
Abstract
Multimodal therapy approaches, such as combining chemotherapy agents with cellular immunotherapy, suffers from potential drug-mediated toxicity to immune effector cells. Overcoming such toxic effects of anticancer cellular products is a potential critical barrier to the development of combined therapeutic approaches. We are evaluating an anticancer strategy that focuses on overcoming such a barrier by genetically engineering drug-resistant variants of immunocompetent cells, thereby allowing for the coadministration of cellular therapy with cytotoxic chemotherapy, a method we refer to as drug-resistant immunotherapy (DRI). The strategy relies on the use of cDNA sequences that confer drug resistance and recombinant lentiviral vectors to transfer nucleic acid sequences into immunocompetent cells. In the present study, we evaluated a DRI-based strategy that incorporates the immunocompetent cell line NK-92, which has intrinsic antitumor properties, genetically engineered to be resistant to both temozolomide and trimetrexate. These immune effector cells efficiently lysed neuroblastoma cell lines, which we show are also sensitive to both chemotherapy agents. The antitumor efficacy of the DRI strategy was demonstrated in vivo, whereby neuroblastoma-bearing NOD/SCID/γ-chain knockout (NSG) mice treated with dual drug-resistant NK-92 cell therapy followed by dual cytotoxic chemotherapy showed tumor regression and significantly enhanced survival compared with animals receiving either nonengineered cell-based therapy and chemotherapy, immunotherapy alone, or chemotherapy alone. These data show there is a benefit to using drug-resistant cellular therapy when combined with cytotoxic chemotherapy approaches.
Introduction
Immunotherapies using either autologous or allogeneic immunocompetent cellular products as well as their bioengineered counterparts with enhanced antineoplastic properties have shown clinical effectiveness (Morgan et al., 2006; Kohn et al., 2011; Porter et al., 2011). However, the administration of intensive chemotherapy, which involves escalations in the frequency of drug treatments or in dose intensifications, in combination with cellular immunotherapy can be detrimental not only to the infused cellular products but can also lead to severe toxicities to bone marrow cells. Genetic engineering of these cells by the introduction of vectors designed to express cDNA sequences that confer drug resistance is a strategy to maintain the antitumor effectiveness of immunocompetent cells during the administration of chemotherapy. Several genes have been designed to confer chemoresistance, and among them P140KMGMT, a mutation of the O 6-methylguanine DNA methyltransferase (MGMT) cDNA that encodes human alkylguanine transferase (hAGT), a DNA repair protein, has been widely investigated for its ability to confer resistance to DNA-alkylating agents such as temozolomide. Various chemoprotective approaches using ectopic expression of P140KMGMT, either as a single transgene or in combination with a second transgene, have been evaluated and shown to confer protection, albeit to various degrees, to bone marrow cells (Sawai et al., 2001; Zielske et al., 2003; Gerull et al., 2007; Larochelle et al., 2009; Trobridge et al., 2009; Beard et al., 2010; Maier et al., 2010; Giordano et al., 2011; Esser et al., 2012).
We exploited the ability to generate chemoresistant immune cells to evaluate an anticancer strategy that we have termed drug-resistant immunotherapy (DRI), whereby drug-resistant immunocompetent cells are administered in conjunction with chemotherapy. In an evaluation of such a DRI strategy, we bioengineered immunocompetent NK-92 cells, which have intrinsic antitumor properties, to express the P140KMGMT transgene (Dasgupta et al., 2010). Using in vitro cytotoxicity assays and the myelogenous leukemia cell line K562, it was demonstrated that bioengineered immunocompetent cells can survive the toxic effects of temozolomide, which resulted in an increase in effectiveness of cell killing during an in vitro chemotherapy challenge. We also previously reported the feasibility of a DRI strategy using bone marrow cells bioengineered to express a single transgene encoding a dihydrofolate reductase variant (L22YDHFR) that confers resistance to antifolates, such as trimetrexate and methotrexate (McIvor, 1996; Spencer et al., 1996; Sweeney et al., 2002; Budak-Alpdogan et al., 2004). Tumor-bearing mice transplanted with drug-resistant bone marrow cells were subjected to combined administration of antifolate-based chemotherapy, trimetrexate (TMTX), along with anti-CD137-based immunotherapy, which resulted in complete tumor eradication (McMillin et al., 2006). In this paper, we evaluate the antitumor effectiveness of a DRI strategy that incorporates genetically engineered NK-92 cells that coexpress the transgenes P140KMGMT and L22YDHFR as a treatment strategy for neuroblastoma, a tumor that is derived from primitive cells of the sympathetic nervous system and is the most common extracranial solid tumor of childhood. To determine whether drug resistance provides a benefit to immunotherapy, the dual drug-resistant immunocompetent cells were used for in vivo studies, using NSG mice and an aggressive neuroblastoma cell line, IMR5. We show here that a DRI treatment consisting of genetically engineered immune cell therapy followed by a dual chemotherapy regimen of temozolomide and trimetrexate controlled the growth of aggressive neuroblastoma, thereby showing the therapeutic potential of using such a strategy to treat cancer.
Materials and Methods
Cell culture
NK-92 cells were obtained from American Type Culture Collection (Manassas, VA) and were maintained in the culture medium AIM V (Invitrogen/Life Technologies, Carlsbad, CA) supplemented with 20% fetal bovine serum (FBS), 1% penicillin–streptomycin, and human recombinant interleukin-2 (IL-2, 100 units/ml) (cat. no. 202-IL; R&D Systems, Minneapolis, MN). Neuroblastoma cell lines IMR5 and SKN-SH (kindly provided by K. Goldsmith, Emory University, Atlanta, GA) were cultured in RPMI cell culture medium (Invitrogen/Life Technologies), supplemented with 10% FBS and 1% penicillin–streptomycin.
Generation of dual chemoresistant vector
The cDNAs encoding P140KMGMT and L22YDHFR were codon optimized. The two sequences were connected via an internal ribosome entry site (IRES) element derived from encephalomyocarditis virus (ECMV). The entire bicistronic cassette, P140KMGMTIRESL22YDHFR, was synthesized (GenScript, Piscataway, NJ) and cloned into the simian immunodeficiency virus (SIV)-based lentiviral expression vector pCL20-CMV-fVIII (Doering et al., 2009), using PmeI and NotI restriction sites. The bicistronic construct was placed under the control of a cytomegalovirus (CMV) promoter.
