
Abstract
Achieving high-efficiency tumor targeting after systemic delivery is a considerable challenge facing oncolytic gene therapists. Efficient retargeting should be combined with efforts to improve in vivo safety, reduce hepatotoxicity, minimize off-target interactions, and improve antitumoral potency and efficacy. We previously described the successful retargeting of adenovirus serotype 5 (Ad5) to αvβ6, an integrin that is highly overexpressed in numerous human carcinomas. In this study, we have further modified this construct by introducing mutations that ablate coxsackievirus–adenovirus receptor (CAR) binding and putative interactions with factor IX (FIX)/C4b-binding protein (C4BP). We have found that the resulting vector, Ad5-477dlTAYTA20, displays a desirable in vivo safety profile. This vector does not agglutinate human erythrocytes, fails to cause thrombocytopenia after intravenous delivery, has limited induction of proinflammatory cytokines, and results in low-level toxicity (aspartate aminotransferase/alanine aminotransferase) when compared with Ad5-EGFPWT. Furthermore, it has reduced accumulation in Kupffer cells (1 hr) and limited hepatocyte transduction at later time points (24 and 96 hr). The parental vector, Ad5-EGFPA20, also displayed many of these desirable properties. As a result of the improved safety profile of both A20-modified vectors, we escalated the dose from 2×1010 to 4×1010 viral particles in an antitumoral efficacy study. We observed improvements in reducing percent tumor growth at early time points (96 hr) when compared with Ad5-EGFPWT, although increasing the dose did not affect the therapeutic outcome beneficially. On completion of the experiment, we detected increased E1A staining in the tumors of all A20-treated groups and we determined that E1A expression was localized largely within αvβ6 + tumor cells. However, in spite of apparently efficient tumor transduction, this did not result in enhanced antitumoral efficacy as the virus failed to disseminate effectively throughout the tumor mass, presumably due to physical intratumoral restrictions. This highlights a remaining challenge that needs to be overcome before such vectors can be developed for future cancer gene therapy applications.
Introduction
To overcome the issue of poor in vivo transduction, extensive attempts have been made to retarget Ad vectors to tumor cell markers (Coughlan et al., 2010). The integrin αvβ6 represents an attractive target for tropism-modified therapeutic Ads because expression in humans is restricted to epithelial remodeling events, such as wound healing and carcinogenesis, and it is largely undetectable in normal human epithelia (Breuss et al., 1995). Conversely, it is overexpressed in numerous human carcinomas (Ahmed et al., 2002; Sipos et al., 2004; Thomas et al., 2006; Hazelbag et al., 2007), where expression correlates with increased tumor cell migration, invasion, and advanced disease states (Ahmed et al., 2002; Bates et al., 2005; Elayadi et al., 2007). Conveniently, αvβ6-specific targeting peptides currently exist and have been validated extensively in vivo (Elayadi et al., 2007; Hausner et al., 2007; Saha et al., 2010). In contrast, the native Ad5 receptor, the coxsackievirus–adenovirus receptor (CAR), has a broad expression profile in human tissue (Bergelson et al., 1998). CAR also is thought to be poorly expressed in certain primary human tumor specimens (Jee et al., 2002; Rauen et al., 2002), and its downregulation has been associated with tumor progression and poor prognosis (Matsumoto et al., 2005; Anders et al., 2009). Such studies suggest that CAR may be dispensable for cancer-targeting strategies. In support of this, the finding that human, but not murine, erythrocytes express CAR on their surface emphasizes the importance of developing CAR-independent retargeting strategies, or at least the use of relevant in vivo models, with which to study tumor delivery (Carlisle et al., 2009; Seiradake et al., 2009).
However, CAR-binding ablation strategies alone have not proven useful in limiting the extensive liver transduction of Ad5 in vivo, nor in altering the overall biodistribution of the vector (Alemany and Curiel, 2001; Martin et al., 2003; Nicol et al., 2004). It has become clear that coagulation factors, notably FX, mediate hepatocyte transduction via heparan sulfate proteoglycans (HSPGs) after intravenous delivery (Parker et al., 2006; Waddington et al., 2008). Shayakhmetov and colleagues previously described an Ad5 mutant, Ad5mut, featuring a set of fiber mutations (Y477A and a TAYT deletion), which they proposed abrogated binding to factor IX (FIX) and C4b-binding protein (C4BP) (Shayakhmetov et al., 2005). When compared with Ad5, this vector was reported to display significantly reduced liver transduction and toxicity, low-level cytokine induction, and a failure to colocalize with Kupffer cells (KCs) after intravenous delivery.
We previously have described Ad5-EGFPA20, an HI loop-modified, αvβ6-retargeted vector, which showed improved tumor uptake accompanied by reduced liver transduction and hepatotoxicity when compared with Ad5 after intravenous delivery (Coughlan et al., 2009). Herein, we sought to improve on this prior construct by introducing an additional set of mutations to ablate CAR binding, in addition to putative fiber–FIX/C4BP interactions (Shayakhmetov et al., 2005). In the present study, we have investigated whether Ad5-477dlTAYTA20 retains or improves on the desirable in vivo characteristics of the parental vector Ad5-EGFPA20. We have assessed its safety profile by measuring the release of proinflammatory cytokines, its interaction with/effect on various hematological cells, its accumulation in the liver at early and late time points after intravenous injection, and the induction of hepatotoxicity (aspartate aminotransferase [AST]/alanine aminotransferase [ALT]). We have also determined its therapeutic efficacy and compared it with Ad5-EGFPWT and Ad5-EGFPA20, using a xenograft tumor model.
Materials and Methods
Cell lines
Human carcinoma cell lines BT-20 and DX3-β6 and their growth requirements have been described (Coughlan et al., 2009). Human breast adenocarcinoma line MCF10-CA1a (a kind gift from S. Santner, Karmanos Cancer Institute, Detroit, MI) was grown in Dulbecco's modified Eagle's medium (DMEM). Chinese hamster ovary cell lines CHO-K1 (American Type Culture Collection [ATCC], Manassas, VA), CHO-CAR (a kind gift from G. Santis, King's College London, London, UK), and CHO-pgsA745 were cultured in DMEM. CHO-pgsE606 and CHO-pgsF17 cells were grown in Ham's F12 medium. HEK293-β6 and JH293-β6 cells were generated by retroviral transduction, stably introducing β6 cDNA as described previously (Marshall et al., 1991). High β6-expressing cells were selected and maintained in puromycin-containing medium (5 μg/ml) and isolated by fluorescence-activated cell sorting (FACS), using a MoFlo FACS machine (Beckman Coulter, Brea, CA) and Summit version 4.0 software. Sorting was performed at the FACS Laboratory of the Cancer Research UK London Research Institute (London, UK).
Flow cytometry
Surface receptors (CAR/αvβ6) were detected by flow cytometry as previously described (Coughlan et al., 2009). Viral enhanced green fluorescent protein (EGFP) gene transfer and competition gene transfer assays were quantified by acquisition of EGFP fluorescence in FL1-H (fluorescence channel 1 height) by flow cytometry, using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and CellQuest Pro software for analysis. Blood samples were taken from mice 6 and 24 hr postinjection of virus and serum was separated, using Sarstedt CB 300 capillary tubes with clot activator (Sarstedt, Nümbrecht, Germany). Serum levels of IL-6, RANTES (regulated on activation, normal T cell expressed and secreted), interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and IL-12(p70) were quantified by multiplex cytometric bead assay, according to the manufacturer's instructions (FlowCytomix; Bender MedSystems, Austria). Calculations and analysis of results were performed with FlowCytomix Pro 2.2 software.
Construction of expression vectors and recombinant protein purification
The pQE30 expression plasmid, containing Ad5 knob and the A20-modified knob protein, has been described (Coughlan et al., 2009). pQE-KnobA20 was used as a template for mutagenic PCR to introduce the Y477AdlTAYT modification, using primers MutY477A-For (5′-TTCCTGGACCCAGAA
Characterization of recombinant knob proteins
Knob477dlTAYTA20 features a Y477A mutation in the fiber that ablates binding to CAR (Kirby et al., 1999) and a αvβ6-selective peptide ligand within the HI loop. CHO-CAR cells (1×105) were preincubated with increasing concentrations of KnobA20 or Knob477dlTAYTA20 (ranging from 0.001 to 100 μg/105 cells) as described (Coughlan et al., 2009). Knob477dlTAYTA20 was used to inhibit the binding of an αvβ6-specific antibody, 53A.2 (Coughlan et al., 2009). BT-20 cells (low CAR, high αvβ6) were incubated with increasing concentrations of protein (0.0001 to 10 μg/105 cells), after which unbound αvβ6 was detected by flow cytometry (Coughlan et al., 2009).
Generation of Ad genomes, virus production and titration
Fully replication-competent viruses were generated by two-step homologous recombination in yeast, using a strategy described in detail elsewhere (Coughlan et al., 2009). To create the Ad5-477dlTAYTA20 backbone (which also has EGFP in place of E3 6.7K/gp19K), pKnob477dlTAYTA20-TOPO was digested with PflMI and MscI, and the fragment containing the modifications was subcloned into the fiber shuttle vector pIVA20 (Coughlan et al., 2009). All viruses were purified by double CsCl banding. The non-CAR-binding virus, Ad5-477dlTAYTA20, was amplified on HEK293-β6 cells. Infectious titers (plaque-forming units [PFU]/ml) of Ad5-EGFPWT, Ad5-EGFPA20, and Ad5-477dlTAYTA20 were determined by titrating viruses in parallel on quadruplicate plates of JH293-β6 cells (Coughlan et al., 2009). Fluorescently labeled viral particles were generated with an Alexa Fluor 488 protein-labeling kit (Invitrogen, Carlsbad, CA) according to a previously established protocol (Bradshaw et al., 2010). Labeled particle titers were determined with a micro-bicinchoninic acid (BCA) assay (Bio-Rad, Hercules, CA) and viral particle (VP) titers were calculated according to 1 μg=4×109 particles.
