at which viral RNA can dilute out effective targeting by TGS-based RNAs. Furthermore, studies into off-target effects have identified a potential interaction between the small nucleolar RNA pathway and the TGS-based antisense RNA, resulting in activation of p53. Although not overtly toxic to primary cells, this represents a novel interaction between antisense RNAs and a cellular pathway that should be considered when pursuing small antisense RNA-based therapeutics.
Introduction
RNA-based therapeutic approaches are quickly emerging as an adjunctive treatment method for controlling HIV-1. The use and study of gene therapy–based delivery approaches for HIV also remain popular, as this represents a long-term method of treatment in the control of a persistent infection (Rossi et al., 2007; DiGiusto et al., 2010). Although various anti-HIV RNAs are currently being tested in gene therapy–based approaches for HIV, none involve RNAs targeted to HIV in a TGS manner. Previous work has demonstrated that a particular small RNA, termed LTR-362as, is capable of modulating stable epigenetic changes to the HIV-1 long terminal repeat (LTR). Notably, the small RNA-susceptible target loci overlapped the conserved nuclear factor-κB (NF-κB) doublet in the HIV-1 LTR (Suzuki et al., 2005, 2008; Turner et al., 2008). To explore the ability of this small RNA to control HIV-1 in primary human cells, we used the clinically validated lentiviral vector termed HIV7-IGFP (Yam et al., 2006; DiGiusto et al., 2010). We find that while LTR-362as was capable of reducing viral replication in both cell culture and primary human CD4+ T cells, a dose-dependent response was observed. We also find that stable long-term expression of the LTR-362as, although exhibiting minor off-target effects, was relatively well tolerated by the cell. These observations suggest that although thresholds of RNA-based transcriptional regulation exist within the cell, long-term expression of the small RNA does not appreciably affect cellular function and offers promise as a future anti-HIV therapeutic.
Methods and Materials
NIH AIDS Research and Reference Reagent Program
The following reagents were obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program: Sup-T1 cells (catalog no. 100; Dr. James Hoxie) (Smith et al., 1984) and human recombinant interleukin-2 (rIL-2) (catalog no. 136; Dr. Maurice Gately, Hoffmann–La Roche Inc., Nutley, NJ).
Lentiviral vectors
The various small RNAs were cloned into the inducible BLOCK-iT vector systems from Life Technologies (Carlsbad, CA) according to manufacturer's instructions. Either the pENTR/U6/TO (362as and 362S) or the pENTR/H1/TO (sh362) plasmids were used. Primer sets p150/p175 (U6) or p174/p175 (H1) were used to PCR-amplify the promoter/RNA sequences while adding NotI restriction sites. Digested PCR products were ligated into NotI-cut pHIV7-IGFP (a gift from Dr. Jiing-Kuan Yee, City of Hope, Duarte, CA) (Yam et al., 2006), generating pHIVIGFP-362as, pHIV7IGFP-362S (antisense 362 scrambled), and pHIV7IGFP-sh362. All plasmids were confirmed by sequencing [The Scripps Research Institute (TSRI) Nucleic Acids Core Lab]: p150: 5’ TATGC GGCCGCGGTCGGGCAGGAAGAGGG; p174: 5’ TATGCG GCCGCCCCCCCTCGAAGATCTAATATTTGC; p175: 5’ TATGCGGCCGCGACTGATAGTGACCTGTTCGTTGC.
Cell culture
Sup-T1 and Jurkat cells were maintained in RPMI 1640 (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (FBS; Life Technologies) and 50 UI/ml Pen/Strep (Mediatech). Primary CD4+ T cells were maintained in RPMI 1640 supplemented with 10% heat-inactivated FBS (Life Technologies), 50 UI/ml Pen/Strep, and 30 U/ml human rIL-2. Isolated CD4+ T cells were stimulated with anti-CD3/CD28 Dynabeads (Life Technologies) immediately after isolation. New beads were added to restimulate cells at times of HX10 infection.
Detection of the 362as RNA
The small RNA fraction (>100 bp) was isolated from Sup-T1-transduced cells using RNAzol RT (Molecular Research Center Inc., Cincinnati, OH) according to the manufacturer's protocol. A total of 100 ng of isolated RNA was polyA-tailed and reverse-transcribed using the NCode kit (Life Technologies) according to the manufacturer's protocol. The LTR-362as and microRNA-16 (miR-16) were PCR-amplified using KAPA2G Fast PCR ReadyMix (Kapa Biosystems, Woburn, MA) and the following primers: NCode universal quantitative PCR (qPCR) primer (provided with kit), p060 for LTR-362as (5’-GAAAGTCCCCAGCGGAAAG), and the miR-16 detection primer (5’-TAGCAGCACGTAAATATTGGCG).
