
Abstract
von Willebrand disease (VWD), the most common hereditary coagulation disorder, results from mutations in the 52-exon gene for von Willebrand factor (VWF), which encodes an 8.4-kB cDNA. Studies with VWF cDNA plasmids have demonstrated that in vivo gene transfer to the liver will correct the coagulation dysfunction in VWF−/− mice, but the correction is transient. To develop gene therapy for VWF that would mediate long-term expression of the VWF cDNA in liver, we first evaluated segmental pre-mRNA trans-splicing (SPTS) with two adeno-associated virus (AAV) serotype 8 vectors, each delivering one-half of the VWF cDNA. However, although the two vectors functioned well to generate VWF multimers after infection of cells in vitro, the efficiency of SPTS was insufficient to correct the VWF−/− mouse in vivo. As an alternative, we assessed the ability of a lentiviral vector to transfer the intact murine VWF cDNA in vivo directly to the neonatal liver of VWF−/− mice, using generation of VWF multimers, bleeding time, and bleeding volume as efficacy parameters. The VWF lentivirus generated VWF multimers and partially or completely corrected the coagulation defect on a persistent basis in 33% of the treated VWF-deficient mice. On the basis of the concept that partial persistent correction with gene transfer could be beneficial in VWD patients, these observations suggest that lentiviral delivery of VWF cDNA should be explored as a candidate for gene therapy in patients with a severe form of VWD.
Introduction
Like other mendelian genetic clotting disorders, reduction of risk of VWD for bleeding could potentially be treated by gene transfer, whereby the wild-type VWF coding sequence could be transferred to affected individuals, providing persistent expression of the VWF protein to correct the systemic deficiency caused by mutations in the VWF gene (VandenDriessche et al., 2003; Chuah et al., 2011). Like the treatment of other hemophilias by gene therapy, even partial persistent correction by gene transfer could be beneficial in that it would provide a persistent increase in the baseline VWF levels, lowering the risk for bleeding and requiring less exogenous VWF to maintain normal clotting (Menache, 1998; Kay and High, 1999; Ahnstrom et al., 2004; Nichols et al., 2009; Nathwani et al., 2011). The major challenge in developing a gene transfer strategy for VWD is that, at 8.4 kb, the size of the VWF cDNA limits the choice of vectors for in vivo gene transfer. On the basis of our studies (Pergolizzi et al., 2006) and those of others (De Meyer et al., 2008; Marx et al., 2008), we know it is possible to correct the murine knockout model of VWD by in vivo administration of a plasmid encoding VWF administered to the liver. These studies proved it was possible to genetically modify the liver to make functional VWF capable of correcting the coagulation defects in the VWF−/− mouse, but like most other in vivo plasmid-based gene therapy strategies, the correction was transient, lasting only days.
With this background, and the goal of providing at least partial, persistent correction of VWD, we have approached the challenge of gene therapy for VWD using two strategies, each designed to circumvent the challenge of in vivo gene transfer of the large VWF cDNA. First, with the knowledge that hepatocytes genetically modified with the VWF cDNA are capable of producing functional VWF (Pergolizzi et al., 2006; De Meyer et al., 2008; Marx et al., 2008), and that adeno-associated virus type 8 (AAV8) vectors are highly efficient at transferring and expressing genes in the liver (Gao et al., 2002; Sarkar et al., 2004; Thomas et al., 2004; Nathwani et al., 2006, 2011), we devised a strategy to use AAV8 vectors to transfer the murine VWF cDNA to the liver in vivo. To circumvent the size limitation of AAV vectors, we used segmental pre-mRNA trans-splicing, with the VWF cDNA split into 5′ and 3′ segments (Chao et al., 2002; Pergolizzi et al., 2003; Pergolizzi and Crystal, 2004; Dong et al., 2010; Lai et al., 2010; Wu et al., 2010). Assuming that the 5′ and 3′ vectors infect the same cell, the two expression cassettes are designed to express precursor mRNAs that trans-splice at the pre-mRNA level to form an intact murine VWF mRNA. Second, based on the knowledge that the intact VWF coding region could fit into a lentiviral expression cassette, we assessed the ability of a lentiviral vector to transfer the intact murine VWF transgene in vivo directly to the liver (Kumar et al., 2001). In both strategies, we used the VWF knockout mouse as the model, with the generation of VWF multimers, bleeding time, and bleeding volume as the phenotype efficacy parameters (Denis et al., 1998).