Hemagglutination of human erythrocytes
We isolated erythrocytes from human blood samples, obtained from volunteers with approval of the local ethics committee at University of Glasgow. Positive control samples were treated with 0.1% Triton X-100 and negative controls with phosphate-buffered saline (PBS). Hemagglutination was determined according to a previously described method (Cichon et al., 2003).
Viral gene transfer with or without FIX, FX, and modified heparins
Standard viral transduction and cytotoxicity assays were performed exactly as described (Coughlan et al., 2009). CHO-K1 or CHO-CAR cells (1×105) were seeded in 24-well plates and allowed to attach overnight. Triplicate wells were then infected for 2 hr at 37°C at a multiplicity of infection (MOI) of 10 (PFU/cell) with Ad5-EGFPWT, Ad5-EGFPA20, or Ad5-477dlTAYTA20 alone, or mixed with physiological concentrations of FIX (5 μg/ml; Cambridge Biosciences, Cambridge, UK), FX (10 μg/ml; Cambridge Biosciences), FIX/FX in addition to porcine heparin, de-N-sulfated heparin, or de-O-sulfated heparin (25 μg/ml; Sigma-Aldrich, St. Louis, MO), or virus with heparin/modified heparins alone (25 μg/ml). Transduction assays were also performed in CHO-pgsA745, CHO-pgsE606, and CHO-pgsF17 cells, which are defective in heparan sulfate glycosaminoglycan (HS-GAG) synthesis (Esko et al., 1987), O- and particularly N-sulfate HS side-chain synthesis (Bame and Esko, 1989), or lack 2-O-sulfated residues (Bai and Esko, 1996), respectively. After incubation, cells were washed twice in PBS, medium was replaced with fresh culture medium, and transduction was allowed to proceed for ∼22 hr under standard conditions. EGFP fluorescence, detected by flow cytometry, was the end point for these assays.
In vivo animal experiments
Animal experiments using immunocompetent BALB/c male mice (6–8 weeks) were carried out at the facility of the Institut d'Investigació Biomèdica de Bellvitge (IDIBELL), within the Institut Català d'Oncologia (ICO, Barcelona, Spain), in accordance with the regulations of the IDIBELL Ethics Committee for Animal Experimentation. Experiments performed in tumor-bearing animals were carried out at the Barts Cancer Institute under Home Office regulations. For biodistribution studies, animals injected intravenously with 4×1010 VP of Ad5-EGFPWT, Ad5-EGFPA20, or Ad5-477dlTAYTA20 were killed 1, 24, or 96 hr postinjection. Viral genomes from tissue were evaluated by quantitative PCR (qPCR) as described previously (Coughlan et al., 2009). To identify virus-interacting cells in the liver and spleen at early time points postinjection (1 hr), viruses were labeled with fluorescent Alexa Fluor 488 (green). To deplete macrophages, 200 μl of PBS or liposome-encapsulated clodronate (dichloromethylene diphosphonate [Cl2MDP], or clodronate, was a gift from Roche Diagnostics, Mannheim, Germany) was administered 48 hr before delivery of labeled viruses (van Rooijen and van Kesteren-Hendrikx, 2003). Blood samples were obtained 6 or 24 hr postinjection. Hematological profiles (24 hr) and liver transaminases (96 hr), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were determined by the Clinical Veterinary Biochemistry Service at the Facultat de Veterinària, Universitat Autònoma de Barcelona (Barcelona, Spain).
MCF10-CA1a human breast carcinoma cells (2×106) were injected subcutaneously into the left and right flanks of CD1 nude female mice. When tumors reached ∼250 mm3 animals were randomized and matched according to average tumor volumes (n=8 per group). Animals were injected intravenously with a single dose of PBS (control), or with replication-competent virus Ad5-EGFPWT (2×1010 VP), Ad5-EGFPA20 (2×1010 or 4×1010 VP), or Ad5-477dlTAYTA20 (2×1010 or 4×1010 VP). Ad5-EGFPWT at the higher dose of 4×1010 VP could not be administered because of the profound hepatocyte transduction, toxicity, and morbidity induced by day 4 postinjection. Tumor volumes and mouse morbidity (e.g., body weight, tumor ulceration) were monitored three times per week. Tumor volume was calculated according to the equation V (mm3)=π/6×W (width)×L 2 (length). The percentage of growth was defined as [(VX −V 0)/V 0]×100, where VX is the tumor volume on the day of measurement and V 0 is the tumor volume on day 0 (Cascallo et al., 2007). All animals were killed once the tumors of the PBS control group approached 1.4 cm3.
Viral biodistribution and immunohistochemistry
Viral genomes were quantified from tissue as described (Coughlan et al., 2009). For 1-hr experiments with Alexa Fluor 488-labeled virus, immunohistochemistry was carried out on frozen liver and spleen sections, using antibodies to macrophage marker F4/80 (KCs) and the splenic marginal zone macrophage marker MARCO (macrophage receptor with a collagenous structure) (Table 1) as described (Alba et al., 2010). Frozen liver sections (24 and 96 hr postinjection) were stained with an anti-E1A antibody (Table 1) as described previously (Coughlan et al., 2009, 2010). Images were captured with fluorescence filters for fluorescein isothiocyanate (FITC) and tetramethylrhodamine isothiocyanate (TRITC) on an Olympus BX60 fluorescence microscope (×10 or×60 oil immersion objective). Images were acquired and merged, using Cell^M software (Olympus, UK), and processed to adjust brightness and contrast, using ImageJ (National Institutes of Health, Bethesda, MD). Adjustments were applied equally to all compared images.
Ab, antibody; Gt, goat; IHC-Fr, immunohistochemistry frozen; IHC-P, immunohistochemistry paraffin; Ms, mouse; N/A, not applicable; PFA, paraformaldehyde; Rb, rabbit; Rt, rat.
Note: Isotype control antibodies were as follows: mouse IgG (Dako), rat IgG2a (Abcam), rat IgG1 (Vector Laboratories), and rabbit IgG (Santa Cruz Biotechnology).
Antibody was provided as part of the †mouse-on-mouse (M.O.M.) basic kit or the ‡ABC Universal Elite kit (both from Vector Laboratories).
Hematoxylin and eosin (H&E) staining was carried out by the Pathology Service at Barts Cancer Institute (London, UK). Staining for E1A or CD31 in paraffin-embedded MCF10-CA1a tumor xenografts was performed as follows: sections were dewaxed and rehydrated, and antigen retrieval was performed with boiling sodium citrate (pH 6.0). Endogenous avidin–biotin activity was blocked with a commercial kit from Vector Laboratories (Burlingame, CA). Sections were blocked for 30 min at 37°C in blocking buffer (PBS with 1% [w/v] bovine serum albumin [BSA], 10% normal horse serum, and 0.1% [v/v] Triton X-100). Primary anti-E1A (rabbit polyclonal IgG; 13S-5) or anti-CD31 (rat IgG2a; SZ31) antibodies were incubated with sections for 1 hr at room temperature. Secondary detection was achieved with an ABC Universal Elite kit (E1A) or ABC standard kit (CD31), followed by application of a 3,3′-diaminobenzidine (DAB) peroxidase substrate kit (all kits from Vector Laboratories) for chromogenic color development. Nuclei were counterstained with Mayer's hematoxylin, dehydrated by exposure to an alcohol gradient, and mounted on slides. Immunostaining for αvβ6 integrin (mouse IgG; 62G2) was performed as described previously (Clark et al., 2011). Antigen retrieval for αvβ6 integrin or pan-cytokeratin (AE1/AE2) was achieved with enzymatic pepsin or proteinase K, respectively. Both antibodies were used with a M.O.M. blocking kit (Vector Laboratories). Further details on antibodies used are outlined in Table 1. Staining was imaged with an Olympus BX40, QImaging MicroPublisher 3.3 RTV microscope/camera and images were acquired with QCapture Pro 6.0 software.
Quantification of immunohistochemistry
E1A-, αvβ6-, CD31-, and cytokeratin-positive staining was quantified with Image-Pro Analyzer 7.0 software (Media Cybernetics, Bethesda, MD), as recommended (
Statistical analysis
In vitro data show means±SD of triplicate samples and are representative of two independent experiments carried out on different occasions. In vivo data show means±SEM of n=5–8 animals per group (indicated in the figure legends). Statistical significance was determined by unpaired Student t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; NS, not statistically significant [p>0.05]).
Results
Fiber modifications (Y477AdlTAYT) ablate CAR binding but do not affect A20-mediated entry via αvβ6 integrin
Recombinant fiber knob protein blocking of Ad transduction is a well-established assay. KnobWT previously has been shown to inhibit Ad5 with a median inhibitory concentration (IC50) of 0.115 μg/105 cells (Kirby et al., 2000). We tested the ability of KnobA20 and Knob477dlTAYTA20 to block the CAR-mediated entry of Ad5-EGFPWT into CHO-CAR cells (Fig. 1A). Preincubation of cells with KnobA20 inhibited Ad5-EGFPWT transduction at an IC50 of 0.34 μg/105 cells (Coughlan et al., 2009) close to the previously published IC50 for Ad5. Knob477dlTAYTA20 failed to inhibit Ad5-EGFPWT infection significantly at concentrations up to ≤100 μg/105 cells.