Lentiviral production and transduction
Recombinant SIV lentivirus was produced with a four-plasmid system (provided by A.W. Nienhuis, St. Jude Children's Research Hospital, Memphis, TN), which was used to transiently transfect 293T producer cells (Doering et al., 2009; Dasgupta et al., 2010). Transient transfection, using Lipofectamine 2000 (Invitrogen/Life Technologies), was carried out by incubating 293T producer cells at 75% confluency, plated in 10-cm collagen-coated BioCoat culture plates (BD Biosciences Discovery Labware, Bedford, MA). The cells were cultured in growth medium, that is, Dulbecco's modified Eagle's medium (DMEM)–F12 (1:1; Invitrogen/Life Technologies), supplemented with 10% FBS and 1% penicillin–streptomycin. Twenty-four hours posttransfection, the cell culture medium was replaced with fresh medium, and at 48, 72, and 96 hr posttransfection the viral supernatant was collected and filtered (pore size, 0.45 μm). The resulting virus stock was filtered (pore size, 0.22 μm) and stored as aliquots at −80°C until further use.
Transduction of recombinant SIV lentivirus was carried out by incubating cells with virus, in appropriate culture medium supplemented with Polybrene (6 μg/ml; Specialty Media/Millipore, Phillipsburg, NJ). Twenty-four hours posttransduction, culture medium was replaced with fresh medium and the transduced cells were further incubated to reach 70–80% confluency, and at this time point the transduced cells were used for downstream applications.
Determination of titers of recombinant SIV lentivirus
Viral titers were determined by transducing 293T producer cells with either 0.1 or 1 μl of the virus stock, incubating the transduced cells for 72 hr, followed by conducting a real-time polymerase chain reaction (qPCR) as described previously (McMillin et al., 2006; Dasgupta et al., 2010). Two sets of primer pairs were used in separate qPCRs. The sequences of these primer pairs are as follows: (1) MGMT-forward, 5′-CCTGGCTGAACGCTTACTTC-3′ and MGMT-reverse, 5′-CACAGCACCTGCCTTGTAAA-3′; and (2) DHFR-forward, 5′-TGGAAGGGAAACAGAACCTG-3′ and DHFR-reverse, 5′-CACCAGATTGATCCGTCCTT-3′.
Survival curve analysis and determination of synergy
Survival curves were generated by measuring cell viability, using a trypan blue exclusion method. All drugs were freshly prepared on the day of use. Each experiment was performed a minimum of three times. The interaction between the drugs was calculated with dose effect analysis software (CalcuSyn; Biosoft, Cambridge, UK), which is based on Chou–Talalay combination indices (CIs) where CI<1 indicates synergy; CI=1 indicates additivity; and CI>1 indicates antagonism (Chou, 2010). CI values were generated with this software. Briefly, IMR5 cells were exposed to either temozolomide or trimetrexate and to combinations of the two drugs, and the drug-treated cells were incubated at 37°C. Seventy-two hours later, cell viabilities were determined, as described previously, and median drug effects were computed to determine the nature of interactions.
Determination of cellular cytotoxicity
Cytotoxicity assays were conducted by mixing various amounts of effector and target cells placed in a 96-well plate to achieve different E:T ratios (in triplicate) followed by a 4-hr incubation. The amount of lactate dehydrogenase (LDH) released to the supernatant as a result of cytolysis of target cells was measured in an LDH release assay according to the instructions of the manufacturer (Roche Applied Science, Indianapolis, IN). The following controls were always included in the assay: (1) Untreated effector cells (at all the concentrations as described in Results) as well as target cells to measure any spontaneous LDH release, (2) lysis reagent-treated target cells to measure the maximal amounts of LDH that can be released without any added effector cells, and (3) cell growth medium to measure background LDH levels. The percentage of cellular cytotoxicity was determined by using the following equation: % cytotoxicity={[(experimental release – spontaneous releaseeffector) – spontaneous releasetarget]/(maximal releasetarget – spontaneous releasetarget)}×100.
Cellular cytotoxicity in the presence of anti-NKG2D antibodies was determined by the addition of 40,000 NK-92 cells to 1 μg of the anti-NKG2D antibody (cat. no. sc-23869; Santa Cruz Biotechnology, Santa Cruz, CA). After 2 hr of incubation, the effector NK-92 cells (E) were mixed with 4000 target IMR5 cells (T) previously plated in a 96-well plate, to achieve an E:T ratio of 10:1. The cytotoxicity assay was performed by methods described previously.
Analysis of UL16-binding protein-2 expression
Temozolomide (50 μM) was added to IMR5 cells, which were incubated for 5 hr. At every hourly interval, drug-treated cells were collected, centrifuged at 500×g for 5 min, and washed three times with an isotonic phosphate-buffered saline (PBS) buffer (supplemented with 0.5% bovine serum albumin [BSA]) to remove any growth factors that may have been present in the culture medium. Cells were suspended in the same buffer to a final concentration of 4×106 cells/ml and 25 μl of cells was incubated with 10 μl of allophycocyanin-conjugated anti-ULBP-2 (UL16-binding protein-2) reagent (cat. no. FAB1298A; R&D Systems) for 30 min at 4°C. At the end of incubation, the cells were washed and resuspended in 200 μl of PBS and flow cytometric analysis was performed with an LSRII flow cytometer system (BD Biosciences, San Jose, CA). Untreated cells, not labeled with antibodies, served as negative control.
Animal studies
NOD/SCID γ-chain knockout (NSG) mice were purchased from Jackson Laboratory (Bar Harbor, ME) and were maintained in a specific pathogen-free environment. Mice were cared for according to the established principles of the Institutional Animal Care and Use Committee (IACUC) and all animal protocols were approved by the IACUC. Approximately 7-week-old mice were each inoculated subcutaneously via the right flank with 1×107 IMR5 cells. Mice were visually monitored and tumor growth was measured with calipers, and treated when tumors reached approximately 25 mm2, typically 10–12 days after inoculation. The chemotherapeutic drugs temozolomide (Schering, Kenilworth, NJ) and trimetrexate glucuronate (MedImmune Oncology, Gaithersburg, MD) were dissolved in PBS and water, respectively. Both drugs were injected intraperitoneally into the animals. Analysis of complete blood cell counts (CBCs) was performed weekly once the treatments started. Fifty microliters of blood was collected retro-orbitally and analyzed with a veterinary hematology system (Heska, Loveland, CO). Animals were killed once the tumor burden reached approximately 200 mm2.
Kaplan–Meier analysis
The survival probabilities of each cohort were determined by Kaplan–Meier log-rank test to analyze whether differences in survival were statistically significant. Statistical analysis was performed with SigmaPlot, version 11 (Systat Software, Chicago, IL).