Lentiviral vector production, transductions, and MPA selection
Low-titer lentiviral vectors were produced by cotransfection of 10 μg of pMDLg/pRRE, 2.5 μg of pRSV-Rev, 5 μg of pMD2.G, and 15 μg of the various HIV7-IGFP vectors into 293T cells using calcium phosphate as previously described (Morris et al., 2004b). The vectors were titered on 293T cells and analyzed for green fluorescent protein (GFP) expression by flow cytometry (FACSCalibur II, Becton, Dickinson and Company, Franklin Lakes, NJ) to determine infectious particles (IP) per milliliter. Sup-T1 and Jurkat cells were tranduced at a multiplicity of infection (MOI) of 1 IP/cell by spinoculation at 1,000 g for 30 min. Cells were placed under 2 μM mycophenolic acid (MPA; Sigma-Aldrich, St. Louis, MO) selection 48 hr post transduction for 8 days, after which enrichment of GFP-positive cells was confirmed by flow cytometry.
High-titer lentiviral vectors were produced by the Preclinical Vector Core of the NIH Gene Therapy Resource Program administered by NHLBI Gene Therapy Group. Stimulated CD4+ cells were transduced at MOIs of 20 or 100 IP/cell. Seventy-two hours post transduction, cells were placed under 0.5 μM MPA selection. GFP enrichment was analyzed by flow cytometry.
RNA/DNA isolation and analysis
Viral RNA and total cellular RNA were isolated according to the manufacturer's instructions using the QIAamp Viral RNA Mini kit and the RNeasy Mini kit, respectively, automated by the QIAcube (QIAGEN, Valencia, CA). Cellular DNA was isolated using the QIAamp DNA mini kit automated by the QIAcube. All RNA samples subject to quantitative real-time PCR (qRT-PCR) were prepared according to the following procedure: Isolated RNA in nuclease-free water was DNase-treated using the Turbo DNA-free DNase Kit (Life Technologies) according to the manufacturer's instructions. Following treatment, samples were standardized and subject to reverse transcription PCR using Mu-MLV (Life Technologies) according to instructions. Controls not subject to reverse transcription did not receive the reverse transcriptase enzyme. All qRT-PCR was carried out using Kapa Sybr Fast universal qPCR mix (Kapa Biosystems, Woburn, MA) and an Eppendorf Mastercycler ep realplex. The following primers were used: p128 (HIV F): 5’-AGGGATGG AAAGGATCACCAGCAA-3’; p129 (HIV R): 5’-CCCACCTC AACAGATGTTGTCTCA-3’; p172 (β-actin F): 5’-AGGTCAT CACCATTGGCAATGAG-3’; p173 (β-actin R): 5’-TCTTTG CGGATGTCCACGTCA-3’; p113 (DNA intron F): 5’-AGCC CTCAGGGAGCTTACGATTTA-3’; p114 (DNA intron R): 5’-AACCCTTCATCACTCTCCTTTGGC-3’.
Thermal cycling parameters started with 3 min at 95°C, followed by 40 cycles of 95°C for 3 sec and 60°C for 30 sec. Specificity of the PCR products was verified by melting-curve analysis.
Serial passage of Sup-T1 cells
Transduced Sup-T1 cells were plated at a density of 2×105 cells/ml and infected in triplicate with HX10 at an MOI of 0.001 or 0.01 [azidothymidine (AZT)-treated] TCID50/cell. Twenty-four hours post infection, cells were washed with Dulbecco's phosphate-buffered saline and replated in 2 ml of RPMI. For AZT-treated cells, AZT was added to a final concentration of 1 μM. Fresh AZT was added every 2 days for the duration of the experiment. Supernatant samples for p24 analysis and cell pellets for RNA/DNA isolation for both experiments were collected every 3 days post infection and stored at −80°C until analyzed. Cells were split 1:10 every 3 days.
Serial passage of CD4+ T cells/population doubling
MPA-selected CD4+ cells were plated at a density of 5×105 cells/ml in 2 ml of RPMI with CD3/CD28 beads at a ratio of 1 bead:2 cells. Cells were infected in triplicate with HX10 at an MOI of 0.001 TCID50/cell and washed 24 hr post infection. Every 3 days, supernatant samples were collected for p24, and total live cells were determined by trypan blue staining using the Countess Automated Cell Counter (Life Technologies). Population-doubling assays represent the average of the triplicate infected cultures. Cells were then split and replated at a density of 5×105 cells/ml in 2 ml of RPMI. Flow-sorted CD4+ cells were plated at a density of 5×105 cells/ml in 1 ml of RPMI with CD3/CD28 beads at a ratio of 1 bead:2 cells. Experimental conditions were the same as stated above.
p24 analysis
Samples were neutralized by lysis in Triton X-100 at a final concentration of 0.5%. Viral p24 analysis was performed by the Translational Virology Core at the University of California–San Diego Center for AIDS Research (AI36214) (with support from the VA San Diego Healthcare System and the Veterans Medical Research Foundation) using the PerkinElmer Extended Curve p24 ELISA assay.