Materials and Methods
Segmental pre-mRNA trans-splicing 5′ and 3′ expression cassettes
All the plasmids were prepared with mini, maxi, or mega kits (Qiagen, Valencia, CA) according to the manufacturer's instructions. The coding sequence of murine VWF was amplified by reverse transcription-PCR from the full-length C57BL/6 mouse VWF mRNA. The 5′ and 3′ segmental pre-mRNA trans-splicing (SPTS) constructs were designed so that the resulting two pre-mRNAs would hybridize and the nuclear spliceosome apparatus would trans-splice the resulting hybridized pre-mRNAs into mature, full-length murine VWF mRNA (Song et al., 2009). A total of seven expression cassettes were constructed, including two for the 5′ SPTS fragment and four for the 3′ SPTS fragment. The engineered segmental pre-mRNA trans-splicing junction was located inside exon 28 (between nucleotides 4293 and 4294).
Both of the 5′ SPTS expression cassettes contained a cytomegalovirus (CMV) promoter/enhancer, a 5′ fragment of murine VWF cDNA (exon 1 through the 5′ part of exon 28), an intron from intron 6 of human vascular endothelial growth factor gene, a foreign 50-bp hybridization binding domain (BD), and a polyadenylation signal [poly(A); Fig. 1]. The hybridization BD, a sequence described by Tahara and colleagues (2004), was used to ensure efficient splicing with a complementary hybridization BD in the 3′ SPTS expression cassettes. The 5′A and 5′B constructs were similar, but to limit the size of the expression cassette, the 5′B construct was designed with the intron shortened to 27 bp from 517 bp in the 5′A construct together with a shorter rabbit β-globin poly(A) segment (SPA, 49 bp) (Levitt et al., 1989; Tahara et al., 2004), compared with the 224-bp bovine growth hormone (BGH) poly(A) segment in the 5′A construct.
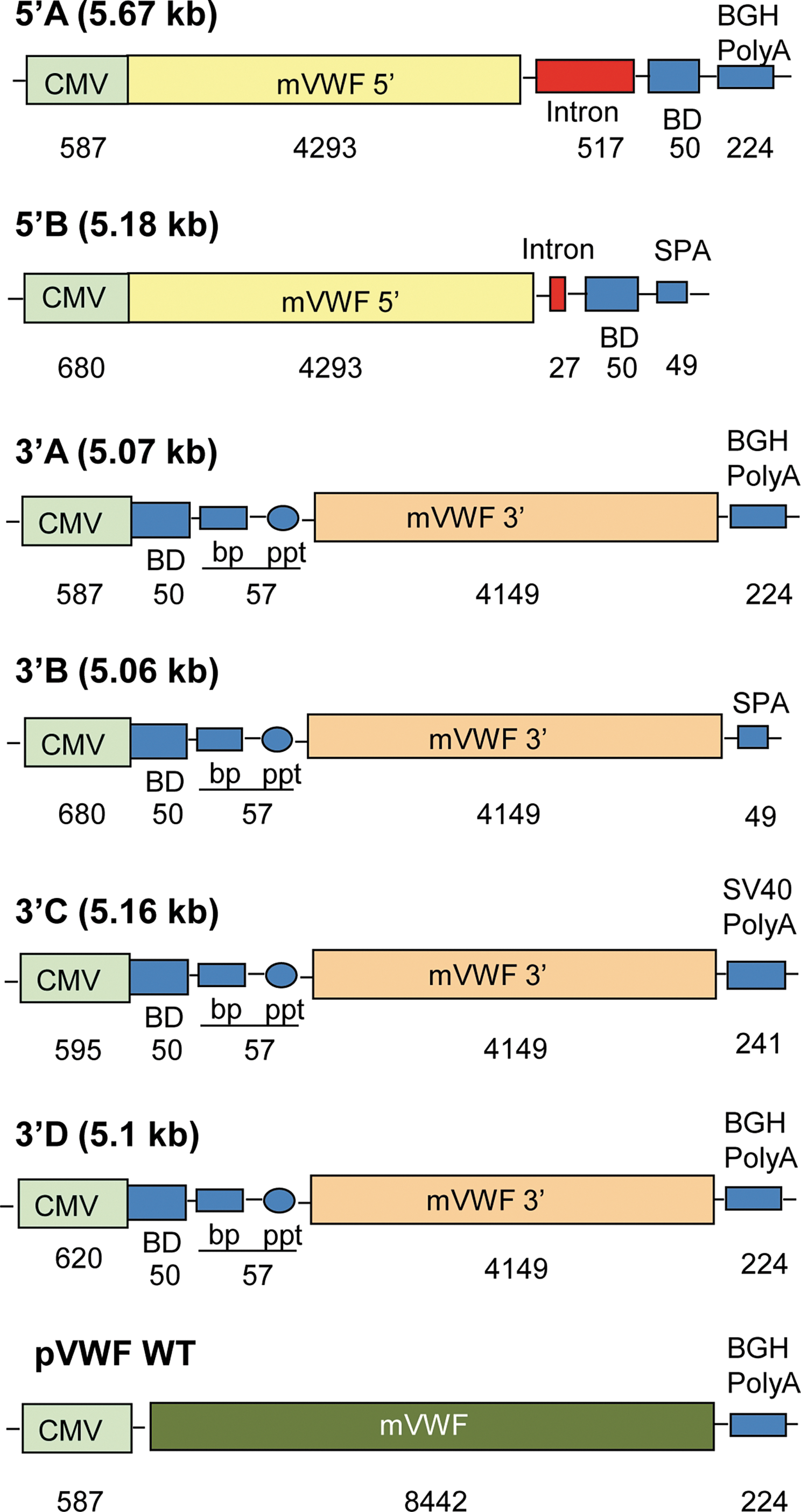
5′ and 3′ construction expression cassettes for segmental pre-mRNA trans-splicing. Shown are two 5′ constructs (A and B) and four constructs (A to D). 