
Abstract
Spinal muscular atrophy (SMA), an autosomal recessive neuromuscular disorder, is the leading genetic cause of infant mortality. SMA is caused by the homozygous loss of Survival Motor Neuron-1 (SMN1). In humans, a nearly identical copy gene is present, SMN2. SMN2 is retained in all SMA patients and encodes the same protein as SMN1. However, SMN1 and SMN2 differ by a silent C-to-T transition at the 5’ end of exon 7, causing alternative splicing of SMN2 transcripts and low levels of full-length SMN. SMA is monogenic and therefore well suited for gene-replacement strategies. Recently, self-complementary adeno-associated virus (scAAV) vectors have been used to deliver the SMN cDNA to an animal model of disease, the SMNΔ7 mouse. In this study, we examine a severe model of SMA, Smn –/–;SMN2 +/+, to determine whether gene replacement is viable in a model in which disease development begins in utero. Using two delivery paradigms, intracerebroventricular injections and intravenous injections, we delivered scAAV9-SMN and demonstrated a two to four fold increase in survival, in addition to improving many of the phenotypic parameters of the model. This represents the longest extension in survival for this severe model for any therapeutic intervention and suggests that postsymptomatic treatment of SMA may lead to significant improvement of disease severity.
Introduction
In this report, we use a severe animal model of SMA referred to as the SMN2 model (Monani et al., 2000). In this model, the human SMN2 gene has been added to a mouse Smn null background (Smn –/–;SMN2 +/+) (Monani et al., 2000). These mice are born symptomatic, gain minimal body weight, and live approximately 4–6 days. Recently, this model has been shown to exhibit nonneuronal defects, including cardiac dysfunction and disrupted hippocampal development (Shababi et al., 2010b; Wishart et al., 2010). Due to the severity of the model, improving its phenotype via therapeutics has proven to be difficult. To date, the best improvement in the SMN2 model has been modest increases in weight and survival (Baughan et al., 2009; Coady and Lorson, 2010).
We chose to use the SMN2 model as a means to determine whether a symptomatic model of disease could be completely rescued using scAAV9-SMN. We delivered scAAV9-SMN at P1 to SMA mice via intracerebroventricular (ICV) injections and intravenous (IV) temporal vein injections. Serotype 9 was selected based on its ability to transduce a variety of neuronal lineages, including motor neurons (Duque et al., 2009; Foust et al., 2009). In both treatment groups, SMN protein levels were significantly increased in the brain and spinal cord, and treated animals survived significantly longer than untreated SMA mice. Additionally, ICV delivery of the vector resulted in a greater extension of life span compared with SMA animals that received the same dose via IV injection. Collectively, this study demonstrates the most profound improvement seen in the SMN2 model and illustrates that postsymptomatic treatment of SMA can significantly lessen disease severity.
Materials and Methods
Genotyping and mouse handling
Animals were handled according to the University of Missouri Animal Care and Use Committee approved protocols. Genotyping was performed as previously described (Coady and Lorson, 2010). SMA mice, both treated and untreated controls, were raised with two SMN heterozygous siblings to control for litter size.
Tissue collection
Dissections were as done as follows: The vertebral column was separated from the torso, and the spinal cord was excised and immediately frozen in liquid nitrogen. The brain was removed from the skull and divided into four equal sections, and each section was immediately frozen.
Muscle fiber size analysis
The hind limbs of each animal were removed at the highest point possible. The foot was bent at a 90° angle, and the tissue was fixed in 4% paraformaldehyde overnight at 4°C to control for differences in muscle stretching. After sectioning, fibers were stained with hematoxylin and eosin and quantitated as previously described (Avila et al., 2007).
Western blotting
Spinal cord or brain tissues were harvested at P5 from heterozygous or SMA pups (either scAAV9-SMN treated or untreated controls) and immediately frozen, as described above. Westerns were quantitated using Fujifilm MultiGauge software. Mouse monoclonal anti-SMN antibody (BD Biolabs; 1:2,000) and anti-IP90 polyclonal rabbit antibody (1:2,000) were used for SMN and calnexin detection, respectively (Mattis et al., 2006; Shababi et al., 2011).