Results
Generation of drug-resistant immunocompetent NK-92 cells by lentiviral transduction
The cDNA sequences encoding P140KMGMT and L22YDHFR variants were synthesized as codon-optimized sequences for efficient expression in human cells and cloned into an SIN-SIV lentiviral expression vector, generating the bicistronic construct P140KMGMTL22YDHFR (Fig. 1A). Viral titers on 293T cells ranged from 5×107 to 8×107 transducing units/ml. Genetic engineering of NK-92 cells was performed by transducing cells twice, at a 12-hr interval, with SIV at a multiplicity of infection (MOI) of 20 for each transduction. Proviral copy number was quantitated by real-time PCR analysis as described previously (Dasgupta, et al., 2010). The transduced NK-92 cells had a P140KMGMTL22YDHFR copy number of 3±0.5 copies/cell.
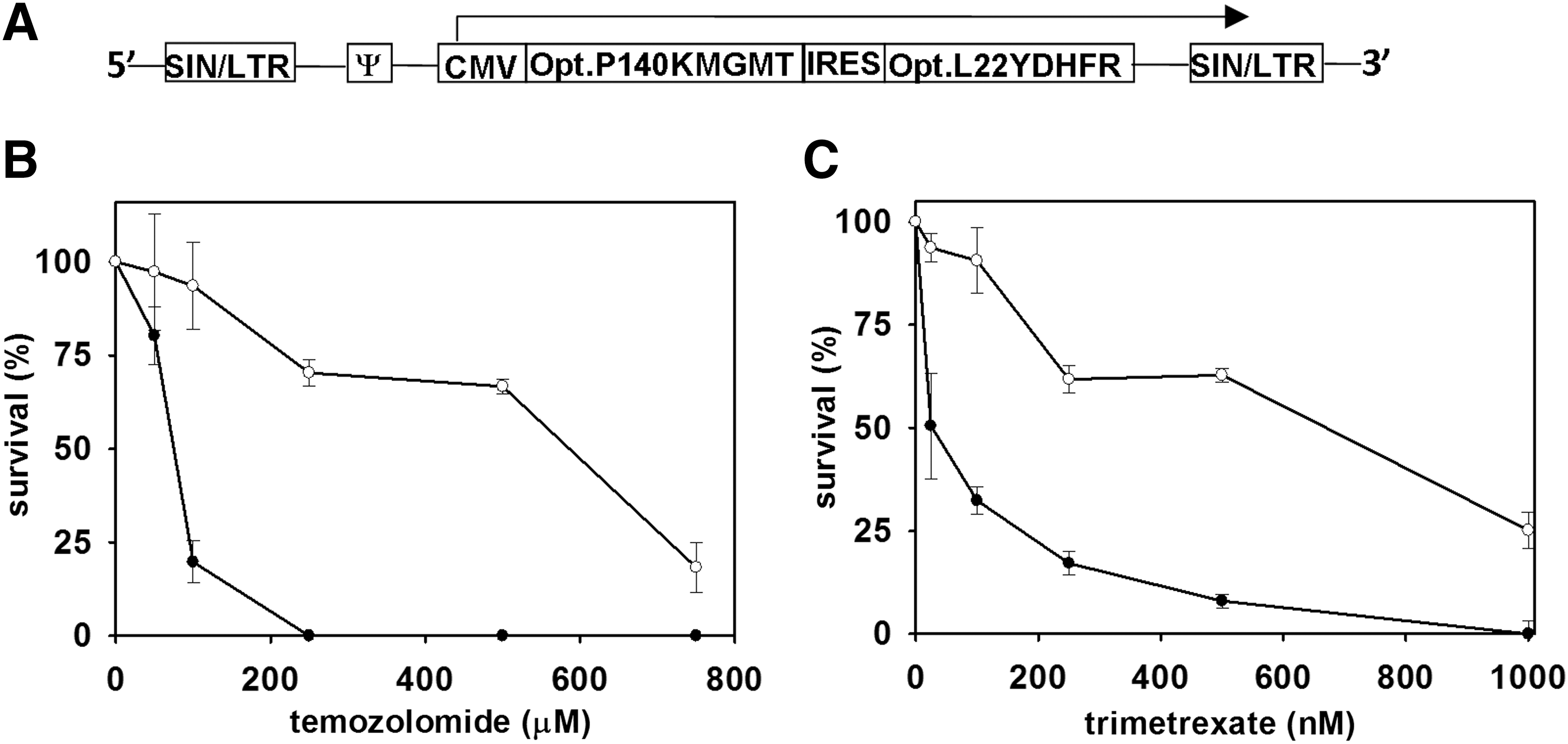
Survival curve analysis of genetically engineered and nonengineered NK-92 cells. NK-92 cells were genetically engineered by lentivirus-mediated transfer of a bicistronic construct that encodes the optimized sequences for P140KMGMT and L22YDHFR.
Resistance to the alkylating drug temozolomide, by expression of the codon-optimized P140KMGMT cDNA, was determined by incubating transduced cells with drug at increasing concentrations, ranging from 50 to 800 μM. Cells were exposed to the drug for 72 hr and then replated and cultured for an additional 4 days, at which time cell viability was determined (Fig. 1B). The effectiveness of the transferred codon-optimized L22YDHFR cDNA sequence was determined by exposing the transduced cells to increasing concentrations of the antifolate drug trimetrexate, ranging from 25 to 1000 nM, and cell viability was determined 72 hr after the addition of the drug (Fig. 1C). Survival curves were generated with respect to untransduced control cells to determine the degree of resistance conferred to the genetically modified cells. Compared with untransduced cells, modified cells were resistant to both drugs, with median inhibitory concentration (IC50) values for temozolomide of 589 and 76 μM for genetically engineered and nonengineered cells, respectively. The trimetrexate IC50 values were 660 and 25 nM for genetically engineered and nonengineered cells, respectively. Thus, compared with untransduced cells, the genetic engineering of NK-92 cells conferred approximately 8- and 26-fold resistances to temozolomide and trimetrexate, respectively.
Neuroblastoma cell lines are sensitive to alkylating and antifolate drugs
The neuroblastoma cell line IMR5, which represents an aggressive human neuroblastoma cell line, was incubated with increasing concentrations of temozolomide after preincubation with or without O 6-benzylguanine (O6BG) and separately with trimetrexate. Cell viabilities were determined 72 hr after the addition of drug and survival curves were generated to assess the response of IMR5 cells to each drug (Fig. 2A and B). On the basis of the survival analysis, IC50 values for temozolomide, with or without O6BG, and trimetrexate were 3.3 μM (without O6BG), 3.6 μM (with O6BG), and 25 nM, respectively. Thus IMR5 cells were sensitive to both temozolomide and trimetrexate. Moreover, O6BG did not potentiate temozolomide-mediated killing of the tumor cells. Similar results were obtained for a second neuroblastoma cell line, SK-SH (data not shown).