Flow cytometry
Cells (5×105) were collected and fixed in 1% formaldehyde post staining for all experiments. CD4 was stained using anti-CD4-APC (MHCD0405; Life Technologies). Stained/GFP-positive cells were analyzed using a BD FACSCalibur II. Cell sorting was performed by the TSRI Flow Cytometry Core using a BD FACSAria.
Microarray analysis
Sup-T1 cells were transduced in triplicate with either HIV7-IGFP or HIV7IGFP-362as at MOIs of 1 IP/cell and selected with 2 μM MPA for 8 days, followed by 8 days of culture without MPA. RNA was isolated using TRIzol Reagent (Life Technologies) according to the manufacturer's protocol. Isolated RNA was then run through RNeasy Mini columns (QIAGEN) to remove any trace phenol contamination. RNA samples were run on HuGene-1_0-st-v1 Affymetrix chips by the TSRI DNA Array Core. Data normalization was performed using RMA Express 1.0 (
SNORD45b and 362as methylation detection
RNA isolated for microarray analysis was subject to the NCode protocol according to instructions or to RT-PCR as described above (see RNA/DNA isolation and analysis). SNORD45b was detected by qRT-PCR using the NCode universal qPCR primer and p187 5’-GCT GAA TCT AAA GTT GAT GTG AGT TCT-3’ or with p182 (SNORD F) 5’-GGT CAA TGA TGT AAT GGC ATG T-3’ and p183 (SNORD R) 5’-GAA CTC ACA TCA ACT TTA GAT TCA GC-3’. To examine LTR-362as methylation, the small RNA fraction of Sup-T1-transduced cells isolated for LTR-362as detection was subject to RT-PCR using the NCode universal RT primer in varying concentrations of dNTPs (1, 0.1, and 0.01 mM). LTR-362 expression relative to miR-16 expression was determined by qRT-PCR with the same primers used for LTR-362as detection.
Western blots
Cells were lysed in RIPA buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with Ambion Turbo DNase and RNase A (Life Technologies) for 30 min on ice. Lysed cells were spun for 10 min at 13,000 rpm to pellet cell debris. Protein-containing supernatants were assayed for protein concentrations by BCA assay (Pierce, ThermoFisher, Rockford, IL) according to the manufacturer's instructions. Proteins were separated by 4–12% NuPage Bis-Tris acrylamide gel (Life Technologies) and transferred onto Immun-Blot PVDF membranes (Bio-Rad, Hercules, CA). Membranes were blocked with Tris-buffered saline (TBS) and 5% milk. For p21, blots were blocked in TBS with 0.5% Tween-20 (TBST) with 5% bovine serum albumin. Membranes were probed with anti-p53 (mouse monoclonal clone DO-1, 1:1,000 dilution; Santa Cruz Biotechnologies, Santa Cruz, CA) or anti-GAPDH (mouse monoclonal, 1:5,000 dilution; Millipore, Temecula, CA) in TBS with 0.5% Tween-20 and 5% milk overnight at 4°C. For p21, membranes were probed with anti-p21 Waf1/Cip1 (mouse monoclonal clone DCS60, 1:2,000 dilution; Cell Signaling Technology, Boston, MA) and anti-β-actin (rabbit monoclonal clone 13E5, 1:1,000 dilution; Cell Signaling Technology) in TBST overnight at 4°C. Primary antibody binding was detected with horseradish peroxidase–conjugated secondary antibodies and HyGlo Quick Spray chemiluminescent substrate (Denville Scientific, Metuchun, NJ). Blots were exposed to HyBlot CL film (Denville Scientific) for autoradiography. Membranes were stripped as needed in 25 mM glycine, pH 2.0, and 1% SDS for 30 min at room temperature, followed by reblocking in TBS with 5% milk. Expression was quantified using ImageJ software (