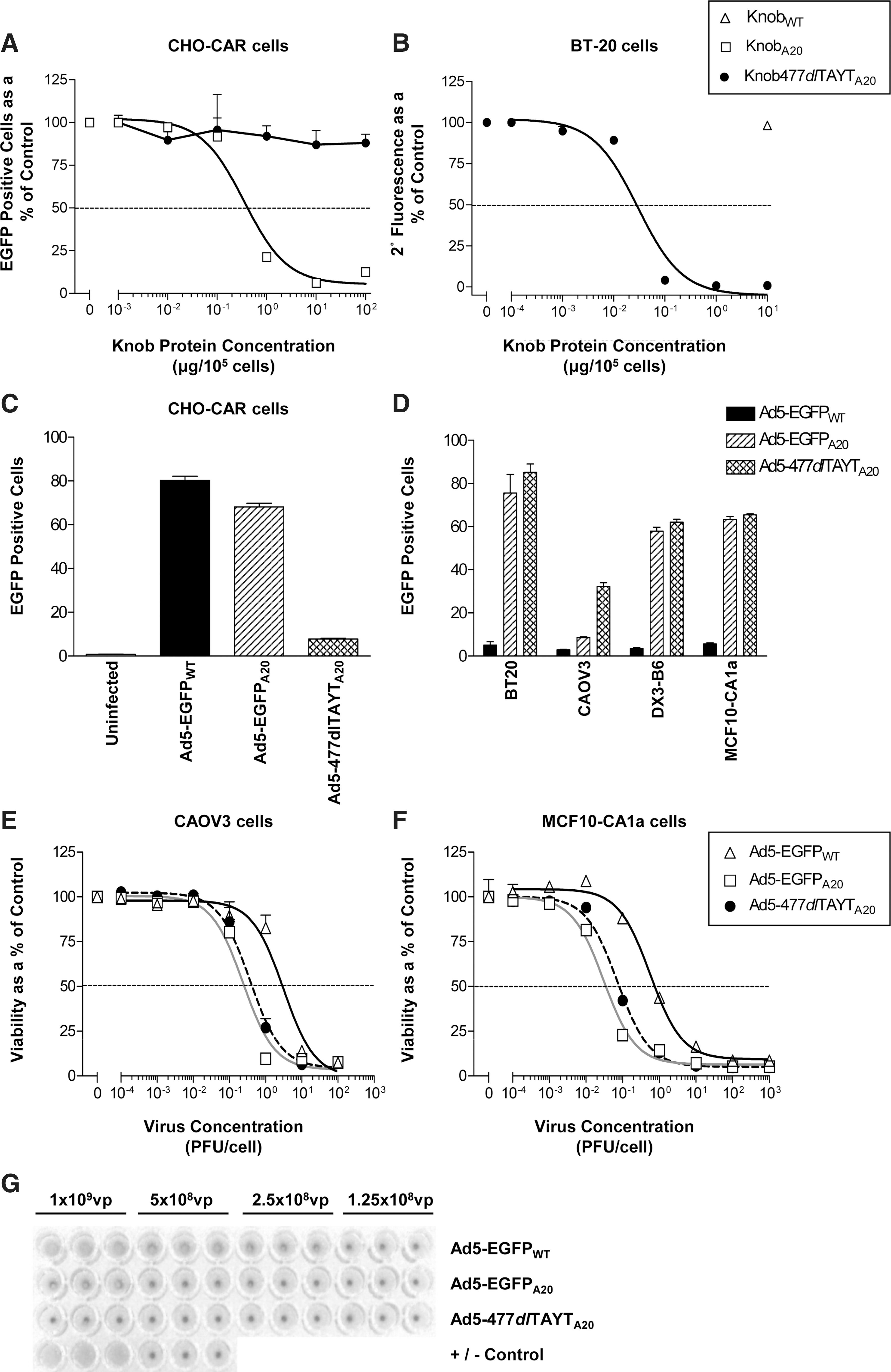
In a separate experiment, Knob477dlTAYTA20 was tested for its ability to bind αvβ6 in vitro (Fig. 1B) and inhibit the subsequent binding of an αvβ6-specific antibody (53A.2). We have previously described that KnobA20 inhibits binding of 53A.2 to αvβ6 with an IC50 of 0.03 μg/105 cells (Coughlan et al., 2009). Knob477dlTAYTA20 inhibited 53A.2 binding at an identical concentration, indicating that the introduced mutations did not affect the interaction between the A20 peptide and αvβ6 integrin. KnobWT negative control was unable to block 53A.2 binding at a high concentration (10 μg/105 cells) and did not differ from untreated cells (p=0.168).
We confirmed that the CAR-binding capacity of the Ad5-477dlTAYTA20 virus was ablated by assessing its transduction on CHO-CAR cells (Fig. 1C). As expected, Ad5 transduction was high (∼80%) whereas in contrast, Ad5-477dlTAYTA20 levels were low (∼10%), confirming that the CAR-binding capacity was ablated in this virus. CAR-mediated transduction with Ad5-EGFPA20 was slightly impaired relative to Ad5-EGFPWT, suggesting that the A20 insertion interferes to some degree with the fiber knob:CAR interaction.
Using a panel of low-CAR, high-αvβ6 human carcinoma cell lines (Table 2), the transduction efficiency of Ad5-477dlTAYTA20 was compared with those of Ad5-EGFPWT and Ad5-EGFPA20 (Fig. 1D). Ad5-477dlTAYTA20 displayed comparable, or superior, transduction efficiency compared with Ad5-EGFPA20, and both were significantly better than Ad5-EGFPWT at an MOI of 10 (PFU/cell). We also verified that Ad5-477dlTAYTA20 exhibited enhanced tumor cell killing relative to Ad5-EGFPWT (Fig. 1E and F). Comparative cytotoxicity profiles (MTT assay), obtained 120 hr postinfection, showed that Ad5-EGFPA20 and Ad5-477dlTAYTA20 had lower EC50 values (Table 3) compared with Ad5-EGFPWT. Collectively, these data confirmed that the A20 peptide, when combined with the introduced mutations, retained its full ability to interact with αvβ6.
CAR, coxsackievirus–adenovirus receptor; Geo. mean, geometric mean fluorescence.
Percent positive statistics (1×104 gated events) were collected by flow cytometry, using single-parameter histograms (FL1-H). Negative control isotype IgG fluorescence values were subtracted from the geometric mean fluorescence of the test antibody (Rmc B/53A.2). Isotype controls: mouse IgG1 for anti-CAR (RmcB) and rat IgG2a for anti-αvβ6 (53A.2).
EC50 values represent half the maximal effective concentration of virus per cell. Viability was assessed by MTT assay and results analyzed by nonlinear regression fitted to a sigmoidal curve. Values expressed as plaque-forming units (PFU)/cell.
It is now known that Ad5 agglutinates human erythrocytes via CAR (Nicol et al., 2004; Carlisle et al., 2009; Seiradake et al., 2009). This fact has prompted some concerns over the relevance of performing tumor-targeting studies in mice, as murine erythrocytes do not express CAR. Ad5-477dlTAYTA20 did not agglutinate human erythrocytes, even at the highest concentration of virus (Fig. 1G), presumably as a result of the introduced Y477A mutation at a critical CAR-binding residue. With the exception of the highest dose, Ad5-EGFPA20 also failed to agglutinate human erythrocytes, possibly because of its slightly reduced interaction with CAR (see Fig. 1A and C).
In vitro characterization of viral entry with or without coagulation factors and modified heparins
Coagulation factor-mediated hepatocyte transduction has been shown to result from engagement with HSPGs, preferentially those with sulfated side chains (Bradshaw et al., 2010). FIX/FX-mediated infectivity enhancement was assessed, using HSPG-expressing CHO-K1 cells (Fig. 2A–C) and CHO cells with deficiencies in the synthesis of HS side chains (Fig. 2D). Transduction of the Ad5:FX complex can be inhibited by preincubation of CHO-K1 cells with porcine, but not modified, heparins (Bradshaw et al., 2010). Transduction assays on CHO-K1 cells compared Ad5-EGFPWT (Fig. 2A), Ad5-EGFPA20 (Fig. 2B), and Ad5-477dlTAYTA20 (Fig. 2C)±FIX/FX and porcine heparin, de-N-sulfated heparin, and de-O-sulfated heparin.