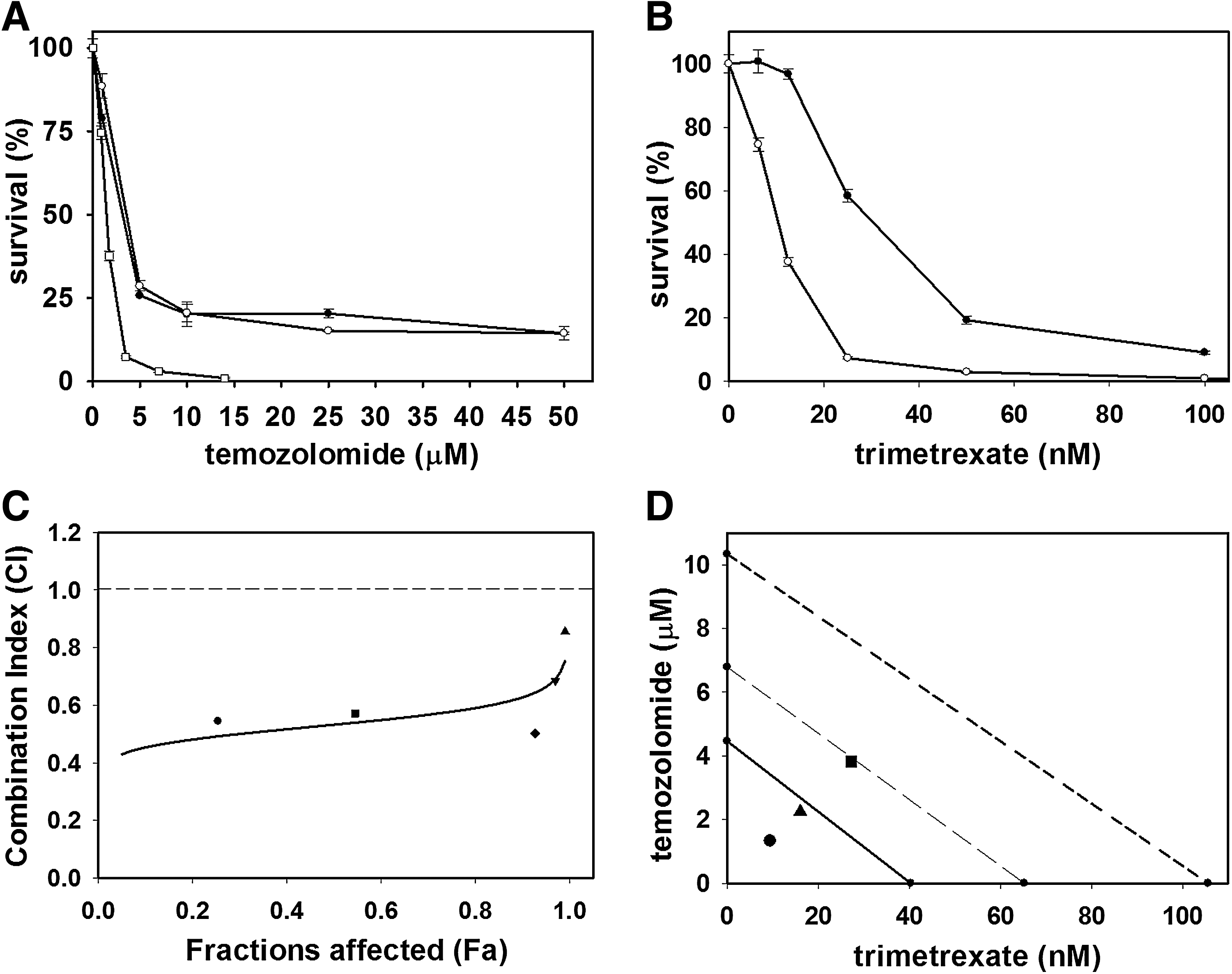
Analysis of the response of neuroblastoma cells to temozolomide and trimetrexate.
Subsequent to the individual drug assays, IMR5 cells were evaluated for their response to combinations of both drugs added simultaneously to culture. The quantitative measure of the degree of interaction between the two drugs was evaluated by the combination analysis method developed by Chou and Talalay (Chou, 2010). Dose–effect curves were computed by performing survival analysis of IMR5 cells incubated with various concentrations of either of the two drugs individually as well as with combinations of both the drugs at fixed ratios. In this assay the following concentrations were used: (1) temozolomide at 0.875 μM, 1.75 mM, 3.5 μM (IC50), 7 μM, and 14 μM; (2) trimetrexate at 6.25 nM, 12.5 nM, 25 nM (IC50), 50 nM, and 100 nM; and (3) combinations of both drugs at 0.875 μM and 6.25 nM (combination 1), 1.75 μM and 12.5 nM (combination 2), 3.5 μM and 25 nM (combination 3), 7 μM and 50 nM (combination 4), and 14 μM and 100 nM (combination 5) of temozolomide and trimetrexate, respectively. The survival curves representing these dual drug combinations are shown in conjunction with the survival analysis with either temozolomide only or trimetrexate, respectively (Fig. 2A and B). The combination index (CI) calculated with CalcuSyn software from the dose–effect curves were 0.544 (combination 1), 0.572 (combination 2), 0.502 (combination 3), 0.685 (combination 4), and 0.855 (combination 5). These CI values are indicated in the median drug effect graph, represented by the median–effect plot (F a vs. CI), which indicates synergy at all the previously given combinations (Fig. 2C). In addition, the combination index values at various equipotent effective dose (ED) combinations were calculated as 0.5323, 0.5775, and 0.6272 at ED50, ED75, and ED90, respectively. The data points from these combinations were plotted as classical isobolograms, constructed with various ED values, and depicted strong synergistic effects (Fig. 2D).
Drug-resistant NK-92 cells efficiently kill IMR5 neuroblastoma cells
Previous reports demonstrated that natural killer (NK) cells can lyse neuroblastoma cells (Klöss et al., 2007). Here, we evaluated whether NK-92 cells, derived from NK cells, can kill IMR5 cells in vitro. Various numbers of effector cells (NK-92) were mixed with a fixed number of target cells (IMR5) at effector-to-target ratios of 2.5:1, 5:1, and 10:1 (Fig. 3A). The effectiveness of tumor cell lysis by the effector cells was determined in a 4-hr cytotoxicity assay, which showed that NK-92 cells efficiently lyse IMR5 cells. We next determined whether genetic engineering of the immunocompetent NK-92 cells altered their cytotoxic effectiveness. Various numbers of either genetically engineered or nonengineered NK-92 cells were mixed with a fixed number of target cells at various effector-to-target ratios. Using a 4-hr cytotoxicity assay, the killing effectiveness of the effector cells was compared with that of drug-resistant cells (Fig. 3A). We showed that the genetically engineered immunocompetent cells displayed a cytolytic ability similar to that of the nonengineered cells. Similar results were obtained for a second neuroblastoma cell line, SKN-SH (data not shown).