Results
Numerous studies have shown that targeting of small noncoding RNAs to promoter regions can result in epigenetic modifications, such as histone and DNA methylation, and lead to a permanent and heritable shutdown of gene expression (for review, see Turner and Morris, 2010). To date, no studies have examined the ability of TGS-based therapeutics to target and transcriptionally silence HIV in primary human cells. Toward this goal, the previously characterized LTR-362 antisense RNA (LTR-362as) was introduced into the self-inactivating HIV-1-based lentiviral vector containing a drug-resistant mutant of the inosine monophosphate dehydrogenase 2 (IMPDH2) gene (HIV7-IGFP; Fig. 1A) (Yam et al., 2006). IMPDH is an enzyme involved in the de novo purine synthesis pathway that converts inosine monophosphate to xanthosine monophosphate, critical for deoxyguanosine triphosphate synthesis (Fig. 1B) (Allison and Eugui, 2000). Isoform 2 (IMPDH2) is preferentially expressed in activated lymphocytes and is five times more sensitive to MPA, an inhibitor of IMPDH (Fig. 1B) (Allison and Eugui, 2000). Notably, MPA is also a clinically relevant compound under investigation as an accompaniment to HIV-1 antiretrovirals (Chapuis et al., 2000; Coull et al., 2001; Hossain et al., 2002). In addition to the LTR-362as (small antisense RNA), a short hairpin version variant of the LTR-362 (LTR-sh362) was also introduced (Fig. 1A). The empty parental vector served as the primary control, due to consistent observations that a scrambled RNA variant (362S) caused off-target effects in human cells (data not shown). The transduced cells treated with MPA showed robust enrichment of GFP expression after 8 days (Fig. 1C), indicating efficient selection of cells expressing the IMPDH2 mutant gene. This allowed the establishment of stable cell lines in which small RNA expression could be reproducibly detected (Fig. 1D).

Selection and detection of the LTR-362.
To assess viral suppression, LTR-362as-expressing and control Sup-T1 cell lines were infected in triplicate and monitored for 9 days. LTR-362as cell lines demonstrated over 10-fold lower p24 levels as compared with control lines at day 9, with one LTR-362as replicate showing viral breakthrough at day 9 (Fig. 2A). RNA from shed virus of the breakthrough replicate was isolated and assessed for mutational analysis of the LTR-362as target site. Previous studies examining small double-stranded RNAs targeted to the coding regions of HIV have observed rapid viral mutation and loss of effective small interfering RNA (siRNA) targeting (ter Brake et al., 2006). In the case of the LTR-362as, no mutations were detected in the viral RNA, suggesting that breakthrough of virus was not due to evolution of the virus to avoid targeting at this particular loci (Supplementary Fig. S1A; Supplementary Data are available online at