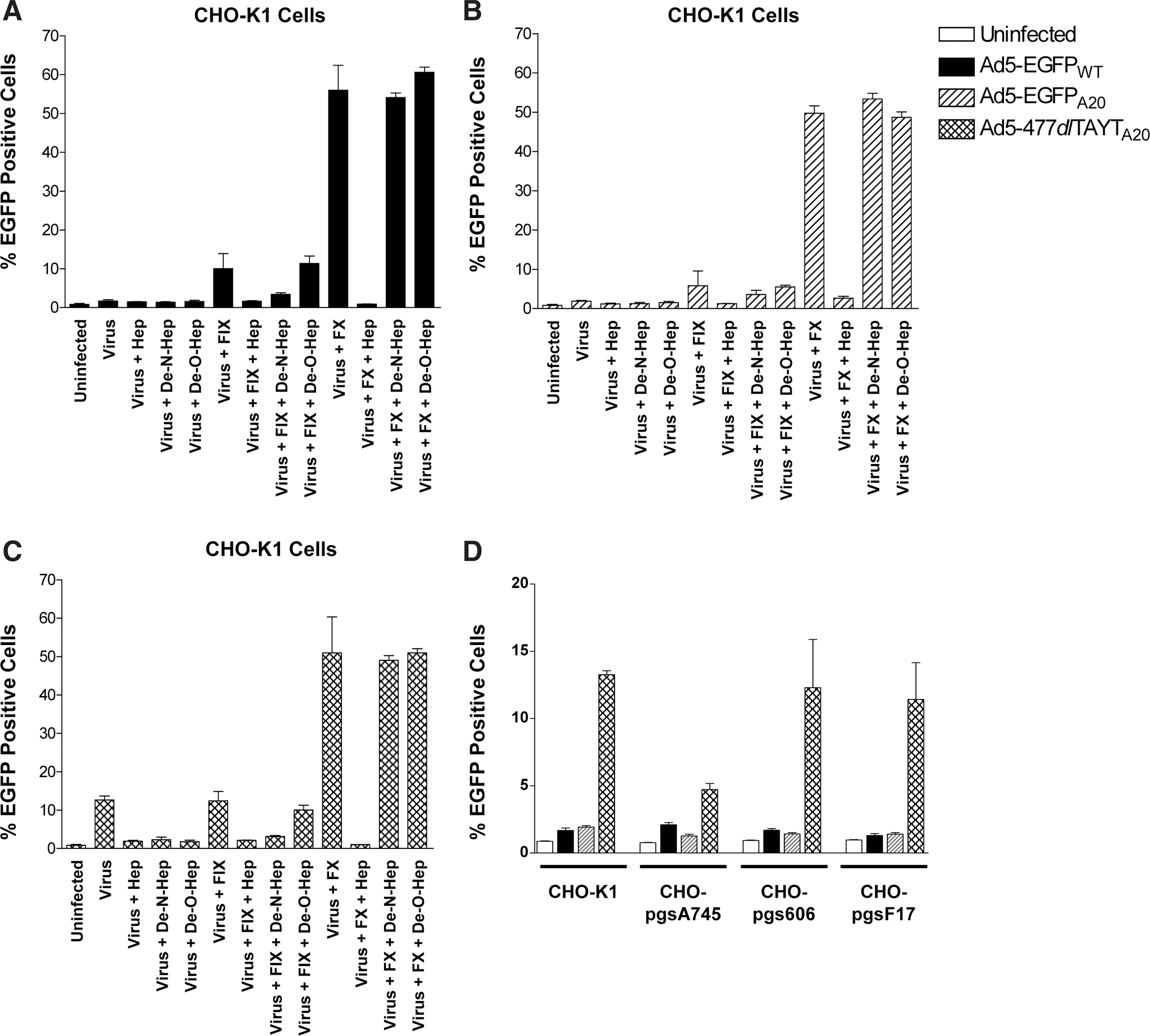
Comparative transduction of
FIX and FX enhanced transduction of Ad5-EGFPWT ∼5-fold and >30-fold, respectively (Fig. 2A), and this was ablated by both heparin and de-N-sulfated heparin, but not by de-O-sulfated heparin. The FX effect was completely inhibited by heparin, but not by de-N- or de-O-sulfated heparin. Transduction of CHO-K1 cells with Ad5-EGFPA20 was enhanced marginally (∼2-fold) by FIX, although this was not found to be statistically significant (Fig. 2B). FX significantly enhanced the transduction of Ad5-EGFPA20 (∼30-fold increase; p<0.0001) and, similar to Ad5-EGFPWT, this effect was inhibited by heparin but not by de-N- or de-O-sulfated heparin. Unexpectedly, Ad5-477dlTAYTA20 mediated ∼15% transduction in CHO-K1 cells (Fig. 2C), compared with <2% transduction levels detected with Ad5-EGFPWT or Ad5-EGFPA20 (Fig. 2A and B). This was inhibited completely when cells were coincubated with heparin, de-N-sulfated heparin, and de-O-sulfated heparin. FX enhanced the entry of Ad5-477dlTAYTA20 in a manner similar to Ad5-EGFPWT and Ad5-EGFPA20. No significant infectivity enhancement was observed on coincubation of Ad5-477dlTAYTA20 with FIX (Fig. 2C; p=0.11).
The transduction of each virus was assessed in CHO cell derivates (CHO-pgsA745, CHO-pgsE606, and CHO-pgsF17), which are deficient in HSPGs or defined side-chain modifications (Fig. 2D). Ad5-477dlTAYTA20 entry was reduced dramatically, in CHO-pgsA745 cells, which are completely deficient in HSPGs, whereas transduction was not affected by the absence of O- or N-sulfated HS side chains (CHO-pgsE606) or 2-O-sulfated residues (CHO-pgsF17).
Investigation of virus accumulation at early time points (1 hr) after intravenous delivery
Ad5-EGFPA20 displays reduced hepatotropism compared with Ad5-EGFPWT, 72 hr postinjection (Coughlan et al., 2009). These prior studies were performed in tumor-bearing immunodeficient mice with no examination of liver uptake at earlier time points. Here, we injected immunocompetent mice with 4×1010 VP of Alexa Fluor 488-labeled viruses: Ad5-EGFPWT (Ad5-488), Ad5-EGFPA20 (AdA20-488), or Ad5-477dlTAYTA20 (477YT-488). Viral genome distribution was quantified and virus colocalization was established 1 hr postinjection. We previously suggested that the reduced liver tropism we observed with Ad5-EGFPA20 may be due to differential interactions with scavenging macrophages after intravenous delivery (Coughlan et al., 2009). To investigate this possibility, separate groups of animals were pretreated with clodronate (Cl2MBP)-containing liposomes to eliminate macrophages.
We observed significantly fewer Ad5-EGFPA20 (>10-fold fewer; p<0.024) and Ad5-477dlTAYTA20 (>60-fold fewer; p<0.015) genomes in the liver when compared with Ad5-EGFPWT (Fig. 3A). However, pretreatment with Cl2MBP did not alter the early uptake of any virus in the liver relative to untreated groups. In untreated animals (–Cl2MBP) virus (green) colocalized with F4/80+ (red) hepatic macrophages (Fig. 3C, left), although the density of viral particles within these KCs appeared less concentrated for both Ad5-EGFPA20 and, especially, Ad5-477dlTAYTA20. Quantification of Alexa Fluor 488-labeled viral particles per field of view in multiple liver sections, using ImageJ image analysis software, confirmed the reduced accumulation of both A20 viruses in the liver (Fig. 3B). In the livers of pretreated animals (+Cl2MBP), F4/80+ macrophages were effectively eliminated and the pattern of viral particle distribution was broad and diffuse (Fig. 3C, right). We detected Ad5-EGFPWT and Ad5-EGFPA20 particles scattered over the surface of the liver parenchyma, although with considerably fewer Ad5-EGFPA20 particles. Ad5-477dlTAYTA20 was not detected in any liver sections of Cl2MBP-treated animals. These data were also further confirmed by image analysis and quantification (Fig. 3B).
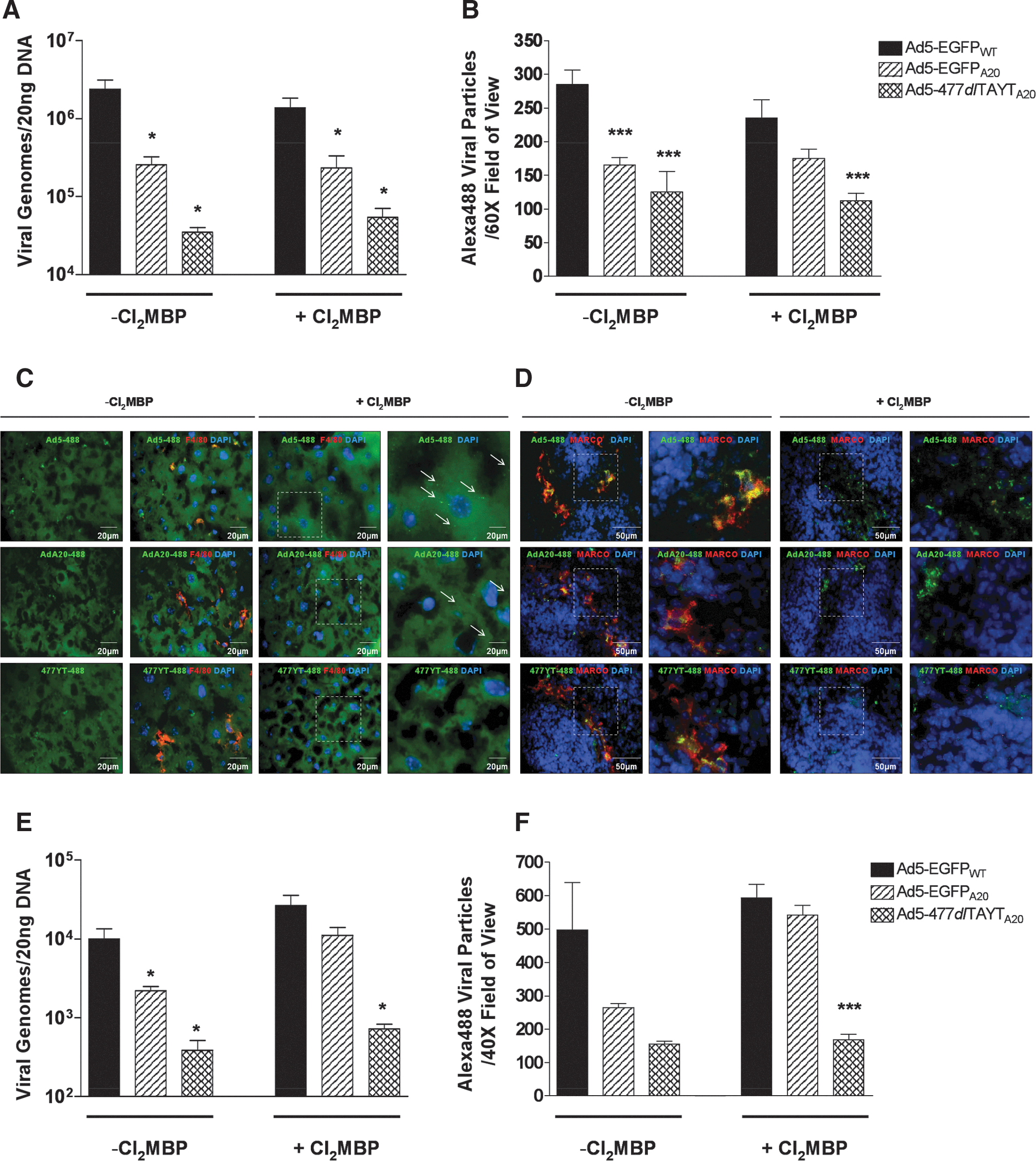
Analysis of viral genome accumulation in liver and spleen 1 hr postinjection (4.0×1010 VP/mouse). BALB/c immunocompetent mice were injected intravenously with PBS, 4×1010 VP of Alexa Fluor 488-labeled Ad5-EGFPWT (Ad5-488), Ad5-EGFPA20 (AdA20-488), or Ad5-477dlTAYTA20 (477YT-488). To deplete macrophages, separate groups of mice were pretreated with PBS (–Cl2MBP) or clodronate liposomes (+Cl2MBP) 48 hr before virus injection. Absolute viral genomes were quantified by TaqMan real-time quantitative qPCR, using a probe to detect the hexon region of the genome.