5′A: The expression cassette (5.67 kb) includes (5′ to 3′): 587-bp cytomegalovirus (CMV) promoter/enhancer, 4293-bp 5′ fragment of the murine von Willebrand factor (VWF) cDNA (exon 1 to 5′ of exon 28), 517-bp intron from human vascular endothelial growth factor (hVEGF) intron 6, 50-bp hybridization binding domain (BD), and 224-bp bovine growth hormone (BGH) polyadenylation signal [poly(A)]; 5′B: 5.18-kb, 680-bp CMV promoter/enhancer, 4293-bp 5′ fragment of murine VWF cDNA, shortened 27-bp hVEGF intron 6, 50-bp hybridization BD and shortened 49-bp poly(A) (SPA) from rabbit β-globin gene; 3′A: 5.07-kb, 587-bp CMV promoter/enhancer, 50-bp hybridization BD that complements the BD of the 5′ cassette, optimized branch point (BP), polypyrimidine tract (PPT), 4149-bp 3′ fragment of murine VWF cDNA (3′ of exon 28 to exon 52), and 224-bp BGH poly(A); 3′B: 5.06-kb, 680-bp CMV promoter/enhancer, 50-bp hybridization BD that complements the BD of the 5′ cassette, BP, PPT, 4149-bp 3′ fragment of murine VWF cDNA, and a shortened 49-bp poly(A) (SPA); 3′C: 5.16-kb, 595-bp CMV promoter/enhancer, 50-bp hybridization BD that complements the one of 5′ cassette, BP, PPT, 4149-bp 3′ fragment of murine VWF cDNA, and 241-bp simian virus 40 (SV40) poly(A); 3′D: 5.1-kb, 620-bp CMV promoter/enhancer, 50-bp hybridization BD that complements the BD of the 5′ cassette BP, PPT, 4149-bp 3′ fragment of murine VWF cDNA, and 224-bp BGH poly(A); pVWF WT, the assay positive control plasmid, is 9.25-kb, 587-bp CMV promoter/enhancer, 8442-bp full-length murine VWF cDNA, and 224-bp BGH poly(A). All constructs were in the pcDNA3.1(+) backbone and then subcloned into AAV2 packaging plasmid (Kaludov et al., 2002). Color images available online at
All four of the 3′ SPTS expression cassettes included a CMV promoter/enhancer, a 50-bp hybridization binding domain complementary to the one in the 5′ cassette, an optimized branch point, a polypyrimidine tract, a 3′ fragment of murine VWF cDNA (3′ part of exon 28–exon 52), and a poly(A) sequence. The differences among the 3′ SPTS constructs were the poly(A) segments and a minor difference in the length of the CMV promoter. All three 3′ expression cassettes were cloned into AAV2 backbone plasmids (Kaludov et al., 2002). The 3′B construct contained a 49-bp poly(A) segment based on the rabbit β-globin gene; the 3′C construct contained a 241-bp simian virus 40 (SV40) poly(A); and the 3′A and 3′D construct contained the 224-bp BGH poly(A) segment (from pcDNA3.1; Invitrogen, Carlsbad, CA).