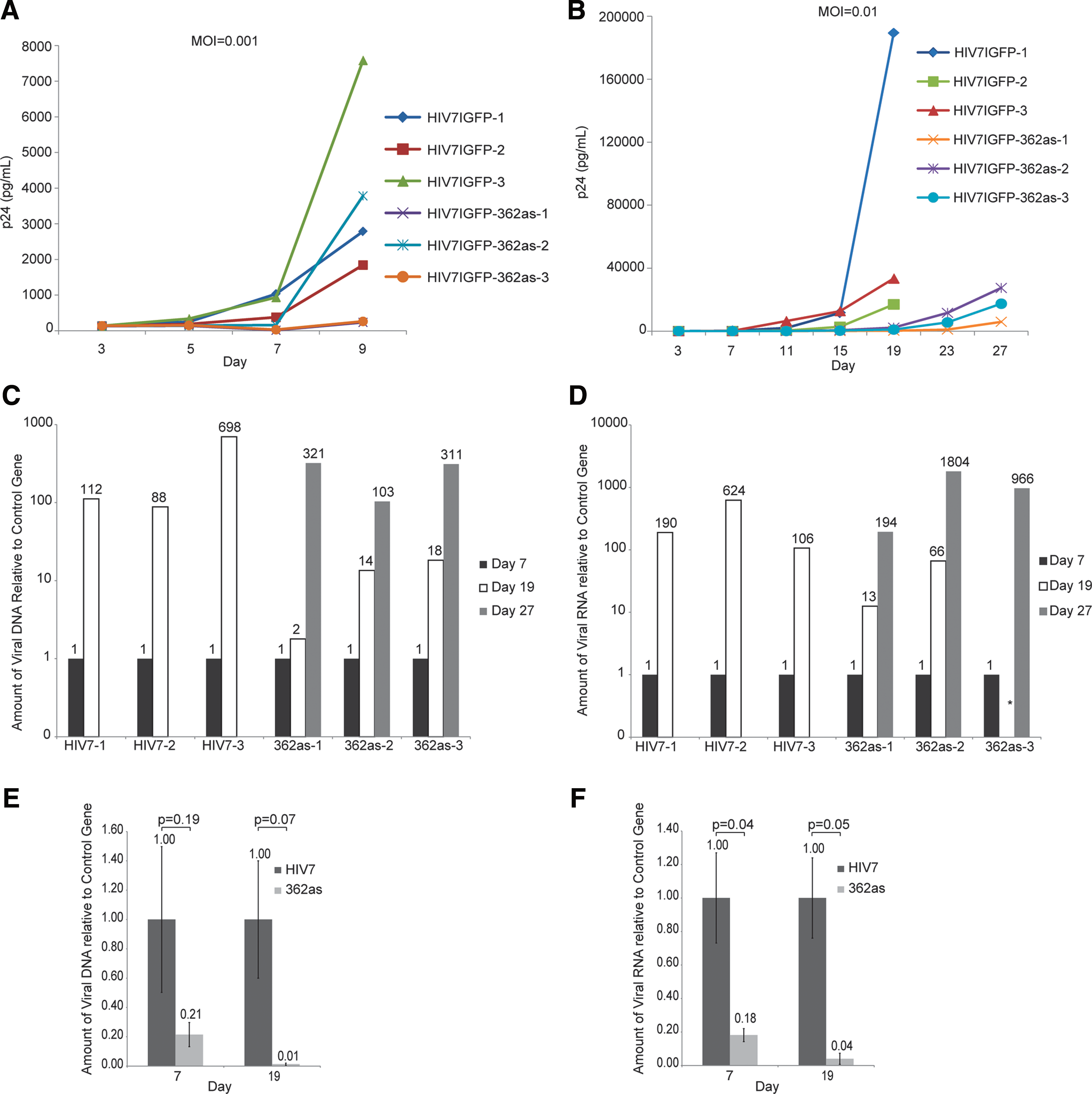
Targeting by LTR-362 in combination with AZT.
To determine if LTR-362as targeting was diluted over time due to increasing levels of viral replication in cell-culture conditions, a small-molecule antiretroviral drug was used to slow the active rate of replication. LTR-362as and control Sup-T1 cell lines were infected in triplicate with HIV and followed by treatment with AZT (1 μM, 24 hr post infection). Under these conditions, LTR-362as-transduced cells were capable of controlling HIV for 27 days, 8 days longer than controls (Fig. 2B). Suggestive of a model in which shed virus is continually reinfecting cells in culture, analysis of DNA isolated from both control and 362-expressing cells showed steadily increasing amounts of viral DNA relative to a two-copy control gene over time (Fig. 2C). It should be noted that the method of DNA isolation could also result in amplification of nonintegrated forms of HIV. Notably however, the levels of viral DNA were reduced in LTR-362as-treated cells as compared with the control at day 19 and similar to the controls by day 27 (Fig. 2C). These observations correlated with similar levels of p24 and viral RNA between these two time points (Fig. 2B and D). The observations did not appear to be the result of differences in the initial rate of infection (Fig. 2E and F) and suggest that a threshold of viral replication exists at which point the LTR-362as becomes ineffective in controlling viral replication. As cell-culture models tend to foster high replication of HIV-1, we wondered whether similar observations would be found in primary human cells.
Primary human CD4+ T cells were transduced with the LTR-362as or control vector (HIV7IGFP), MPA-selected (Supplementary Fig. S2), and infected with HIV. Untransduced, non–MPA-selected CD4+ cells were also infected in parallel. No suppression of viral p24 was observed in the LTR-362-expressing CD4+ cells (Fig. 3A). Rather, LTR-362as CD4+ cells lost GFP expression at twice the rate of the control HIV7IGFP cells, suggesting that a negative selection against 362as-expressing cells was occurring (Fig. 3B). Furthermore, a loss of 362as-expressing cells was observed within 72 hr of stimulation with anti-CD3/CD28 beads, after which cell doubling recovered (Fig. 3C). This negative population doubling was also found in uninfected CD4-362as cells relative to the controls, suggesting that viral infection was irrelevant (Fig. 3D). These data indicated that expression of the LTR-362as in primary cells was resulting in a potentially toxic secondary effect. To determine possible secondary targeting of host transcripts by LTR-362as, microarray experiments were carried out (Supplementary Fig. S3). Only two genes showed significant changes (up-regulation) in expression in the LTR-362as-expressing samples as compared with the control: advillin and SNORD45b (Fig. 4A).