Lower levels of Ad5-EGFPA20 and Ad5-477dlTAYTA20 genomes were detected in the spleen compared with Ad5-EGFPWT (Fig. 3E). Evidence has highlighted the importance of the spleen as a major site for the induction of innate immune responses directed toward intravenously delivered Ads (Zhang et al., 2001; Koizumi et al., 2007). Therefore, vectors that have limited splenic uptake may elicit reduced inflammatory responses and subsequent toxicity. In animals pretreated with Cl2MBP liposomes there were slight increases in the level of qPCR-detected virions. As reported elsewhere (Di Paolo et al., 2009a), we detected Ad5-EGFPWT and Ad5-EGFPA20 colocalized with MARCO+ scavenging macrophages, which reside in the marginal zone (MZ) of the spleen (Fig. 3D, left). Pretreatment with Cl2MBP completely eliminated the MARCO+ MZ macrophage population (Alba et al., 2010), but diffuse Ad5-EGFPWT and Ad5-EGFPA20 viral particles were clearly detectable in a similar anatomical location to the MZ, surrounding the white pulp (Fig. 3D, right). Viral particles did not appear to colocalize with 4′,6-diamidino-2-phenylindole (DAPI), suggesting that virions were largely extracellular at this time point. Images were quantified by ImageJ analysis for the number of Alexa Fluor 488-labeled viral particles in multiple sections from various animals (Fig. 3F).
Assessment of viral genome distribution and E1A viral gene expression 24 hr after intravenous delivery (4.0×1010 VP/mouse)
Absolute genome copy numbers in the liver were quantified as described previously (Fig. 4A). Approximately 17-fold fewer Ad5-EGFPA20 and ∼27-fold fewer Ad5-477dlTAYTA20 genomes were detected in liver compared with Ad5-EGFPWT (p<0.0001 for both). We also detected ∼2-fold fewer Ad5-EGFPA20 and Ad5-477dlTAYTA20 genomes in spleen relative to Ad5-EGFPWT (p=0.013 and p=0.007, respectively). Substantially lower levels of E1A expression also were detected in the livers of groups that had received the A20-retargeted viruses (Fig. 4B).

Quantification of inflammatory (6 hr) and hematological (24 hr) profiles after intravenous delivery of virus (4.0×1010 VP/mouse)
We wished to obtain inflammatory and hematological profiles for each virus after intravenous delivery. Intravenous delivery of Ad5 has been shown to induce rapid innate immune responses, with the release of various proinflammatory cytokines and chemokines (Lieber et al., 1997; Zhang et al., 2001; Shayakhmetov et al., 2005; Di Paolo et al., 2009a) ∼6–12 hr postinfection (Lieber et al., 1997). Ad5-EGFPWT elevated serum cytokine/chemokines significantly 6 hr postinjection (Fig. 4C). IL-6 was elevated ∼22-fold (p<0.0001) in mice treated with Ad5-EGFPWT whereas the levels of IL-6 induced by Ad5-EGFPA20 and Ad5-477dlTAYTA20 were substantially lower. RANTES and IFN-γ levels were elevated above baseline in all virus-treated cohorts (p<0.01). However, Ad5-EGFPWT induced substantially higher levels of RANTES (∼13-fold elevation; p<0.0001) and IFN-γ (∼1400-fold; p<0.0001) compared with Ad5-EGFPA20 and Ad5-477dlTAYTA20 (∼4-fold elevation [p<0.004] and ∼7-fold elevation [p=0.001], respectively). No significant elevation of TNF-α levels occurred for any virus (p>0.05 for all), as reported by others (Zhang et al., 2001).
Systemic administration of Ad5 induces acute transient thrombocytopenia, which occurs between 5 min and 24 hr postinjection in mice (Othman et al., 2007). A full hematological profile for each of the treatment groups (24 hr) was assessed (Fig. 4D). Interestingly, only Ad5-EGFPWT induced thrombocytopenia (∼2-fold reduction in total platelet count; p=0.003), whereas the total platelet counts of animals treated with Ad5-EGFPA20 or Ad5-477dlTAYTA20 were not altered, being similar to the PBS control group (p=0.228 and p=0.213, respectively). Total monocyte and lymphocyte levels were reduced in all virus-treated cohorts. Total neutrophil levels were reduced in the Ad5-EGFPWT-treated group. No significant differences were observed in overall basophil or eosinophil populations (data not shown).
Quantification of viral genome distribution and toxicity 96 hr postinjection (4.0×1010 VP/mouse)
We chose a late time point postinjection for analysis of toxicity and acute hepatic injury. Separate groups of animals were killed 96 hr postinjection, and their livers and spleens were harvested for analysis (Fig. 5A). Absolute viral genomes were quantified by qPCR. As expected, significant amounts of Ad5-EGFPWT were detected in the liver but, in agreement with findings at 1 and 24 hr, we detected ∼7.5-fold fewer Ad5-EGFPA20 (p=0.002), and ∼5.5-fold fewer Ad-477dlTAYTA20 (p=0.002) genomes. Ad5-EGFPA20 and Ad5477dlTAYTA20 genome levels in the spleen were also ∼2.6-fold lower than Ad5-EGFPWT (p=0.0008 and p=0.007, respectively). No differences were noted between Ad5-EGFPA20- and Ad5-477dlTAYTA20-treated cohorts (p=0.929). By immunohistochemistry, we observed a clear reduction in levels of E1A expression in the livers of Ad5-EGFPA20- and Ad5-477dlTAYTA20-treated groups, compared with the Ad5-EGFPWT group (Fig. 5B), correlating with our findings at 24 hr.