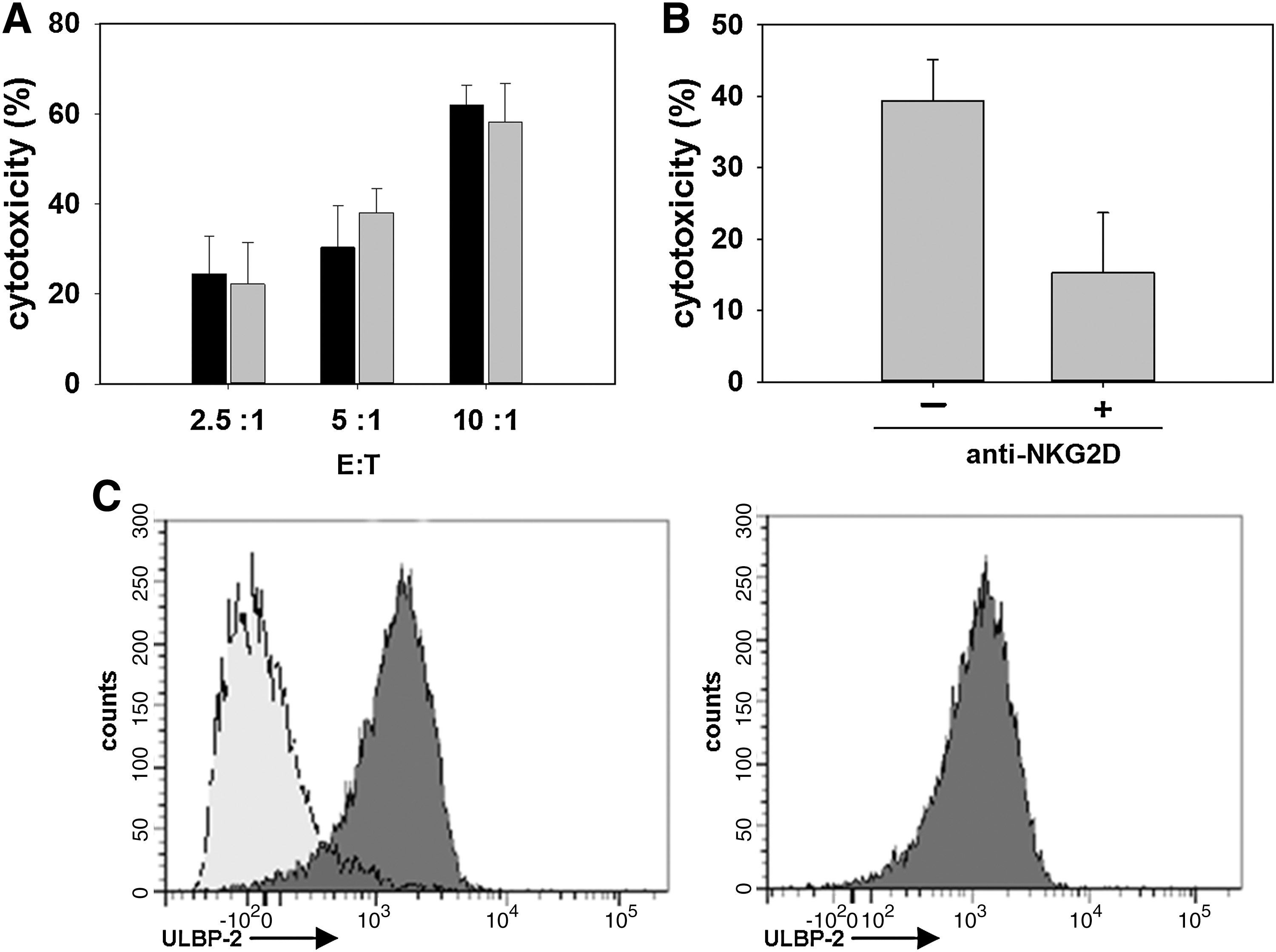
Determination of the cytotoxicities of nonengineered and genetically modified NK-92 cells toward IMR5 cells.
NK-92 cells kill neuroblastoma cells through the NKG2D receptor
On the basis of our finding that NK-92 cells can efficiently lyse neuroblastoma cells, we investigated the mechanism of interaction between immune effector cells and tumor cells. The antitumor immunity of NK cells is mediated by the cell surface expression of NKG2D receptors, which can recognize a repertoire of NKG2D ligands such as ULBP-2 (UL16-binding protein-2) expressed on a variety of tumor cells. To determine whether inhibition of the function of NKG2D receptors expressed on the surface of NK-92 cells alters their cytotoxicity toward neuroblastoma cells, immunocompetent cells were initially incubated with an anti-NKG2D antibody. After 2 hr of incubation, a fixed number of either antibody-treated or untreated NK-92 cells was mixed with a fixed number of target cells, and both the effector and target cell combinations were incubated for an additional 4 hr. Last, the killing effectiveness of antibody-treated NK-92 cells was compared with the cytolytic ability of untreated immunocompetent cells (Fig. 3B). Compared with untreated effector cells, NKG2D antibody-treated NK-92 cells exhibited reduced cytotoxic activity, indicating that inhibition of the binding of NKG2D receptors expressed on NK-92 cells abrogates the cytotoxicity of NK-92 cells toward IMR5 cells.
We next evaluated the extent of ULBP-2 ligand expression on the IMR5 cell line during a chemotherapy challenge with temozolomide. IMR5 cells were cultured in 50 μM temozolomide for 5 hr. At hourly intervals, ULBP-2 expression was monitored by flow cytometry and compared with that of untreated cells. The expression of ULBP-2 in untreated cells and cells after drug treatment is shown (Fig. 3C). ULBP-2 expression in cells cultured in the absence of drug was similar to ULBP-2 expression in cells incubated with drug for 5 hr. Thus, neuroblastoma cells express the NKG2D ligand, ULBP-2, during a temozolomide chemotherapy challenge.
Drug-resistant immunotherapy controls IMR5 tumor growth in immunodeficient mice
Having established that genetically engineered, dual drug-resistant NK-92 cells can recognize and lyse neuroblastoma cells in culture, we evaluated the effectiveness of combining genetically modified NK-92 cell-mediated therapy with chemotherapy in vivo. IMR5 cells were used to establish xenografted neuroblastoma tumors in NSG mice. A treatment schedule was followed whereby animals received treatments on days 1, 3, and 5 during week 1, followed by treatments on days 8 and 10 during week 2 and finally a single treatment on day 15 in week 3 with respect to the development of palpable tumors (approximately 25 mm2) (Fig. 4). The animals received systemic administration (via tail vein injection) of immune cell-mediated therapy, which was followed 2 hr later by intraperitoneal administration of chemotherapy. We independently tested several combined therapy regimens, composed of either low, medium, or high doses of immune cell therapy or chemotherapy.