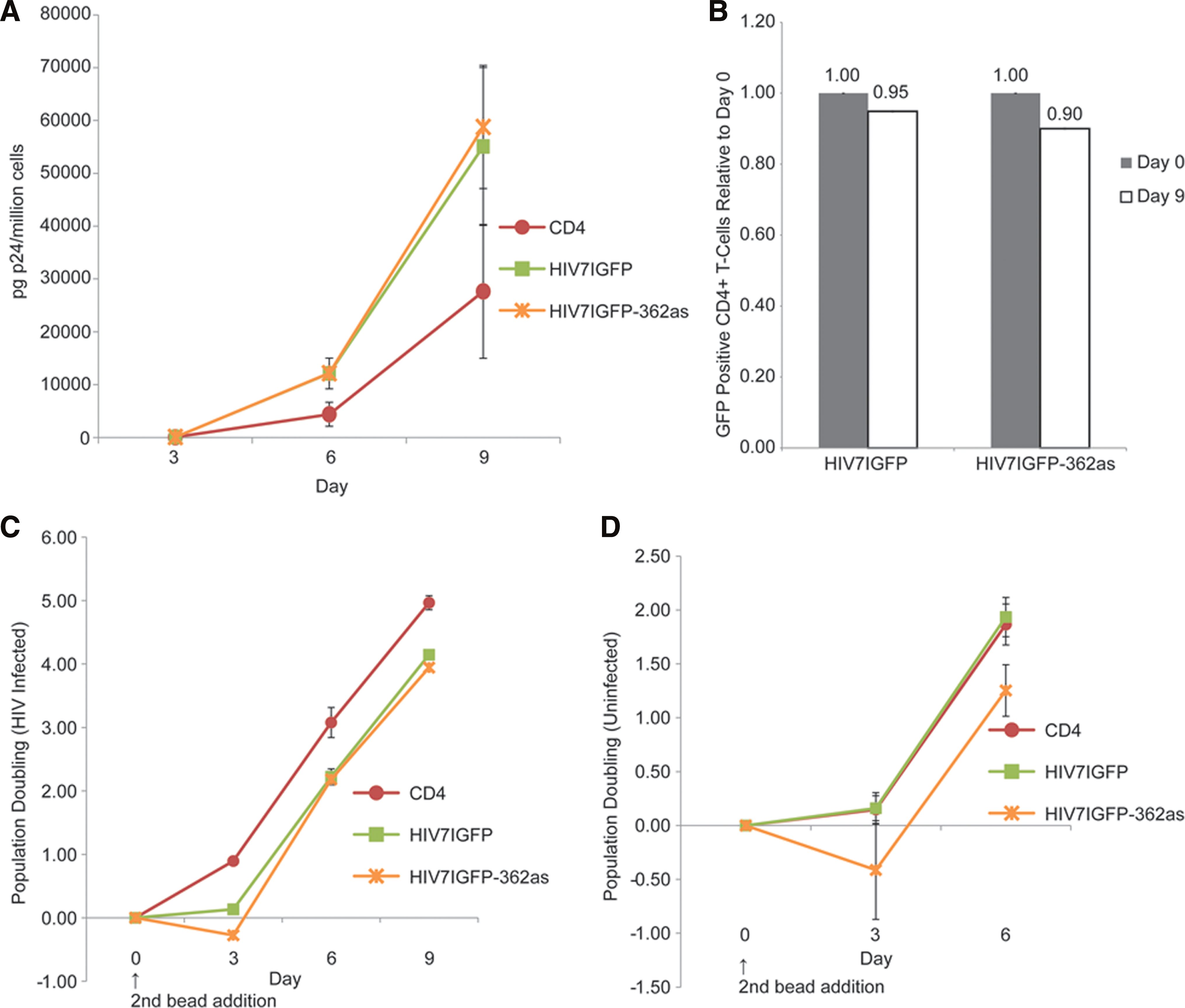
Targeting of HIV by LTR-362as in primary T-cells.

Off-target effects of LTR-362as.
Advillin is an actin-binding protein not shown to be highly expressed in leukocytes (Marks et al., 1998). Furthermore, the transcript shown to be up-regulated was not the annotated advillin mRNA and, as such, we chose not to pursue this target. SNORD45b, a 71-nt noncoding RNA, is a member of the box C/D small nucleolar RNA (snoRNA) family (Kiss-László et al., 1996). Box C/D proteins contain two conserved elements, box C (RUGAUGA) and box D (CUGA), which exist at the 5’ and 3’ ends, respectively (Huang et al., 2011). These RNAs exist in the cell as parts of a ribonucleoprotein (RNP) complex containing Nop1p (fibrillarin), Nop56p, Nop58p, and Snu13p (Huang et al., 2011) and are involved in directing site-specific 2’-O-methylation of ribosomal RNA (rRNA) by binding of the snoRNAs to complementary sequences in the rRNA (Kiss-László et al., 1996). SNORD45b is also closely related to SNORD45a and SNORD45c, as all contain overlapping rRNA binding sites, suggesting a degree of redundancy in the snoRNA system (Fig. 4B and C) (Kiss-László et al., 1996). To determine if SNORD45b was interacting with the LTR-362as RNA and possibly directing RNP-mediated methylation of LTR-362, we performed reverse transcription on purified SupT1-362as cellular RNA using differing concentrations of dNTPs. At low concentrations of dNTPs, it has previously been shown that the reverse transcriptase enzyme is inefficient at transcribing past sites of 2’-O-methylation whereby RT-PCR at normal levels of dNTPs (1 mM) is not affected (Rebane et al., 2002; Huang and Yu, 2010). A notable 25% reduction in LTR-362as detection was observed at lower concentrations (Fig. 4D), suggesting that the LTR-362as may be methylated.