AAV8-based SPTS 5′ and 3′ vectors
Preliminary in vitro studies of murine VWF segmental pre-mRNA trans-splicing in AAV plasmid-transfected cells demonstrated that cells transfected with the 5′B + 3′C AAV plasmids yielded higher VWF levels compared with the 5′B + 3′B or 5′B + 3′D pair. On the basis of these observations, the 5′B and 3′C AAV plasmids were selected to generate the AAV8-based SPTS 5′ and 3′ vectors AAV8-VWF 5′ and AAV8-VWF 3′. The vectors were produced by a three-plasmid strategy as previously described (De et al., 2006), including (1) 5′B or 3′C expression AAV plasmids; (2) pAAV8.2, a packaging plasmid that provides the AAV Rep proteins derived from AAV2 needed for vector replication as well as the viral structural (Cap) proteins VP1, VP2, and VP3 derived from AAV8 (the Cap proteins determine the serotype of the AAV vector); and (3) pAdΔF6, an adenoviral helper plasmid that provides adenoviral helper functions of E2, E4, and VA RNAs. To produce the AAV8-VWF SPTS vectors, the 5′B or 3′C expression plasmid (600 μg), pAAV8.2 (600 μg), and pAdΔF6 (1.2 mg) were cotransfected with PolyFect (Qiagen) into HEK-293orf6 cells (American Type Culture Collection [ATCC], Manassas, VA); these cells contain the adenoviral E1 and E4 regions required for optimal adenoviral helper function (De et al., 2006). Seventy-two hours posttransfection, the cells were harvested, and a crude viral lysate was prepared by four freeze–thaw cycles and clarified by centrifugation. The AAV8-VWF 5′ and 3′ vectors were purified by iodixanol gradient and QHP anion-exchange chromatography (De et al., 2006). The purified vectors were concentrated with an Amicon Ultra-15 100K centrifugal filter device (Millipore, Billerica, MA) and stored in phosphate-buffered saline (PBS, pH 7.4) at −80°C. The vector genome titers were determined by quantitative TaqMan real-time PCR analysis, using a CMV promoter-specific primer–probe set (Applied Biosystems, Foster City, CA) (De et al., 2006).