Production of the scAAV9-SMN virus
scAAV9-SMN was produced via triple transfection in HEK 293T cells using polyethyleneimine prepared to a working concentration of 1 mg/ml, pH 5.0 (Grieger et al., 2006). The scAAV plasmid was constructed to express the open reading frame of SMN1 cDNA (NCBI accession number NM_000344) under the control of the chicken β-actin promoter. The vector was purified by two cesium chloride density-gradient ultracentrifugation steps and dialyzed against HEPES buffer. Viral particles were titered by quantitative PCR using SYBR Green.
In vivo injections
On P1, SMA (Smn –/–;SMN2 +/+) mice were injected with 2×1011 viral particles of scAAV9-SMN. Due to the volume restrictions of the ICV technique, only ∼7 μl can be injected into the ventricle at one time; therefore, two injections were given to obtain a titer of 2×1011 viral particles. Animals chosen for ICV injections were injected twice on P1, with at least 1 hr between injections. Animals chosen for IV injection received a single injection on P1. Injections were visualized for accuracy by the addition of filter-sterilized green food dye.
Results
SMN protein is significantly elevated in the CNS of ICV and IV treated SMA mice
To determine whether scAAV9-SMN delivery is able to correct a severe model of disease after the initiation of disease symptoms, we used the SMN2 model (Smn –/–;SMN2 +/+). These severe SMA mice are born symptomatic, having a marked decrease in motor neurons and weighing less than wild-type animals. The disease is so severe that many of the SMA embryos are absorbed in utero (Shababi et al., 2010b). We began by administering 2×1011 viral particles of scAAV-SMN via ICV and IV temporal vein injections. Injections were administered within 24 hr of birth, and animals were weighed daily until death. Because SMA is primarily a central nervous system (CNS) disease, increasing SMN protein levels in the brain and spinal cord is desirable. To determine if ICV and IV administration of scAAV9-SMN increased SMN levels, we performed Western blots on extracts derived from brain and spinal cord tissue of scAAV9-SMN treated animals, untreated controls, and unaffected animals. Initially, it was important to provide a comparison with the untreated SMA animals; therefore, due to the short life span of this model, we selected a relatively early time point, P5. As expected, SMN levels are very low in untreated animals in both the brain and spinal cord extracts (Fig. 1). In each of the brain and spinal cord extracts from ICV and IV treated animals, significantly higher levels of total SMN protein were detected at P5 compared with untreated controls. In the brain (Fig.1A), SMN protein expression in the heterozygous animals (Smn +/–;SMN2 +/+) was nearly five fold higher than that in untreated SMA animals (Smn –/–;SMN2 +/+), whereas ICV and IV treated SMA animals displayed a five fold increase over untreated SMA controls. In the spinal cord, ICV and IV treated extracts contained nearly six and four fold more SMN protein over untreated SMA levels, respectively. These results demonstrate that the scAAV9-SMN vector can be efficiently delivered and used to express high levels of SMN in disease-relevant tissues, even in the context of an advanced symptomatic stage of disease.