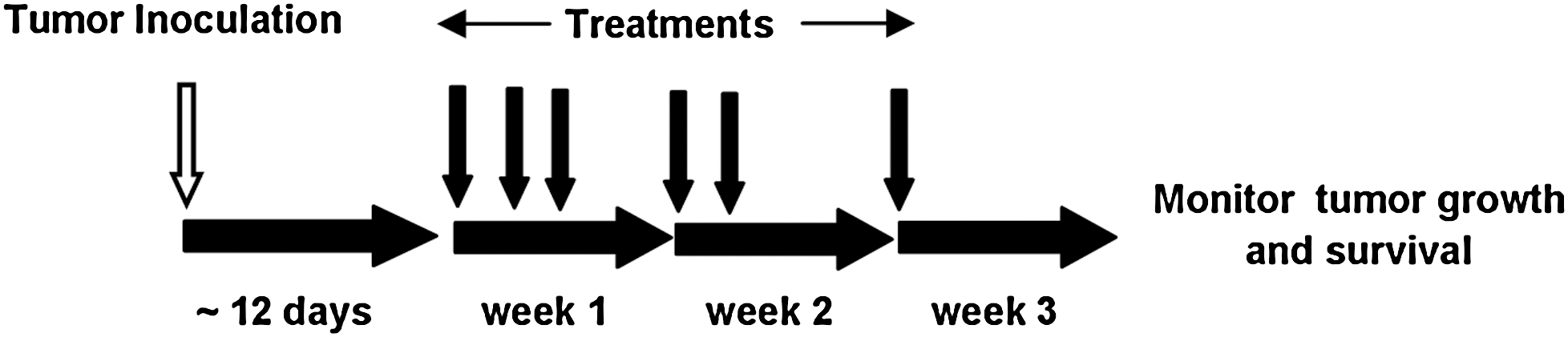
Schematic of treatment. IMR5 cells were injected (open arrow) into NSG animals: After the tumors became palpable (∼25 mm2), treatments consisting of either chemotherapy, immunotherapy, or immunotherapy and chemotherapy were administered weekly as shown with solid arrows (see text for details). Immunotherapy consisted of either bioengineered drug-resistant cells or nonengineered cells. The day treatments were initiated was counted as day 1.
Low-dose treatment
The low-dose treatment modality was composed of an immunotherapy regimen of 1×106 NK-92 cells per injection, and a chemotherapy regimen composed of temozolomide (50 mg/kg) and trimetrexate (15 mg/kg). These drug doses resulted in only a transient and mild decrease in circulating mononuclear cells and platelets (Fig. 5A), and the body weights of the treated animals did not change significantly (data not shown). A total of five cohorts of mice were individually challenged with (1) drug-resistant NK-92 cell therapy followed by low-dose chemotherapy (DRI treatment), (2) nonengineered NK-92 cell therapy followed by chemotherapy, (3) drug-resistant NK-92 cell therapy alone, (4) chemotherapy alone, or (5) no treatment (Fig. 5B). Administration of chemotherapy alone attenuated IMR5 cell growth during treatment compared with nontreated animals. However, once treatment ended these mice were killed at an average of 22 days because of uncontrolled tumor growth, compared with 12 days for nontreated mice. Administration of immunotherapy only also provides some benefit, but all mice treated only with immunotherapy were killed on average by day 24 because of excessive tumor burden. However, all DRI-treated mice had significant reduction in tumor growth for 70 days from the start of treatment, but were killed on average by 106 days because of tumor growth. In contrast, mice treated by naive, nonmodified NK-92 cell-based therapy and chemotherapy under identical conditions did not respond well to therapy and were killed by day 24, which was surprisingly similar to the effect in the mice treated only by chemotherapy. The overall survival rates of the individual mouse cohorts were evaluated by Kaplan–Meier analysis (Fig. 5C). The DRI-treated animals had significantly increased survival compared with the survival of all other treatment groups (p≤0.001).
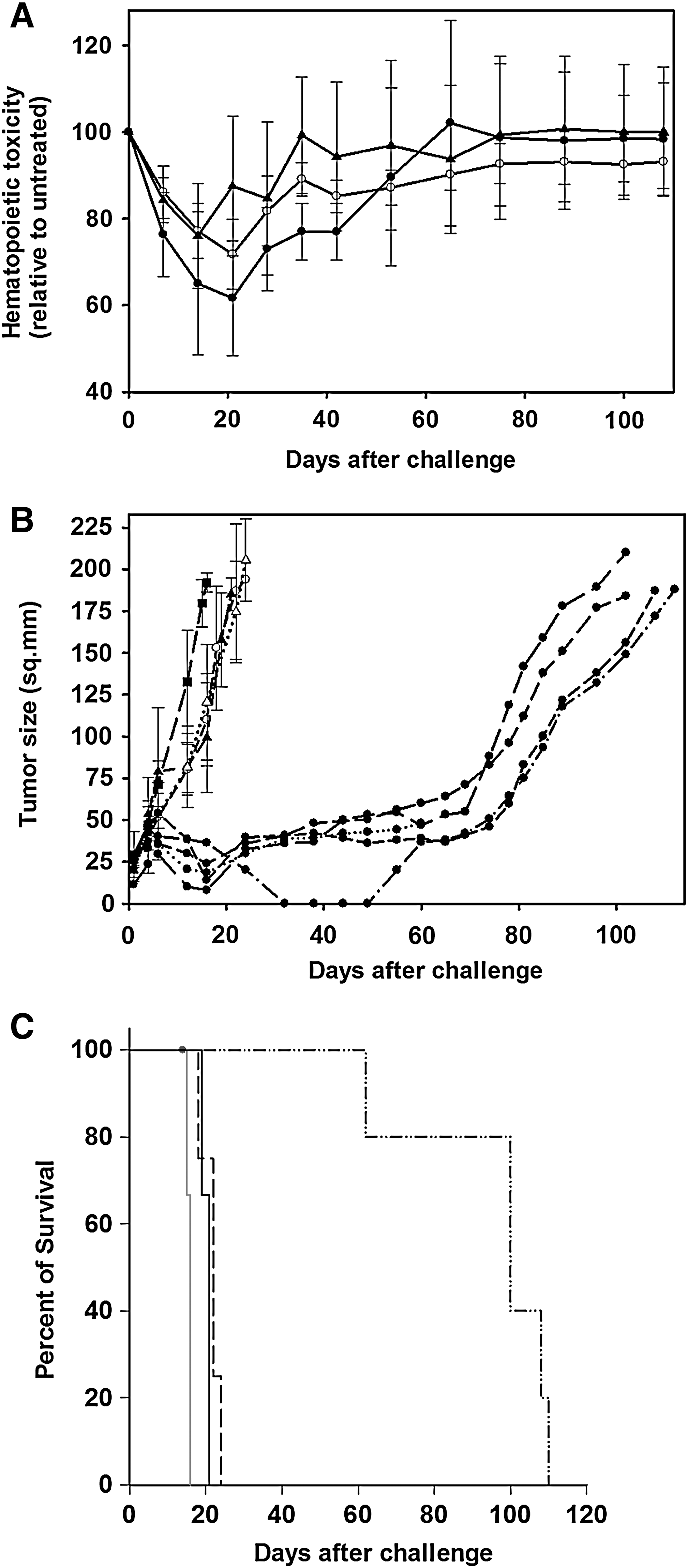
Evaluation of low-dose drug-resistant immunotherapy (DRI) treatment in NSG mice.