These observations suggested that the LTR-362as could interact, and potentially interfere, with the small nucleolar RNA pathway, indicating a potential cause of the toxicity observed in primary CD4+ T cells (Fig. 3). Ribosomal stress has been shown to result in the release of ribosomal proteins L5, L11, and L23, which can bind and inhibit the activity of MDM2, the protein responsible for the ubiquitination and degradation of p53 (Bhat et al., 2004; Dai et al., 2004; Jin et al., 2004; Gilkes et al., 2006; Hölzel et al., 2010). Stabilization and activation of p53 can result in cell-cycle arrest or transcription of proapoptotic genes, leading to cell death (Bhat et al., 2004; Dai et al., 2004; Jin et al., 2004; Gilkes et al., 2006; Hölzel et al., 2010). To determine if ribosomal stress was resulting in the loss of LTR-362as-expressing CD4+ cells, p53 expression in MPA-selected CD4+ cells prior to and after stimulation with anti-CD3/CD28 beads was analyzed. A twofold increase of p53 levels was observed at early time points in LTR-362as-treated CD4+ cells as compared with untransduced CD4+ cells (Fig. 5A). This increase in p53 was also observed to a lesser extent in Sup-T1 cells (Fig. 5B). However, MPA has also been shown to activate the ribosomal stress pathway and result in the induction of p53 (Sun et al., 2008), explaining the observation of elevated p53 levels in MPA-selected control CD4-HIV7 cells as compared with untransduced cells. To ensure that the elevated p53 activation observed in LTR-362as-treated cells was due to RNA expression and not MPA selection, CD4+ LTR-362as and LTR-sh362 cells were subject to fluorescence-assisted cell sorting (FACS) and were then assessed by western-blot analysis. Similar levels of p53 activation were observed only in cells transduced at the high MOIs (Fig. 5C; 100 IP/cell). Furthermore, levels of p21, a gene tightly regulated by p53 (El-Deiry et al., 1993), were increased in cells expressing both forms of the LTR-362, but not in control cells (Fig. 5D). The possibility of multiple integration events is likely in cells transduced with higher concentrations of vector particles, suggesting that p53 activation is dependent on the level of the LTR-362 expressed in the cell. Supporting this notion was the observation that the most profound anti-HIV-1 effects in both LTR-362as and LTR-sh362 CD4+ cells were observed at high MOI (Fig. 5E), whereas no antiviral effect was observed at the lower MOI (data not shown). Interestingly, CD4+ cells transduced with the LTR-sh362 construct exhibited reduced population doubling as compared with both the HIV7 control and the LTR-362as (see Supplementary Fig. S4A). Previous studies have demonstrated that high expression of short hairpin RNA (shRNA) constructs can lead to oversaturation of the endogenous microRNA (miRNA) pathway and result in cellular toxicity (Grimm et al., 2006). These observations suggest that expression of the 362 RNA as a hairpin may negatively impact CD4 cell growth, whereas expression as an antisense RNA avoids this pathway of toxicity. Although expression of both forms of the LTR-362 RNAs results in activation of p53, it remains possible that the pathway of activation is different between the two forms.

p53 expression in LTR-362-transduced CD4+ T-cells.
Discussion
The use of RNA-based modalities as therapeutics against HIV and other diseases continues to be a major trend in drug development. We have previously studied the mechanistic aspects of a small antisense RNA, the LTR-362as, which targets a conserved region within the HIV-1 LTR (Suzuki et al., 2005, 2008; Turner et al., 2008). The LTR-362as and the siRNA promA, an RNA shifted only 4 bp upstream from the LTR-362as, have been shown to remodel the chromatin surrounding the HIV-1 LTR by increasing both histone and DNA methylation and resulting in the loss of NF-κB recruitment (Suzuki et al., 2005, 2008; Turner et al., 2008). This continuing study on the LTR-362as represents the first assessment of the ability of TGS-based small RNAs to target HIV-1 in primary human cells. We show here that both the small antisense RNA and short hairpin form of the LTR-362 are capable of a modest suppression of viral replication in HIV-1-infected human CD4+ T cells. Effective targeting by the LTR-362as is dose-dependent, as increasing levels of viral replication can slowly overwhelm targeting.