Lentiviral vectors
Three different transgenes were subcloned into self-inactivating lentiviral (SIN18) backbone plasmids (Naldini and Verma, 2000; Follenzi and Naldini, 2002) in order to create the lentiviral vectors. Because there are differences in the platelet receptor-binding sites between human and murine VWF, the murine VWF cDNA was used for all the experiments. The full-length 8.4-kb murine VWF cDNA, derived from the C57BL/6 mouse, was subcloned into lentiviral plasmids under the control of either the CMV or phosphoglycerate kinase (PGK) promoter/enhancer. Included 3′ of the VWF gene was the woodchuck hepatitis posttranscriptional regulatory element (WPRE). The U3 region was extensively deleted, but the polyadenylation sequence was retained in the 3′ long terminal repeat (LTR). The mouse VWF cDNA was cloned from the C57BL/6 mouse mRNA and the sequence was confirmed to be the wild-type murine VWF coding sequence, identical to that reported by Chitta and colleagues (2007) (GenBank AY208897).
To further assess the promoters for the lentiviral vectors in vivo, the human α1-antitrypsin (hAAT) reporter gene was used to help assess expression in C57BL/6 mice. The hAAT cDNA, encoding a secreted protein not recognized as foreign to C57BL/6 mice, was subcloned downstream of both the CMV and PGK promoters (LentiCMV.hAAT and LentiPGK.hAAT). All transgenes and their plasmids were sequenced and found to be without mutations or polymorphisms.
Production of replication-defective lentivirus was accomplished by a four-plasmid (transgene plasmid, pMDL-RRE, pRSV-Rev, and pVSV-G, at a 4:2:1:1 ratio) cotransfection of the HEK-293T cell line (ATCC), using calcium phosphate precipitation (Follenzi and Naldini, 2002). Lentivirus was harvested after 72 hr by collecting the cultured medium and concentrating it by centrifugation at 20,000 rpm for 2 hr at 4°C. Viral particle pellets were resuspended in Hanks' balanced salt solution and frozen at −80°C. All lentiviral preparations were quantified by p24 antigen levels (HIV-p24 ELISA kit; PerkinElmer, Waltham, MA), and titered by means of limiting dilution infections in additional 293T cells, and TaqMan PCR was performed with viral gag gene primers. Titer determinants were performed by real-time PCR (TaqMan) against the NIH3T3 cell line with one integrated lentiviral genome (a gift from M. Sadelain, Sloan-Kettering Institute, New York, NY). Infectious titers ranged from 5×107 to 8×108 transducing units/ml.
Evaluation of vector function
The ability of the plasmid constructs to transfer the murine VWF cDNA in vitro was tested by assessing the ability of the vector to direct the cells to produce VWF multimers in vitro. HEK-293 cells (6×106 cells) were transfected with plasmids, using PolyFect (Qiagen). To avoid possible nonspecific binding of virus to fetal bovine serum proteins in the medium, the cells were washed once with PBS before addition of transfection medium and serum-reduced culture medium (Opti-MEM; Invitrogen) was used. Sixty to 96 hr later, the transfected cell supernatants were harvested and concentrated by passage through Amicon Ultra-4 100K centrifugal filter (Millipore) for analysis of VWF multimers.