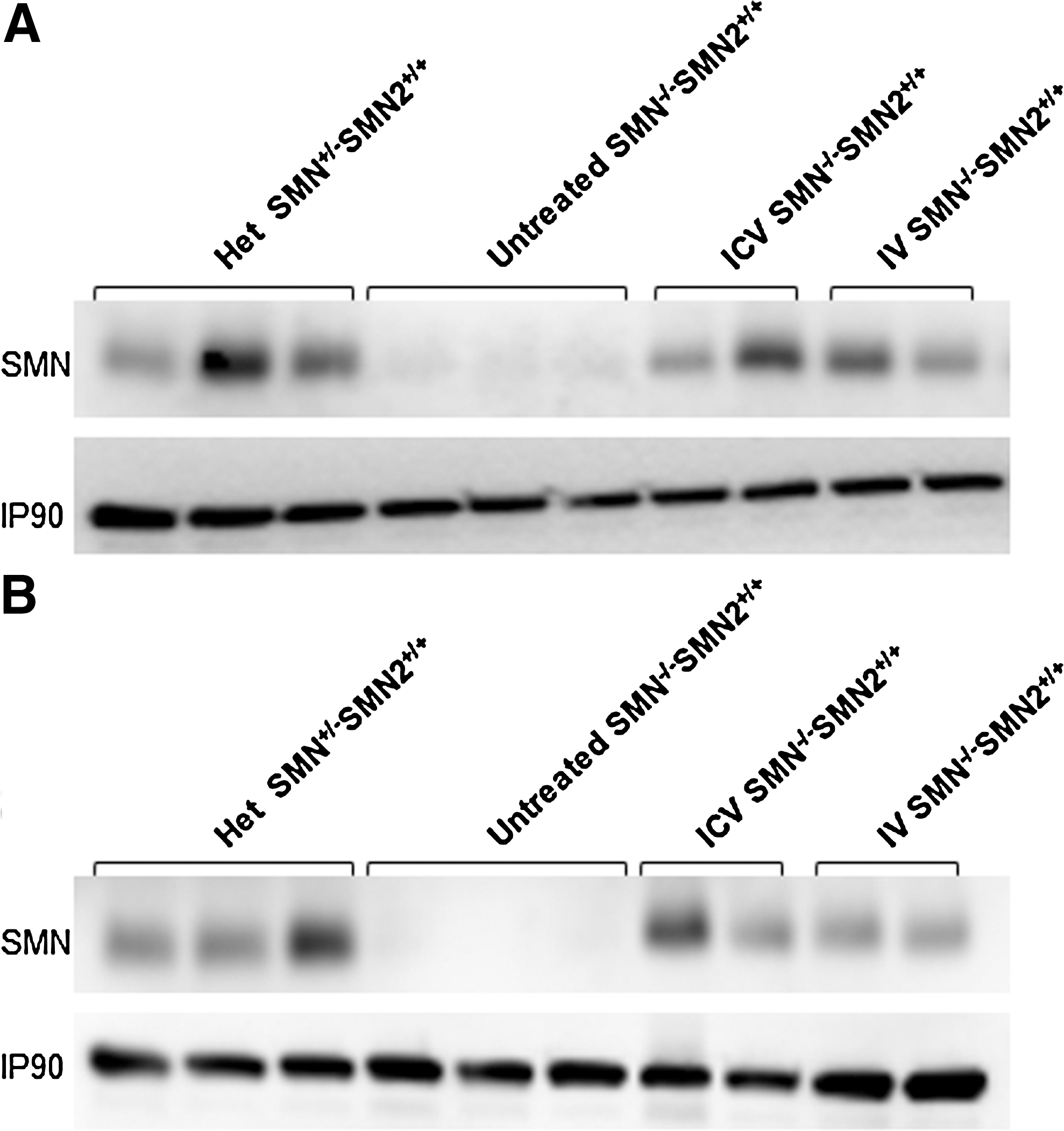
SMN protein expression was increased to near-normal levels following treatment with scAAV9-SMN. Western blots of
Muscle fiber size is moderately increased in treated animals
To determine if the increase in SMN resulted in improved muscle morphology in the treated animals, we harvested animals at P5 to examine the average size of their tibialis anterior muscle fibers. Although there is minimal improvement in IV treated animals, ICV treated animals display a slight improvement in muscle fiber area (Fig. 2A). From the muscle sections, we were able to see that the tibialis anterior muscle was generally smaller in size in the untreated animals as compared with that in the treated and unaffected animals (Fig. 2B–E). Despite the improvement in muscle fiber size in the treated mice, gross motor function was not dramatically improved as animals living past 10 days were able to walk only short distances before fatiguing (data not shown).