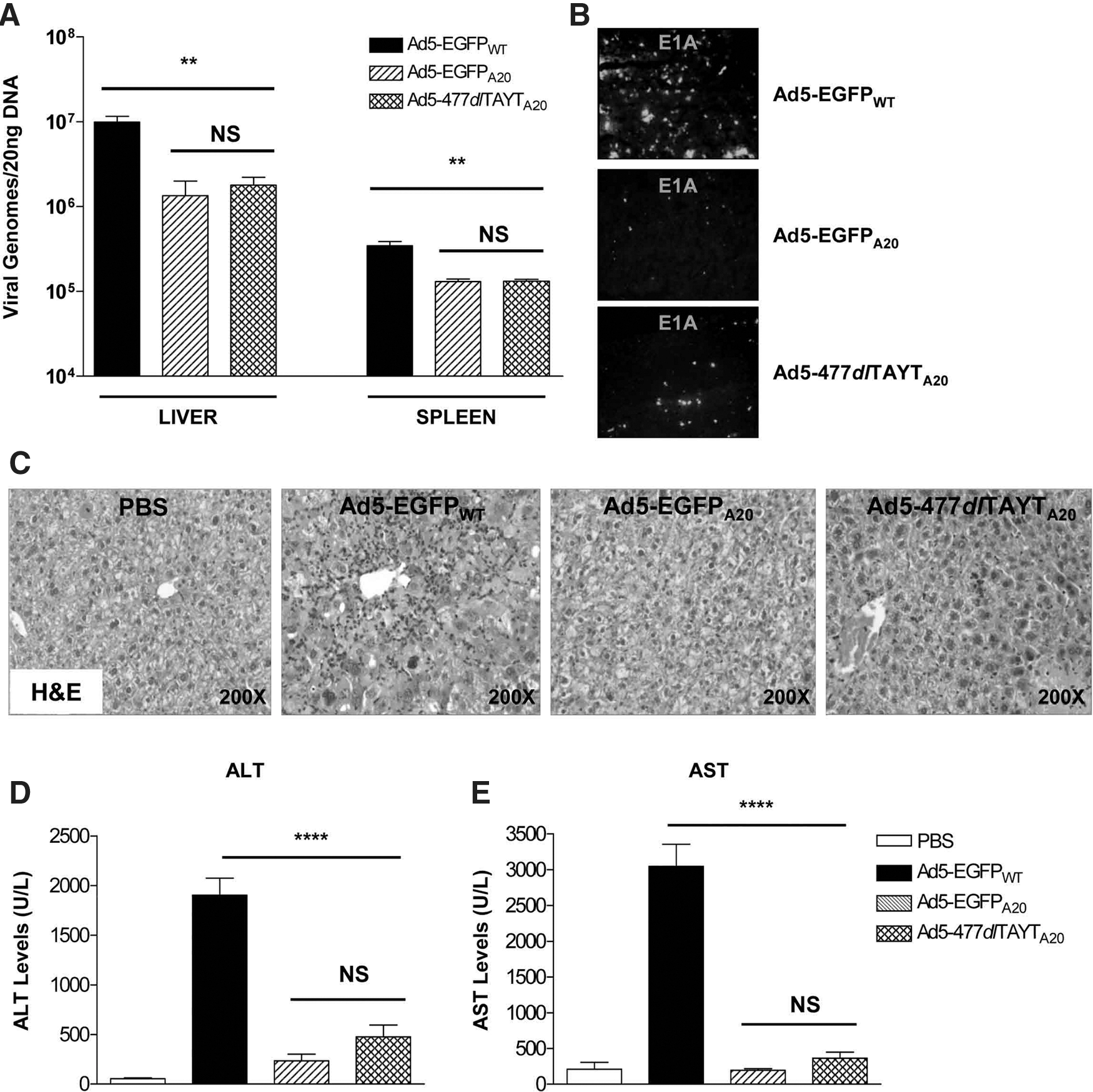
H&E staining of liver sections revealed distinct histological differences between Ad5-EGFPWT- and Ad5-EGFPA20/Ad5-477dlTAYTA20-treated groups (Fig. 5C). The livers of mice administered Ad5-EGFPWT showed striking hepatocyte atypia, with ballooning degeneration of cells and individual cell necrosis. A prominent periportal lymphocytic inflammatory infiltrate was present in the livers of animals that received Ad5-EGFPWT but not those that received Ad5-EGFPA20 or Ad5-477dlTAYTA20. Liver transaminase levels were altered significantly between the Ad5-EGFPWT group and A20-retargeted groups (Fig. 5D and E). The Ad5-EGFPWT-treated group displayed markedly elevated ALT levels, ∼35-fold above basal PBS levels (p<0.0001). In comparison, the elevation of ALT in the Ad5-EGFPA20- and Ad5-477dlTAYTA20-treated groups was only ∼4.5-fold (p=0.027) and ∼9-fold (p=0.008), respectively. AST levels were also similarly affected by each virus (Fig. 5E). Ad5-EGFPWT treatment induced high levels of AST, ∼15-fold above the basal level (p<0.0001). However, neither the Ad5-EGFPA20 nor Ad5-477dlTAYTA20 group presented an elevated AST level compared with the basal PBS level (p>0.05 for both).
Comparison of antitumoral efficacy of intravenously delivered Ad5-EGFPWT, Ad5-EGFPA20, and Ad5-477dlTAYTA20 and measurement of therapeutic outcome in MCF10-CA1a xenograft model
We tested the antitumoral efficacy of Ad5-EGFPWT, Ad5-EGFPA20, and Ad5-477dlTAYTA20 (2×1010 VP) after intravenous delivery in immunodeficient mice, using a subcutaneous MCF10-CA1a xenograft model (Fig. 6A and B). In previous experiments, Ad5-EGFPA20 and Ad5-477dlTAYTA20 were well tolerated in immunocompetent mice at 4×1010 VP. Therefore, we also decided to test whether we could improve the therapeutic outcome by using this increased dose of vector.

Antitumoral efficacy in MCF10-CA1a tumor xenograft model after single intravenous delivery of virus. MCF10-CA1a xenografts were grown on the flanks of CD1 nude mice to an average volume of 250 mm3. Animals were injected intravenously with 2×1010 VP (all viruses), 4×1010 VP (Ad5-EGFPA20 or Ad5-477dlTAYTA20 only), or PBS, and tumor volumes were recorded three times weekly until the experiment terminated (day 14 postinjection). Absolute tumor volumes are shown in
The appropriate end point for solid tumor models is tumor growth inhibition or tumor growth delay (Yarnold et al., 1986; Wallace, 2000; Teicher, 2006). Using percent tumor growth as a measure of therapeutic outcome, we found that treatment with Ad5-477dlTAYTA20 slowed tumor growth compared with PBS control (Fig. 6B). At 4 days postinjection, both Ad5-477dlTAYTA20-treated groups displayed retarded tumor growth compared with the Ad5-EGFPWT-treated animals (p=0.02). Absolute tumor volumes also differed at this time point; the Ad5-EGFPWT group had an average volume of ∼428 versus ∼372/340 mm3 for low-dose and high-dose Ad5-477dlTAYTA20, respectively. Tumors of the group that received the lower dose of 2×1010 VP continued to exhibit slower growth than other treatment groups and differed significantly from Ad5-EGFPWT again on day 9 postinjection (p=0.048). Again, the tumor volumes of both Ad5-477dlTAYTA20 groups were reduced in comparison with the Ad5-EGFPWT group (∼507 and ∼496 mm3 vs. ∼549 mm3). However, differences in absolute tumor volume did not reach significance because of the variable size of tumors within the treatment groups. We had to terminate the experiment on day 14 postinjection because of the large volume and ulceration of tumors within the PBS control group. We observed that although most treatment groups had an improved therapeutic outcome versus the PBS-treated group, it was disappointing to note that there were no differences between animals that received Ad5-EGFPWT and animals that received A20-retargeted viruses (p>0.05).
We confirmed that the MCF10-CA1a tumors retained high-level expression of the target receptor, αvβ6, by immunohistochemistry (Fig. 6C). We then analyzed tumors from several animals for viral antigen by E1A staining (Fig. 6C). Reassuringly, the pattern of E1A staining in the A20-treated groups correlated with regions of αvβ6 + tumor cells. However, there were distinct differences in the extent of E1A staining, as well as the overall dissemination and distribution of viral gene expression throughout the tumor mass. Overall, we observed greater amounts of E1A staining in the tumors of Ad5-EGFPA20- and Ad5-477dlTAYTA20-treated animals than in those of Ad5-EGFPWT-treated animals, suggesting that these vectors had improved tumor targeting or, at least, increased intratumoral replication in αvβ6 + cells. We confirmed these observations by quantifying the amount of E1A+ staining in tumor images, analyzing ∼30 tumor images from 4–6 different animals for each separate group (Fig. 7A). We found that Ad5-EGFPWT transduced approximately 1.5% of the tumor sections analyzed, whereas at the same dose (2×1010 VP), Ad5-EGFPA20 and Ad5-477dlTAYTA20 transduced ∼3.3 and 2.8%, respectively. After this type of analysis, we found that the higher dose of both A20-retargeted vectors displayed increased tumor transduction when compared with the lower dose, with ∼4.6% E1A expression (both doses) detected versus ∼3.3 and 2.8%.