To assess VWF multimers, agarose ([SeaKem HGT(P)], VWF grade; FMC Bioproducts, Rockland, ME) was dissolved in 40 mM Tris-acetate (pH 8.3), 0.1% sodium dodecyl sulfate, and 1 mM ethylenediaminetetraacetate (EDTA), poured to form a 4-mm-thick 1% gel, and solidified at 4°C. Samples were mixed at a 9:1 ratio with 50 mM NaH2PO4 (pH 7.0), 185 mM iodoacetamide, and 5.0% sodium dodecyl sulfate and were incubated at 37°C for 60 min. After incubation, 0.1 vol of 50% glycerol and 1% bromophenol blue were added to the samples. Electrophoresis was carried out for 30 min at 30 mA followed by 5 hr at 50 mA at 4°C. To transfer the proteins, 0.45-μm polyvinylidene difluoride (PVDF) transfer membrane (Amersham Hybond-P; GE Healthcare, Piscataway, NJ) was presoaked for 2 min in methanol and rinsed with transfer buffer in 2.5 mM Tris (pH 8.8), 19.2 mM glycine, 20% methanol, and 0.01% sodium dodecyl sulfate. Transfer was carried out in a Trans-Blot cell (Bio-Rad, Hercules, CA) in 2.5 mM Tris (pH 8.8), 19.2 mM glycine, 20% methanol, and 0.01% sodium dodecyl sulfate at 75 mA at 4°C overnight. After transfer, the membrane was blocked for 1 hr in 5% nonfat dry milk (Bio-Rad) diluted in 1×PBS. Detection was accomplished by incubation, for 2 hr at 23°C with mild shaking, with a primary rabbit anti-human VWF antibody (Dako, Carpinteria, CA) diluted 1:1000 in 5% milk. After washing with 0.05% Tween-20 in 1×PBS, the membrane was incubated, for 1 hr at 23°C with mild shaking, with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:2000 in 5% milk, and was washed several times with 0.05% Tween-20 in PBS. VWF multimers were visualized with enhanced chemiluminescence (ECL) Western analysis reagents (GE Healthcare) and X-ray film. Densitometric analysis of the VWF multimers in the Western analyses was carried out by scanning the X-ray film with a flat-bed scanner at 300 dpi, and the resulting .tif file was analyzed with Quantity One software (version 4.2.0; Bio-Rad), using Vertical Trace to generate a vertical cross-section of the multimers in each lane.
To assess the function of the AAV constructs in vitro, HEK-293orf cells (8×105 cells) were seeded in 6-well plates and infected with AAV8-VWF 5′, AAV8-VWF 3′, or AAV8-VWF 5′ plus AAV8-VWF 3′ for 60 min at a dose of 2.5×105 genome copies (GC)/cell of each vector in serum-free Dulbecco's modified Eagle's medium (DMEM; Invitrogen). After infection, cells were cultured in Opti-MEM containing 10 μM ZnCl2 and, after 72 hr, the infected cell supernatants were harvested and concentrated by passage through an Amicon Ultra-4 100K centrifugal filter (Millipore). The supernatants were evaluated for the expression of mVWF multimers as described previously.
The lentiviral preparations were tested for transgene expression and activity by infecting human lung A549 cells. For the transduction, 200 ng of p24 per 100-mm plate (3×105 cells) was used to infect the cells in serum-free medium. Cultured media were harvested after 72 hr and tested for VWF expression by Western analysis. Medium was concentrated with Amicon Ultra-4 100K MWCO filters to allow for a small volume to be analyzed by gel electrophoresis.
Murine VWF model
All mice were maintained in pathogen-free environments, and the experiments were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College (New York, NY). VWF knockout mice (VWF−/−) on the C57BL/6 background used as a model of severe VWD were a gift from D.D. Wagner (Harvard Medical School, Boston, MA), who provided breeder pairs of mice (Denis et al., 1998). These mice have a NeoR cassette disrupting exon 4 of the murine vwf locus, generating progeny mice that express a completely dysfunctional VWF molecule, rendering the mice similar in phenotype to humans with severe VWD. The mice exhibit dual defects in hemostasis with highly prolonged bleeding times (>20 min) and pseudohemophilia with markedly reduced factor VIII activity levels (FVIII:C) and activity (10 to 20% normal). Wild-type C57BL/6 mice were used as positive controls.