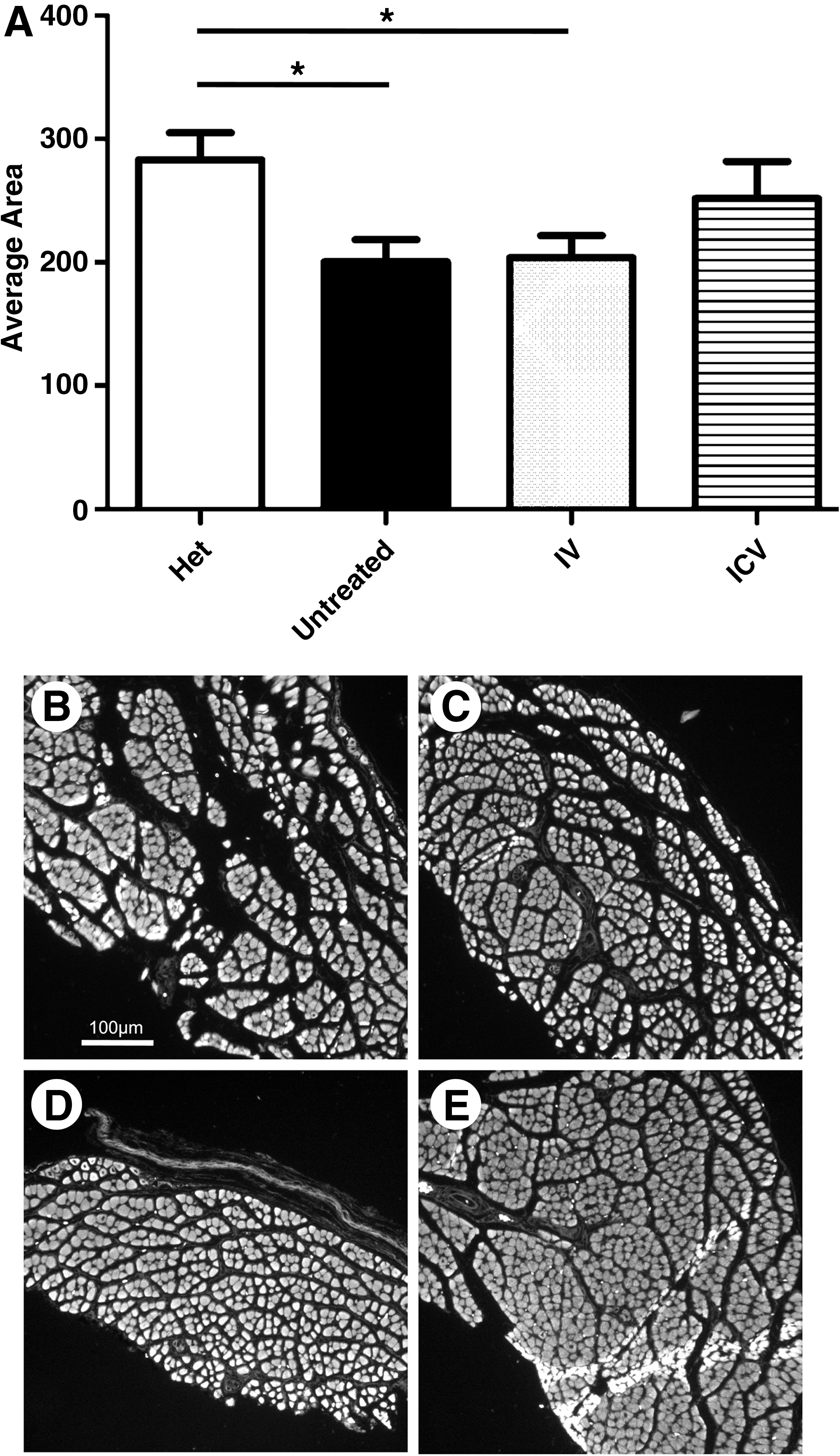
Treatment with scAAV9-SMN resulted in increased muscle fiber size.
scAAV9-SMN–treated animals gain significantly more weight
Untreated SMA animals gain minimal body weight during their life span. They are typically born at ∼1.5 g and reach a maximum weight of ∼2.2 g, averaging a weight gain of ∼19% (Fig. 3). In contrast, ICV and IV treated animals were able to achieve 3-4 g on average, and in some instances exceeded 4 g. From P3 to their peak weight, ICV treated animals gained an average of 85% of their body weight and IV treated animals gained 27%, compared with the ∼19% increase of the untreated SMA controls (Fig. 3B).