Expression of the LTR-362 as a hairpin appears to improve antiviral targeting in CD4+ cells. The LTR-sh362 can act both in a TGS manner by targeting the integrated virus and in a posttranscriptional gene-silencing (PTGS) manner by targeting the 3’ LTR of the viral RNA. It has previously been shown that shRNAs and miRNAs can act through the TGS pathway to remodel chromatin (Castanotto et al., 2005; D.H. Kim et al., 2006, 2008; Weinberg et al., 2006; J.-W. Kim et al., 2007; Hawkins et al., 2009). Thus, the ability of the LTR-sh362 to act through PTGS targeting of the virus may improve antiviral efficacy by additionally reducing viral replication and allowing time for the establishment of TGS (D.H. Kim et al., 2006). However, this presents the increased possibility of viral mutation in response to PTGS-based targeting, which could also impact the ability of the LTR-sh362 to target in a TGS manner. We have observed that the LTR-362as is capable of tolerating up to four mismatches and still maintain some level of TGS-based targeting (Knowling et al., unpublished observations), suggesting that significant mutation would be required, which could impact viral fitness.
The observation of the virus overwhelming the TGS-based RNA effect indicates that a combination of TGS-based RNAs with other anti-HIV therapies, such as small-molecule drugs, or other RNA-based modalities could improve the establishment of TGS. Furthermore, the level of active viral replication in HIV-infected individuals is notably lower than in cell-culture systems, suggesting that the LTR-362 could be more effective in controlling virus than what has been observed in these systems. These observations overall present an interesting finding for the field of TGS and epigenetic-based regulation of HIV-1, suggesting that a critical level of the therapeutic RNA must be expressed relative to the target gene to effectively target in a TGS-based manner. One possible avenue to achieving this balance would be through the use of an HIV-inducible lentiviral vector (Unwalla et al., 2004).
Equally interesting is the observation that LTR-362as interacts with the SNORD pathway, resulting in low-level activation of p53 expression in primary cells. Meanwhile, the LTR-sh362-treated cultures demonstrated a different form of secondary effect, namely, a reduction in population doubling. However, both forms of the LTR-362 were capable of activating p21. Taken together, these data indicate that both forms of the LTR-362 RNAs exhibit adverse cellular effects, which ultimately lead to p53 and p21 activation, but the pathways of activation may be different. Although neither form showed high levels of toxicity, these observations should be carefully considered when introducing small RNAs into T-cell populations, as these toxic effects could build up over time in resting cells and impede proper activation and immune responses. Indeed, other promoter-targeted antisense RNAs have been shown to demonstrate off-target effects (Weinberg et al., 2007; Moses et al., 2010), suggesting that this form of targeting is not immune to the aberrant targeting problems also seen with siRNAs and miRNAs. Such adverse effects can, however, be addressed with the use of selectable vector systems, such as the MPA-selectable system presented here (Yam et al., 2006) or with HIV-1-inducible TGS-targeted small RNAs (Unwalla et al., 2004), allowing greater control of RNA expression levels. Although used primarily for selection of vector-containing cells in this study, the use of drug-selectable gene-therapy vectors has great potential. Current gene-therapy trials have commonly noted a loss of detectable vector-containing cells over time (Levine et al., 2006; DiGiusto et al., 2010). The use of a drug-selectable vector could aid in providing transduced cells with a selective advantage and improve antiviral efficacy of gene therapy–based approaches. Furthermore, the introduction of the IMPDH mutant protein into a mobilization-competent lentiviral vector (Morris and Looney, 2005; Turner et al., 2008) could prove highly efficacious in providing a sustained selective pressure for mobilization and result in increased amounts of protected cells over time in HIV-1-infected individuals. A major challenge will be to examine the potential for selectable vectors in therapeutic settings, and whether these vectors, in combination with other anti-HIV therapeutics currently in use, result in an improved sustained antiviral effect.
Footnotes
Acknowledgments
This work was supported by NIH R01HL083473 and R01AI084406 to K.V.M. and by a fellowship from the George E. Hewitt Foundation for Medical Research to M.A.M. This manuscript is TSRI no. 21491. We would like to acknowledge the Gene Therapy Resource Program (GTRP) of the National Heart, Lung, and Blood Institute, National Institutes of Health for providing the gene vectors used in this study.
Author Disclosure Statement
The authors have nothing to disclose and no conflicts of interest with this body of work.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.