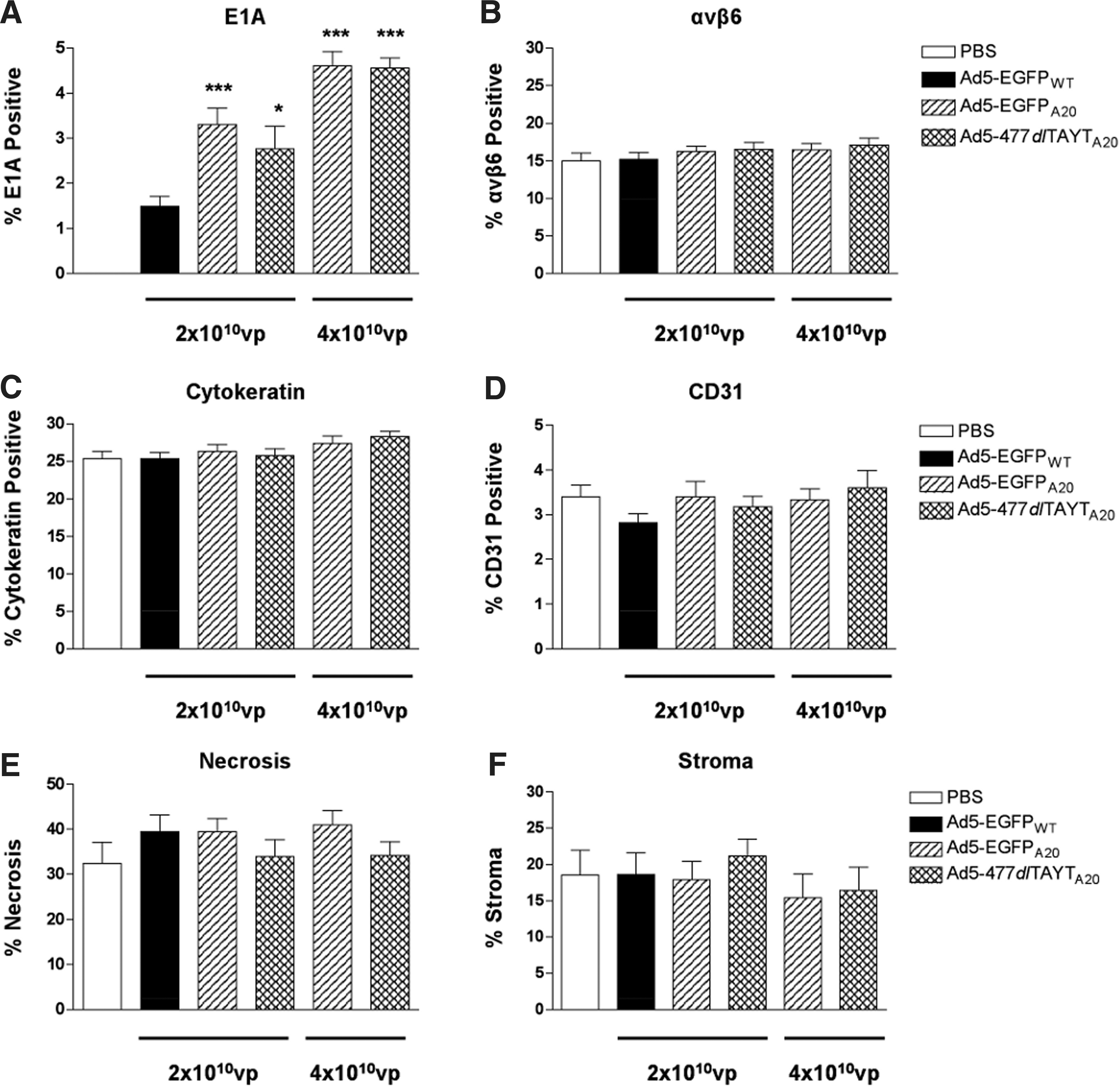
Quantification of immunohistochemistry. Staining positive for
To eliminate the possibility that differences in the levels of αvβ6 expression had affected the efficiency of tumor transduction we also quantified the amount of αvβ6 + staining in tumor images, as described previously (Fig. 7B). There were no differences in the percentage αvβ6 + regions between groups, with ∼16% of the tumors being composed of αvβ6 + carcinoma cells. To assess the cellular composition of the tumor, we also performed immunohistochemistry to detect human cytokeratin (carcinoma cells) and murine CD31 (murine endothelial cells of the tumor vasculature). Approximately 26% of the tumors analyzed were cytokeratin+, demonstrating that there were both αvβ6-positive and αvβ6-negative carcinoma cells within the tumor (Fig. 7C). The amount of tumor vasculature did not differ between groups, with CD31+ cells making up ∼3.3% of the tumors (Fig. 7D). Therefore, we concluded that differences in tumor transduction efficiency due to varying levels of viable tumor cells or differential vascularization were unlikely. In H&E-stained sections, we noted that areas of viable tumor cells were separated by large areas of stromal tissue and extensive regions of necrosis (Fig. 6C). Using image analysis software, we estimated the proportion of necrotic and stromal cells in tumor sections (Fig. 7E and F). On average, necrotic areas and stromal cells made up a large proportion of the tumor, representing ∼37 and 18% of the total tumor area, respectively.
Inefficient intratumoral spread represents a major remaining challenge to the development of oncolytic viral vectors. Stromal cells not only impart physical barriers to virus spread, but cells of murine origin are not conducive to productive adenoviral replication. The latter issue may affect the interpretation of tumor efficacy studies performed in mice. To attempt to overcome these limitations in the future, it will be necessary to combine our fiber modifications with transgenes that promote intratumoral spread.
Discussion
To refine the therapeutic index of intravenously delivered Ad5 vectors, enhance potency, and improve therapeutic outcome, it is widely accepted that transductional retargeting will need to be combined with primary receptor detargeting, strategies to allow evasion of hepatocytes and scavenging macrophages, as well as attempts to limit vector-related toxicity. The intravenous delivery of Ad5 elicits a robust inflammatory response, directed against both viral particles and expressed genes (Lieber et al., 1997; Zhang et al., 2001), which can limit transgene expression and reduce therapeutic efficacy. Characteristic elevations in serum chemokines and cytokines contribute to the profound liver pathology and toxicity associated with Ad5. Furthermore, systemically delivered Ad vectors can cause hemodynamic changes (Schiedner et al., 2003) and induce acute transient thrombocytopenia (Othman et al., 2007), potentially exacerbating preexisting coagulopathic conditions in a clinical setting. Differences between Ad5-mediated agglutination of human and murine erythrocytes limit the translational relevance of murine in vivo studies and may affect the success of tumor-targeting strategies (Carlisle et al., 2009). Therefore, tropism-modified Ad vectors that do not trigger these side effects and have limited off-target interactions should represent more promising candidates for future clinical assessment.
In this study, we monitored the in vitro and in vivo biological characteristics of an αvβ6-retargeted vector, Ad5-477dlTAYTA20, compared with Ad5-EGFPWT and Ad5-EGFPA20. Ad5-477dlTAYTA20 features the Y477dlTAYT modification, reported to eliminate fiber interactions with CAR and FIX/C4BP (Shayakhmetov et al., 2005). The CAR-binding capacity of Ad5-477dlTAYTA20 was ablated and this vector failed to agglutinate human erythrocytes, while retaining transduction efficiency comparable to that of Ad5-EGFPA20 in a panel of αvβ6-expressing human carcinoma cells. Ad5-477dlTAYTA20 also appeared to have acquired some affinity for HSPGs in vitro, using them as additional receptors for attachment/entry. Dysregulation of HSPGs occurs in various pathophysiological conditions, including cancer (Blackhall et al., 2001), and therefore enhanced interaction with HSPGs may be an attractive vector property for targeting tumors in vivo. Relative to other organs, the liver is particularly high in HSPGs with N- and O-sulfated side chains (Vongchan et al., 2005), which have been shown to promote Ad5:FX-mediated transduction in murine models (Bradshaw et al., 2010). However, Ad5-477dlTAYTA20 transduction via HSPGs in CHO-K1 cells was independent of N- or O-sulfation (Fig. 2C) and we detected no increased HSPG-mediated hepatocyte transduction when Ad5-477dlTAYTA20 was administered systemically.
The Ad5 hexon:FX interaction is the major determinant of hepatocyte transduction in murine models (Parker et al., 2006; Kalyuzhniy et al., 2008; Waddington et al., 2008). Significant advances have resulted in the generation of FX-binding ablated vectors that completely avoid the liver, thereby greatly improving Ad toxicity and safety (Alba et al., 2010). Furthermore, it has been shown that FX-binding ablated vectors can be successfully retargeted to CD46-positive lung tissue by pseudotyping the fiber of Ad35 (Alba et al., 2010). Coagulation factors have been shown to enhance the transduction of human carcinoma lines in vitro and also have been implicated in tumor transduction in vivo (Gimenez-Alejandre et al., 2008; Shashkova et al., 2008; Coughlan et al., 2009; Koski et al., 2009; Liu et al., 2009). Depletion of vitamin K-dependent coagulation factors, using warfarin, can be employed as a liver avoidance strategy, with the aim of increasing the blood persistence and bioavailability for the tumor. However, the level of success of this strategy has been controversial, with some reports of enhanced tumor uptake (Shashkova et al., 2008), and others describing negligible changes or even reductions in tumor uptake (Gimenez-Alejandre et al., 2008; Coughlan et al., 2009; Koski et al., 2009). Importantly, reduced tumor uptake was observed with integrin-retargeted Ads that feature the RGD-4C or A20FMDV2 peptide in the HI loop of the fiber, suggesting that integrin-directed retargeting is sensitive to the anticoagulative effects of warfarin in vivo. To date, it is unclear precisely which warfarin-sensitive coagulation factor mediates this effect. Selective depletion of FX alone, using X-bp (factor X-binding protein), has been shown to improve CD46-positive tumor transduction with an Ad5/35 vector (Liu et al., 2009). This is reassuring for the future development of tropism-modified Ads, although further assessment and investigation will be required to determine whether this also applies to integrin-retargeted Ads.
It seems possible that blood factors other than FX may play some undefined role in Ad vector biodistribution or tumor uptake. Shayakhmetov and colleagues first suggested that FIX and C4BP bound to the Ad5 fiber knob domain, and they reported that the Y477AdlTAYT mutation successfully ablated this interaction (Shayakhmetov et al., 2005). Despite lacking any FX-binding ablating modifications within the hexon, the latter vector was shown to have reduced hepatotropism and limited accumulation in KCs after intravenous delivery. Direct binding of FIX to the fiber of Ad5 has not been conclusively confirmed, but it has been hypothesized that a two-site FIX:Ad5 interacting model may be a possibility (Kalyuzhniy et al., 2008). FIX has been shown previously to enhance the transduction of Ad5 in vitro (Kalyuzhniy et al., 2008; Jonsson et al., 2009; Rogee et al., 2010). We report that, unlike Ad5-EGFPWT, fiber-modified vectors Ad5-EGFPA20 and Ad5-477dlTAYTA20 do not permit FIX-mediated infectivity enhancement in vitro. Furthermore, both A20-modified vectors display an altered in vivo phenotype, typified by reduced hepatotropism, limited induction of inflammatory cytokines, and reduced vector-related toxicity.