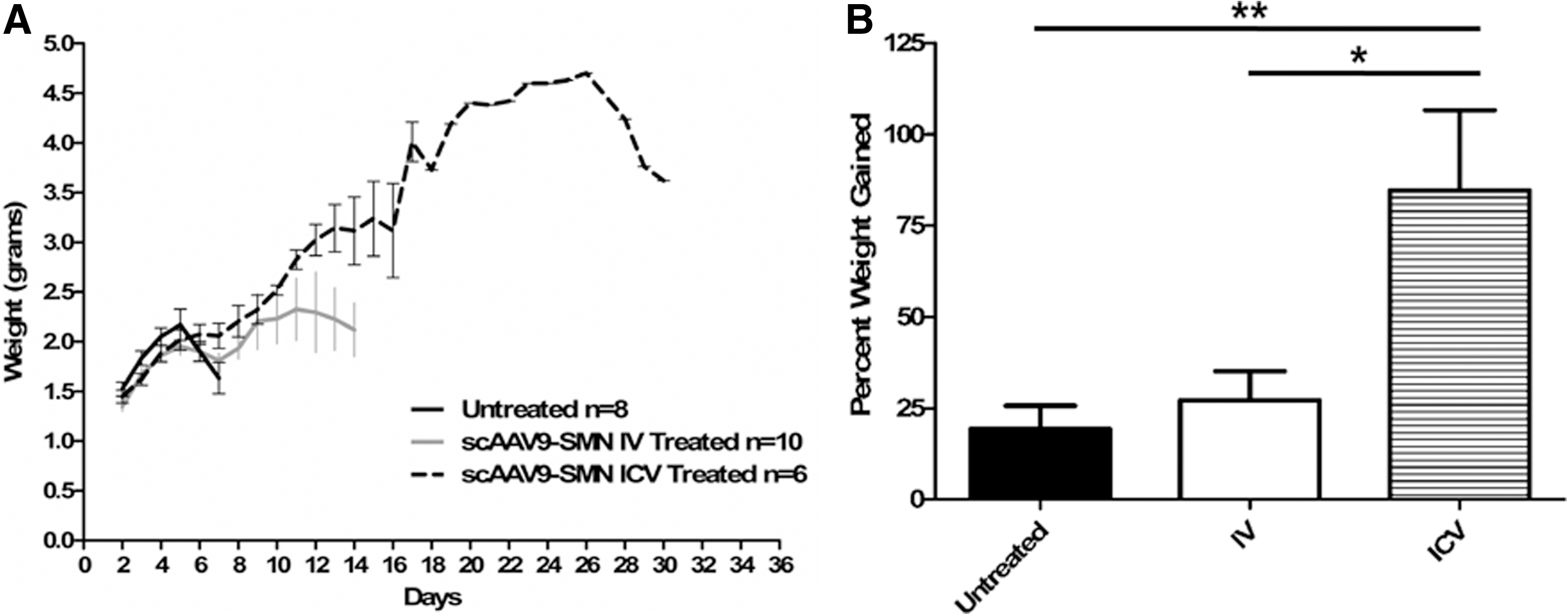
scAAV9-SMN treated animals gained more weight than untreated SMA animals.