Evaluation of AAV8-mediated segmental pre-mRNA trans-splicing VWF expression in vivo
To assess whether the AAV8 vectors could mediate full VWF segmental pre-mRNA trans-splicing in vivo, the AAV8 SPTS vectors were administered to mice via a single tail vein injection. Experiments were performed with male or female VWF−/− mice (n=5 per group). Mice were warmed under a heat lamp and then anesthetized with ketamine plus xylazine (100 and 10 mg/kg, respectively) by the intraperitoneal route. AAV8-VWF 5′, AAV8-VWF 3′, premixed AAV8-VWF 5′ plus AAV8-VWF 3′ at a dose of 2×1011 GC each, or PBS diluted in 100 μl of PBS was injected into the tail vein through an insulin syringe with a 30-gauge needle over a 30-sec period. At 2, 4, and 8 weeks postinjection, blood was collected by tail vein sampling into polypropylene tubes containing 0.1 vol of 3.8% sodium citrate. Plasma was prepared by centrifugation of the blood at 2000×g for 10 min, at 23°C. Plasma was diluted 1:2.5 in dilution buffer (2.5 mM Tris-HCl [pH 8.0] with 50 mM sodium chloride) for the assay of mVWF multimers as described previously.
Evaluation of VWF lentiviral gene expression in vivo
To assess newborn lentiviral intrahepatic administrations, a positive control vector containing the human AAT transgene driven by either the PGK or CMV promoter was administered to neonatal livers. Plasma levels of the AAT transgene product were monitored with an AAT ELISA (ALPCO Diagnostics, Salem, NH) (De et al., 2006). Mouse plasma was diluted 1:50 in 1×sample buffer (from the kit), assayed in anti-AAT antibody-coated microtiter wells, and incubated for 90 min at 25°C. After washing, the captured antigens were detected with an enzyme-linked secondary antibody. Color development was terminated after 15 min in the dark and assessed at 490 nm by a microtiter plate reader (Bio-Rad). The resulting absorbance was measured against a standard of purified AAT (Brantly et al., 1991) and reported as nanograms of human AAT per milliliter of plasma.
VWF lentiviral vector was administered directly into the liver of newborn VWF−/− mice (1 to 2 days old), using 32-gauge needles (1 or 4 μg of p24, 109 particle units [PU]), along with recombinant human VWF protein to prevent internal bleeding (Sferra et al., 2004; Wang et al., 2005). The majority of the injected pups survived without any bleeding or complications and developed into adult mice. The mice were bled at 3, 8, and 12 weeks for VWF antigen detection by Western analysis, and were tested for changes in bleeding time at week 3 or week 12.
For immunohistochemical localization of VWF expression, livers were harvested 3 weeks after LentiPGK.mVWF administration, fixed overnight in 4% paraformaldehyde in PBS (EM Sciences, Hatfield, PA), and paraffin embedded, and 5-μm sections were cut onto ProbeOn slides (Fisher Scientific, Pittsburg, PA). For staining, the sections were cleaned and rehydrated through graded alcohol. The slides were heated in a steamer for 20 min, immersed in citrate buffer (pH 6.0), and then allowed to sit at 23°C for 20 min. Endogenous peroxidases were quenched with 0.3% H2O2 in PBS for 30 min at 23°C. Sections were blocked for 20 min in 5% normal goat serum in PBS. The slides were incubated overnight at 4°C in a humid chamber with rabbit anti-human VWF (Dako). The next day the slides were developed with a Vector ELITE rabbit ABC kit (Vector Laboratories, Burlingame, CA), stained with peroxidase chromogenic substrate AEC, and counterstained with Mayer's hematoxylin.