Furthermore, we tested the treatment effectiveness of intratumoral administration of 1×106 drug-resistant cells followed 2 hr later by intraperitoneal administration of low-dose chemotherapy into tumor-bearing mice. Compared with the systemic administration of immunocompetent cells, the intratumoral DRI treatment was less effective, with animals being killed by day 56 because of excessive tumor burden (data not shown).
Medium-dose treatment
The medium-dose treatment modality consisted of an immunotherapy regimen of 3×106 NK-92 cells per injection, and a chemotherapy regimen of temozolomide (125 mg/kg) and trimetrexate (15 mg/kg). Mice were individually challenged with either DRI treatment or nonengineered NK-92 cell therapy followed by chemotherapy, or chemotherapy only (Fig. 6A). In the DRI-treated cohort, 80% of the DRI-treated mice (four of five) had tumor regression up to 90 days from the start of treatment, with no visible tumor growth in 40% of animals (two of five) on day 115. In contrast, the cohorts of mice treated with either combined therapy of nonengineered NK-92 cells and chemotherapy or chemotherapy only were killed, because of uncontrolled tumor growth, on day 40. The overall survival of the individual mouse cohorts was evaluated by Kaplan–Meier analysis (Fig. 6B). DRI-treated animals had significantly increased survival compared with that of animals treated with the individual treatments as well as nonmodified NK-92 cells and chemotherapy (p=0.003).

Evaluation of medium-dose drug-resistant immunotherapy (DRI) treatment of NSG mice with established human neuroblastoma.
High-dose treatment
In addition, we evaluated high-dose DRI treatment whereby neuroblastoma-bearing mice were challenged with 3×106 genetically engineered cells followed by a high-dose chemotherapy regimen of temozolomide (200 mg/kg) and trimetrexate (30 mg/kg). Although the animals had dramatic tumor reductions by day 4 from the start of treatment, three of four animals were killed by day 30 because of acute drug-related toxicities (data not shown).
Discussion
Bioengineered drug-resistant immunocompetent cells were investigated with respect to their potential use as an alternative to conventional anticancer therapies. Adoptive transfer of ex vivo-expanded and activated autologous T cells and NK cells has met with limited effectiveness as a cancer therapeutic (Law et al., 1995; Burns et al., 2003). However, immunocompetent T cells that are genetically engineered to recognize tumor antigens have shown some success (Morgan et al., 2006; Porter et al., 2011). With respect to NK cells, the focus has shifted to the use of allogeneic cells (Miller et al., 2005). The NK-92 cell line has shown anticancer activity in initial clinical trials (Klingemann, 2005; Arai et al., 2008). In addition, (1) these cells represent a well-characterized immunophenotype with powerful antitumor properties that are independent of MHC restrictions; (2) they express activating receptors and lack most of the inhibitory killer immunoglobulin-like receptors (KIRs) (Middleton et al., 2002), thus retaining their cytotoxicity against cancer cells that upregulate MHC class I molecules; (3) it is relatively easy to generate adequate quantities for in vivo studies; (4) they display mild toxicity to normal cells during adoptive therapy; (5) we have observed that they do not develop malignancies on infusion into NSG mice; and (6) they are readily transduced with recombinant lentivectors. Thus we chose to explore the use of NK-92 cells as a model immunotherapeutic candidate in our proof-of-concept studies to evaluate the effectiveness of a DRI-based antineuroblastoma therapy. High-risk neuroblastoma is rarely curable, even with current multimodal treatment approaches that include increasingly aggressive myeloablative chemo- and radiotherapy. Therefore, new treatment strategies such as DRI should be evaluated for this cancer.
To evaluate genetically engineered drug-resistant NK-92 cells, a cell line was developed with enforced expression of the cDNAs encoding L22YDHFR and P140KMGMT, which confers protection against the chemotherapeutic agents trimetrexate and temozolomide, respectively. Temozolomide has previously been shown to be effective against some neuroblastoma cell lines and in animal xenografts (Houghton et al., 2000; Rosati et al., 2008; Cai et al., 2010). Temozolomide, in combination with other chemotherapy drugs, has also shown clinical activity against relapsed or high-risk neuroblastoma (Rubie et al., 2006; Bagatell et al., 2011; Kushner et al., 2011). We show that the IMR5 cell line, which represents an aggressive neuroblastoma, is highly sensitive to temozolomide. However, although the majority of cells are killed at relatively low drug concentrations, a small fraction of the cells remains unaffected even in the presence of temozolomide at concentrations greater than 400 μM. We also show that IMR5 cells are sensitive to trimetrexate, an antifolate drug that has been tested in a clinical trial against neuroblastoma (Pappo et al., 1993). The nature of interactions between the combinations of trimetrexate and temozolomide in a cell-based assay demonstrated that the drugs acted synergistically to enhance neuroblastoma cell killing, indicating the potential benefits of dual drug chemotherapy in the context of DRI in animal models of neuroblastoma.
NK-92 cells displayed potent cytotoxicity toward IMR5 cells, and such cytotoxic activities increased with an increase in effector cell numbers, as was also shown previously (Klöss et al., 2007). The dual drug-resistant NK-92 cells exhibited significant resistance against both drugs, although the fold resistance to trimetrexate was lower than previously achieved (Spencer et al., 1996). This is not surprising as transgenes located downstream of an IRES element in a bicistronic construct containing DHFR can exhibit lower expression compared with the expression level of the first transgene (Mishra et al., 2009). The dual drug-modified NK-92 cells retained their characteristic tumor-killing properties as we previously reported with NK-92 cells resistant only to temozolomide (Dasgupta et al., 2010).