scAAV9-SMN–treated animals live significantly longer
In addition to being weighed daily, the animals were monitored for survival. The animals that received the scAAV9-SMN via ICV and IV injections lived significantly longer than the untreated controls (Fig. 4A and Supplementary Fig. S1; Supplementary Data are available online at
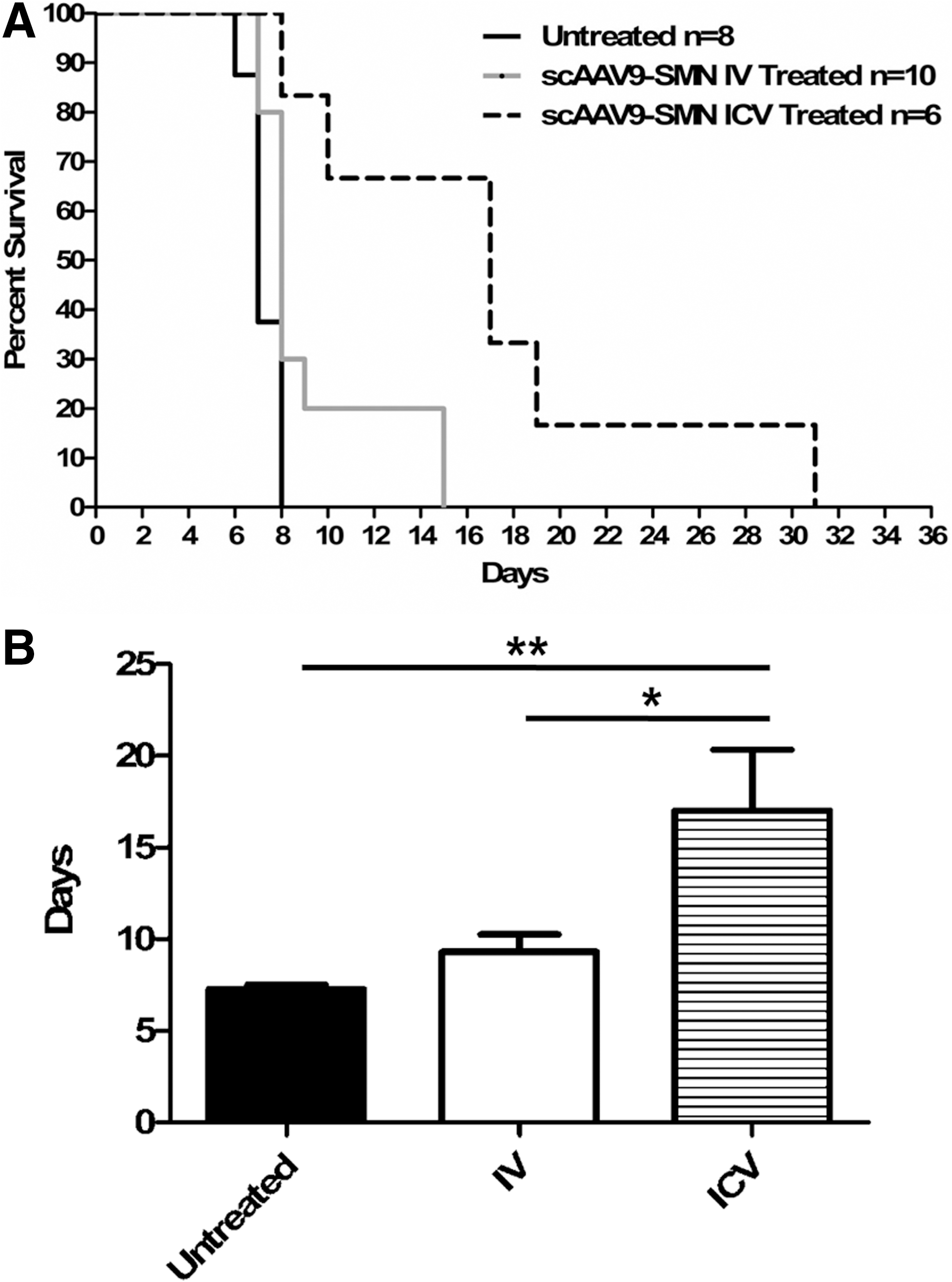
IV and ICV scAAV9-SMN–treated animals lived significantly longer than untreated SMA animals.
Discussion
Increasing SMN protein levels in the CNS is the most straightforward approach when designing SMA therapeutics. Based on the unique opportunities inherent to the existence of SMN2, many therapeutic strategies have focused on modulating SMN2 gene expression, including modulating exon 7 splicing, increasing transcription, stabilizing SMN2 mRNA, and/or increasing the stability of the SMN2-derived protein (Shababi et al., 2010a). Recently, therapeutics designed to increase SMN protein through gene therapy have resulted in dramatic extensions in survival. These studies have demonstrated that supplying exogenous SMN via a viral vector is a highly effective tool for treating an animal model of SMA (Foust et al., 2010; Passini et al., 2010; Valori et al., 2010; Dominguez et al., 2011). To date, all of the published gene therapy studies have been conducted in the SMNΔ7 mouse model, demonstrating that neonatal delivery of scAAV-SMN is able to rescue the SMNΔ7 model to a near-normal weight, phenotype, and a range in survival from ∼65 to 200+ days. In each of these studies, the vector was administered presymptomatically, generally within 48 hrs of birth. Injecting at this early time point allows the viral genome to be expressed before the animals develop overt disease symptoms around P7 (Le et al., 2005). Remarkably, a delay in administering the vector resulted in a profound decrease in efficacy, such that administration of the vector at P5 resulted in a maximum survival of ∼30 days, and delivery after onset of symptoms had no effect (Foust et al., 2010).