Several reports exist of fiber-modified, fiber-pseudotyped, or rare Ad species with reduced liver tropism (Denby et al., 2004; Nakayama et al., 2006; Coughlan et al., 2009; Rogee et al., 2010). Such effects may involve inefficient vector trafficking, increased vector degradation, lack of available functional receptors on the liver cell surface, or vector retargeting to other organs (Shayakhmetov et al., 2004). However, despite reduced hepatic transduction at later time points, many of these vectors accumulate in the liver at levels equivalent to Ad5 at early time points. By contrast, we showed a significantly reduced amount of Ad5-EGFPA20 and Ad5-477dlTAYTA20 viral genomes retained in the liver 1 hr, as well as 24 and 96 hr, postinjection (Fig. 3A). Reasons for the reduced early tropism of the A20-modified viruses currently are unclear.
Accumulation of Ads in the liver at early time points is a result of synergistic mechanisms, involving not only KCs and trapping of viral particles in hepatocytes, but also liver sinusoidal endothelial cells (LSECs) and the space of Disse (Di Paolo et al., 2009b; Ganesan et al., 2011). The exact order of viral particle interaction with these diverse cell types, and the relative importance of each cell type's role in vector clearance, is not fully understood and remains controversial. Colocalization studies with KC marker F4/80 showed that although all viruses accumulated within F4/80+ cells (Fig. 3C), the levels of both A20-modified viruses, especially Ad5-477dlTAYTA20, were visibly reduced. Such data support the original finding that a 477dlTAYT fiber-mutated virus, Ad5mut, had reduced colocalization with KCs in vivo (Shayakhmetov et al., 2005). The precise mechanisms underlying KC-mediated recognition of virus are not clear, although uptake of long-shafted Ads appears to be mediated by the fiber knob, independently of CAR (Shayakhmetov et al., 2004). Haisma and colleagues have demonstrated that Ad5 uptake by primary KCs, via scavenging receptor A (SR-A), can be inhibited by soluble Ad5 fiber knob protein in vitro or preincubation of Ad5 with an anti-knob antibody before injection intravenously, partially reducing accumulation in KCs (Haisma et al., 2009). These findings may provide support for the idea of a fiber knob:FIX interaction acting as a “bridge” for viral particle recognition by hepatic macrophages.
The release of proinflammatory molecules from KCs function as chemotactic signals for infiltrating neutrophils as well as contributing directly to hepatocyte damage (Lieber et al., 1997). These factors have been associated with acute hepatic injury and elevation of liver transaminases ALT and AST (Muruve et al., 1999). Thus, viruses having reduced interaction with KCs at early time points may have limited hepatotoxicity. Here, animals administered Ad5-EGFPA20 or Ad5-477dlTAYTA20 were well throughout the duration of the experiment, with low-level induction of IL-6, RANTES, and IFN-γ (6 hr); no thrombocytopenia (24 hr); limited serum transaminase elevations; and dramatically reduced accumulation of virion DNA in the liver at the time of death (1, 24, and 96 hr). In comparison, the Ad5-EGFPWT-treated groups showed signs of cachexia, significantly elevated serum transaminases, higher levels of viral DNA, and extensive hepatic E1A expression. Histological examination of H&E-stained liver sections revealed distinct differences between the groups (96 hr), with Ad5-EGFPA20- and Ad5-477dlTAYTA20-treated groups showing an absence of periportal inflammatory infiltrate compared with Ad5-EGFPWT-treated cohorts.
We previously published that Ad5-EGFPA20 has improved uptake in αvβ6 + tumor xenografts, 72 hr after intravenous delivery (Coughlan et al., 2009). Ad5-477dlTAYTA20 also displays this property in the same in vivo xenograft model (our unpublished data). Thus, both A20-modified viruses satisfy several of the criteria thought to be required for enhanced antitumoral efficacy, having improved uptake after intravenous delivery, reduced hepatotropism and toxicity, and an improved in vivo safety profile. However, the efficacy results were, largely, disappointing. Although we detected reductions in tumor growth in animals treated with Ad5-477dlTAYTA20 on days 4 and 9 postinjection, these effects were not sustained for the duration of the experiment. On completion of the experiment we did not detect any improvements in efficacy for A20-treated groups compared with the Ad5-EGFPWT group, although there were improvements versus PBS-treated groups. Furthermore, reductions in percent tumor growth were not improved by increasing the dose. We chose a therapeutic dose of 4×1010 VP for the A20-retargeted vectors on the basis of prior evidence that this was well tolerated in immunocompetent animals. At present, we do not know the maximal tolerated dose of the A20-retargeted vectors; however, a dose of 5×1010 VP of wild-type replicating Ad5 generally is considered to be a lethal dose (Cawood et al., 2009). Because our current viruses are fully replication competent, we were cautious in selecting a dose for an efficacy experiment. However, we cannot exclude the possibility that using a higher dose could potentially improve the therapeutic index of these vectors.
We verified that all tumors expressed the target receptor αvβ6, and that overall levels did not differ between animal groups (∼16%). Assessment of E1A expression in the tumors showed that there was efficient E1A expression within the αvβ6 + tumor cells of the A20-treated groups. Interestingly, after quantification of E1A staining in images, both A20 vectors transduced tumor cells more efficiently that Ad5-EGFPWT (∼3.3 and 2.8% vs. 1.5%). Moreover, the amount of E1A expression was further increased in the tumors of animals that received the higher dose of Ad5-EGFPA20 and Ad5-477dlTAYTA20 (∼4.6%). However, E1A staining within the tumors was localized to islands of carcinoma cells separated by large areas of stromal cells and regions of necrosis, suggesting that the virus could not disseminate throughout the tumor mass efficiently.
Poor intratumoral spread of oncolytic Ads is widely reported and thus represents a further challenge to achieving successful oncolytic viral therapy. Solid tumors contain neoplastic cells, stromal fibroblasts, myofibroblasts, endothelial cells, and immune cells, all of which interact with various components of the extracellular matrix (ECM) to affect migration, proliferation, and invasion (Wernert, 1997). Poorly distributed vasculature within the tumor mass can impede the uniform distribution and subsequent spread of therapeutic delivery vehicles. Furthermore, dysregulation of blood vessel formation or architecture, and the absence of functional lymphatics, can contribute to the development of a necrotic and hypoxic microenvironment. The presence of expansive necrotic regions within the tumor mass negates the therapeutic efficacy of oncolytic Ads, which require living cells for replication and progeny production. In addition, the physical nature of the tumor stroma and ECM imposes barrier-like restrictions on the dissemination of virus throughout the tumor. To assess the cellular composition of MCF10-CA1a tumor xenografts, we quantified the overall proportions of carcinoma cells, endothelial cells, stromal regions, and necrotic areas within the tumors. In general, we found that the tumor composition was similar between groups, being largely composed of necrotic areas and stroma, with only ∼26% tumor cells. At present we are examining whether these restrictions within this xenograft model are responsible for limiting antitumoral efficacy. Furthermore, our constructs, in their current form, are not optimized to overcome these barriers and will require further modifications in the future to address these possibilities.
Various Ad-based vector-engineering strategies currently are being employed in an attempt to overcome some of these restrictions. Dramatic improvements in efficacy have been achieved with Ads when enhancing viral release and spread through overexpression of the adenoviral death protein (E3-ADP) or the viroporin-like activity of a truncated, mutant E3-gp19K protein; or by using drugs, such as verapamil, that augment virion release (Doronin et al., 2003; Gros et al., 2008, 2010). Novel Ad-engineering strategies to enhance potency include the incorporation of fusogenic proteins to induce syncytium formation, or enzymatic transgenes that digest/depolymerize components of the extracellular matrix, thereby maximizing viral spread throughout the tumor (Ganesh et al., 2007; Guedan et al., 2008, 2010, 2011). Such agents have also demonstrated efficacy on pre- or coadministration with virus (Ganesh et al., 2008). More recently, in vitro bioselection of mutagenized Ad vectors in cancer-associated fibroblasts has identified vectors with enhanced release and cytotoxicity in stromal cells, resulting in improved antitumoral activity (Puig-Saus et al., 2012).
It is important to note that the vectors used in this current study do not feature any therapeutic transgene, nor do they possess any transcriptional selectivity for tumor cells. They are all completely replication-competent viruses. However, we show that the introduction of defined mutations within the fiber can improve the in vivo safety profile and the therapeutic index. Therefore, both A20-modified vectors, Ad5-EGFPA20 and Ad5-477dlTAYTA20, represent attractive platform vectors for future investigation. Combination of these capsid configurations with modifications that confer replication selectivity (Oberg et al., 2011) or transgenes to enhance intratumoral dissemination, would likely lead to improvements in potency (Guedan et al., 2010, 2011). Furthermore, an assessment of integrin-mediated targeting efficiency in an FX-ablated background also is warranted. Collectively, such modifications, together with use at a maximal dose, potentially could improve therapeutic outcome.
Footnotes
Acknowledgments
L.C. was funded by an EMBO Short Fellowship (ASTF 122-2008), a Society for General Microbiology (SGM) Presidents Research Visit award (PFRV 08/3), and a European Association for Cancer Research (EACR) Travel Fellowship Award. Other grants supporting this work included research funding from Bart's and the London School of Medicine and Dentistry, IDBELL, CR-UK, and the BBSRC (BB/G016844/1 awarded to A.H.B.). The authors thank staff at Barts Cancer Institute (Linda Hammond, Keyur Trivedi, and Mohammed Ikram) and the Institut Catala d'Oncologia (Sònia Guedan, Juan José Rojas, Jordi Martínez-Quintanilla, Francisca Alcayaga, Cristina Puig, and Blanca Luena) for invaluable assistance.
Author Disclosure Statement
The authors declare that they have no conflict of interest.