The exact mechanism by which NK-92 cells exert their cytotoxic effect is only partially understood. It has been established that these cells employ activating receptors such as NKG2D and natural cytotoxicity receptors, such as NKp30, to bind to tumor cell surface-associated molecules (Maki et al., 2001; Brandt et al., 2009). NKG2D recognizes two distinct families of ligands: MHC class I chain-related molecules (MICA and MICB) and UL16-binding proteins (ULBPs). These NKG2D ligands are overexpressed in some malignant cells, and NK cytotoxicity correlates with expression levels of MICA, MICB, and ULBP on tumor cell targets. For example, a decrease in NK cell activity appears to be related to decreased expression levels of the ULBP ligands on acute myeloid leukemia (AML) blasts (Nowbakht et al., 2005). In mouse models, the expression of NK-stimulatory NKG2D ligands induces not only short-term rejection of tumors but also a protective adaptive immune response (Diefenbach et al., 2001). Similarly, mice genetically deficient in NKG2D are more susceptible to spontaneous cancer than are wild-type mice (Guerra et al., 2008). Here we investigated whether genetically engineered, drug-resistant NK-92 cells engage their NKG2D receptors to target neuroblastoma cells and showed that a block in the binding of these receptors abrogates the cellular cytotoxicity of NK-92 cells against IMR5 cells. It is thought that tumor cells express different ligands against the same NKG2D receptor to escape from NK cell-mediated immune surveillance and that tumors vary the expression levels of these ligands in response to biochemical stress (Nausch et al., 2008). To evaluate whether chemotherapy-induced stress to neuroblastoma cells affects their cell surface expression of NKG2D ligands, we measured the amount of ULBP-2 present on the surface of IMR5 cells before and after temozolomide drug challenge. We established that even in the presence of high doses of temozolomide, IMR5 cells continue to express ULBP2 and thus remain susceptible to NK-92-mediated killing. A differential expression pattern of these ligands has been reported in several neuroblastoma cell lines (Raffaghello et al., 2004), and whether any of these ligands are upregulated in response to chemotherapy and thereby synergize with DRI treatment remains to be investigated.
For our initial in vivo DRI study, a dosing schedule was used whereby chemotherapy-induced toxicity was minimal with respect to myelosuppression and killing of genetically engineered NK-92 cells. The dose of temozolomide and trimetrexate was selected on the basis of previously published studies (McMillin et al., 2006; Dinca et al., 2007; Natsume et al., 2008; Robinson et al., 2010). Notably, temozolomide was administered without preadministration of O6BG, as our in vitro data show that O6BG is not required to potentiate temozolomide activity in IMR5 cells. In the current study, administration of dual chemotherapy induced moderate toxicity only during the course of treatment, and the animals regained normal blood counts shortly after the termination of treatment. The antitumor cellular therapeutic agents were administered over a span of 17 days, which can circumvent the possibility of the short-term persistence of immunocompetent cells in vivo and mitigate the benefits of immunotherapy. For the combined immuno- and chemotherapy, cellular therapy was administered before chemotherapy in an effort to enhance the likelihood of the accumulation of these cells within the tumor by the time chemotherapy was administered. In addition, administration of chemotherapy can ablate host immunocompetent cells that may be directed against the exogenously added anticancer immune cells. Also, it has been observed that antitumor responsiveness in patients with melanoma is significantly enhanced by the administration of chemotherapy-based lymphodepleting regimens before the infusion of cellular immunotherapy (Morgan et al., 2006). In the present study, the administration of chemotherapy to NSG animals resulted in a transient decrease in host hematopoietic cells, which may have enhanced the antitumor effectiveness of the transferred genetically engineered NK-92 cells.
The DRI-based antitumor effect was in sharp contrast to the poor antitumor response mediated by combination therapy consisting of nonmodified cell therapy and chemotherapy. The antitumor effectiveness of either chemotherapy only or immune cell therapy only was not beneficial. Thus we conclude that the bioengineered antitumor immunocompetent cells survived drug challenges and the combination of such drug-resistant cellular therapy with chemotherapy can provide therapeutic benefit. The rapid regrowth of tumors beyond an average of 70 days from the start of treatment may be attributed to the expansion of a small fraction of neuroblastoma cells that survived the drug treatment, as we observed in our in vitro chemotherapy assays, where a percentage of neuroblastoma cells survived high doses of temozolomide. Administration of moderately high DRI treatment was effective in controlling tumor growth for more than 90 days and extended the survival of tumor-bearing mice compared with the lower dose group. As expected on the basis of the results with the low-dose treatment cohorts, moderately high doses of drug with nonengineered cell therapy were not effective at controlling tumor growth. Further increasing the dose of chemotherapy in combination with a moderately high dose of engineered cellular products led to dramatic reductions in tumor growth but was met with excessive drug-induced toxicities.
A caveat to our dual chemotherapy regimen is the use of temozolomide and trimetrexate to treat neuroblastoma. The choice of these agents was based on our in vitro results. However, conventional therapy uses temozolomide concurrently with irinotecan, a topoisomerase inhibitor (Wagner et al., 2009; Bagatell et al., 2011). To further translate these proof-of-concept studies to a clinical setting, genetically engineered immunocompetent cells can be generated with a dual drug resistance vector encoding P140KMGMT and TDP-1 (tyrosyl-DNA phosphodiesterase-1), which can confer resistance to both temozolomide and irinotecan. We previously showed that the retroviral expression of TDP-1 confers resistance to the topoisomerase inhibitor, camptothecin (Nivens et al., 2004). In addition, several other avenues be explored to increase the effectiveness of DRI-based strategies. For example, it has been shown that NK-92 cells genetically engineered to express a neuroblastoma cell-specific chimeric antigen receptor (CAR) led to an increase in killing of neuroblastoma cells in culture (Esser et al., 2012). Incorporation of a drug resistance phenotype into these cells can potentially enhance tumor killing by these cells in vivo when combined with a chemotherapy regimen. In addition, DRI-based strategies developed to incorporate adaptive immune response cells, such as cytotoxic T lymphocytes (CTLs), can provide a durable immune response before, during, and after chemotherapy treatment regimens. Such a strategy may involve incorporating engineered T cells expressing specific anticancer T cell receptors (TCRs) (Morgan et al., 2006; Porter et al., 2011). Furthermore, it is possible to incorporate a DRI strategy using hematopoietic stem cells after bone marrow transplantation followed by the combined administration of immunomodulatory agents and chemotherapy. Thus, DRI can provide a needed improvement in the treatment of cancers that are currently difficult to manage.
Footnotes
Acknowledgments
This work was supported by a grant from the National Institutes of Health to H.T.S. (HL087969), and from CURE Childhood Cancer (H.T.S). The authors thank Dr. Nienhuis (St. Jude Children's Research Hospital, Memphis, TN) for the plasmids to generate the SIN-SIV lentivirus vectors.
Author Disclosure Statement
No competing financial interests exist for any author.