Recent genetic experiments examined the requirement of SMN postsymptomatically. Using a Cre-lox system, SMN was induced at various time points pre- and postsymptomatically (Lutz et al., 2011). These results demonstrate that presymptomatic expression of SMN dramatically rescues the phenotype, whereas a significant benefit can still be achieved if induction initiates at P8. This is in contrast to the work with the gene therapy vectors that showed a narrower therapeutic window (Foust et al., 2010). This distinction could be due to several variables between the experimental approaches, including the overall level of SMN induction in disease-relevant tissues, the inability of the vector to efficiently cross the blood–brain barrier at later time points, and the temporal requirements of each organ system to achieve therapeutically relevant levels of SMN expression. As anticipated, in the SMN2 mouse model, our results strongly suggest that the therapeutic window that would lead to a complete rescue of the SMA phenotype is likely inaccessible after birth.
This is the first time ICV and IV administrations have been examined using the same viral vector, scAAV9-SMN. Our results demonstrate that the ICV mediated delivery is more efficient in partially rescuing the SMA phenotype. As SMA is primarily a disease of the CNS, this is not altogether surprising. The ICV injection physically bypasses the blood–brain barrier; therefore, a high dose of vector is present immediately within the CNS. Additionally, scAAV9 has a broad cellular tropism, and a significant fraction of the vector is likely absorbed in peripheral tissues following an IV injection. Therefore, IV injection likely results in less CNS-penetrating vector and a subsequent lesser rescue of the primary motor neuron defect. The Smn –/– ;SMN +/+ model is extremely severe, and it is known that SMN expression is also necessary in the periphery. One possibility to explain the effectiveness of the ICV administration is that a fraction of the virus crosses the blood–brain barrier at P1, providing a low level of transduction in peripheral organs. Previous work with scAAV8 suggests that peripheral organs are important for a complete rescue. For example, when scAAV8-SMN was delivered via ICV injection, the extension in survival was not as long as scAAV9-SMN delivered via IV injection. Although the motor neuron uptake was high for scAAV8, it has been previously documented that cardiac tissue is poorly transduced by AAV8 vectors (Inagaki et al., 2006; Bish et al., 2008). This likely contributes to the shortened life span of the ICV scAAV8-treated animals relative to IV treated animals and highlights the importance of examining multiple delivery methods for a single vector. Our results demonstrate that, in a direct comparison of ICV and IV delivery of scAAV9-SMN, ICV delivery results in a more profound rescue of the SMA phenotype.
Here we demonstrate that injection of scAAV9-SMN provides significant therapeutic benefit in an extremely severe SMA model, indicating that postsymptomatic delivery of scAAV9-SMN can be highly beneficial. Despite this unprecedented extension of survival, we do not observe a full rescue to normal life span like that seen in the SMNΔ7 model. It is possible that in utero delivery would be the only means to achieve a full rescue of the severe model. The extremely low levels of SMN present in this model also appear to impact development in a global manner; therefore, a complete rescue, other than at the genetic level, may not be achievable. From this and the previous studies noted, we conclude that presymptomatic treatment is clearly superior to postsymptomatic treatment. However, it is encouraging to see that postsymptomatic treatment still provides a dramatic reduction in disease severity.
Footnotes
Acknowledgments
We thank John R. Marston for expert technical assistance and Hansjörg Rindt, Ph.D., for his assistance in statistical analysis. This work was supported by grants from the National Institutes of Health (R01 HD054413, R01 NS41584) and a training fellowship for J.J.G. (NIGMS T32 5T32GM008396).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.