VWF bleeding time and volume
To monitor phenotypic changes in the knockout mice after gene transfer, whole blood clotting time was determined to measure the time until cessation of bleeding and total blood loss (Ware et al., 2000). Mouse tails were cut ∼1 cm from the terminal tip and the remainder of the tail was immersed in warmed PBS (heated to 37°C in water bath). The bleeding time was recorded as the time it took for complete cessation of blood flow. Mice that did not stop bleeding by 8 min were cauterized to prevent further blood loss. Immediately after the timed assay, 1 ml of sodium citrate was added to the saline–blood tubes to prevent clotting. The tubes were centrifuged at 2000 rpm for 10 min, and the saline layer was completely aspirated. The amount of packed red blood cells was measured manually with P200 pipets and recorded as “total RBC loss.” Control mice included two wild-type (C57BL/6) and two VWF knockout mice for each bleeding time assay. Wild-type mice routinely stopped bleeding between 1 and 3 min; VWF knockout mice bled with heavy blood loss to 8 min, when the bleeding was stopped by cauterization.
Statistical analyses
All data are expressed as means±SEM (standard error of the mean). In the bleeding time and blood loss assays, comparisons between the treated groups and controls were conducted by nonparametric one-way analysis of variance (ANOVA) using the Kruskal–Wallis test and Mann–Whitney U test (from
Results
To determine whether gene therapy could be used to treat von Willebrand disease in vivo, we developed two viral delivery systems that could accommodate the large VWF cDNA. The first strategy used adeno-associated viral (AAV) vectors with the murine VWF cDNA split at exon 28 into two parts, with splice donor, hybridization, and splice acceptor domains designed to mediate segmental pre-mRNA trans-splicing of the 5′ and 3′ VWF pre-mRNAs to hybridize and then splice to form a full-length VWF mRNA (Pergolizzi et al., 2003; Pergolizzi and Crystal, 2004; Song et al., 2009). The second strategy used lentiviruses carrying the full-length murine VWF cDNA. Both systems used murine VWF cDNA from the C57BL/6 mouse.
Evaluation of mVWF segmental pre-mRNA trans-splicing in AAV vector-infected cells
To test the feasibility of VWF segmental pre-mRNA trans-splicing, 293 cells were transfected with various combinations of the 5′ and 3′ expression cassettes (Fig. 1). As a positive control, VWF Western analysis displayed high expression level multimers from cells transfected with the intact wild-type VWF cDNA (Fig. 2A, lane 10; Fig. 2B, lane 20; Fig. 2C, lane 25). As expected, multimers were not found in mock-transfected cells or cells transfected with any 5′ or 3′ cassette alone (Fig. 2A, lanes 1–5; Fig. 2B, lanes 11–15). However, multimers could be detected in cells transfected with any of the combinations of 5′ and 3′ VWF plasmids (Fig. 2A, lanes 6–9; Fig. 2B, lanes 16–19). Cells transfected with the 5′B + 3′D plasmids (Fig. 2A, lane 8) and 5′A + 3′A plasmids (Fig. 2A, lane 9) showed similar amounts of high molecular weight multimers. The intensity of bands demonstrated that the 5′B expression cassette had comparable expression efficiency as the 5′A construct even with the shorter intron and poly(A) element, which were designed to circumvent the packaging limit of AAV vectors. Cells transfected with the 5′B + 3′B constructs (Fig. 2A, lane 6) and 5′A + 3′B constructs (Fig. 2A, lane 7) showed fewer high molecular weight multimers and less intensity of bands, suggesting that the 49-bp shorter 3′ SPA element likely decreases the efficiency of the 3′ cassette compared with the 224-bp BGH poly(A) element in the 3′D construct. Cells transfected with 5′B + 3′C (Fig. 2B, lane 17) showed more high molecular weight multimers and also higher intensity of bands than cells transfected with 5′B + 3′B constructs (Fig. 2B, lane 16) and 5′B + 3′D constructs (Fig. 2B, lane 18), suggesting that the 3′C construct with the longer 241-bp poly(A) element has higher efficiency compared with the other two 3′ cassettes with either the 49-bp SPA or 224-bp BGH poly(A) element. Thus, the 5′B + 3′C expression cassettes yielded the relatively highest amount and quality of VWF multimers. These constructs were used to generate the AAV8 trans-splicing vectors.
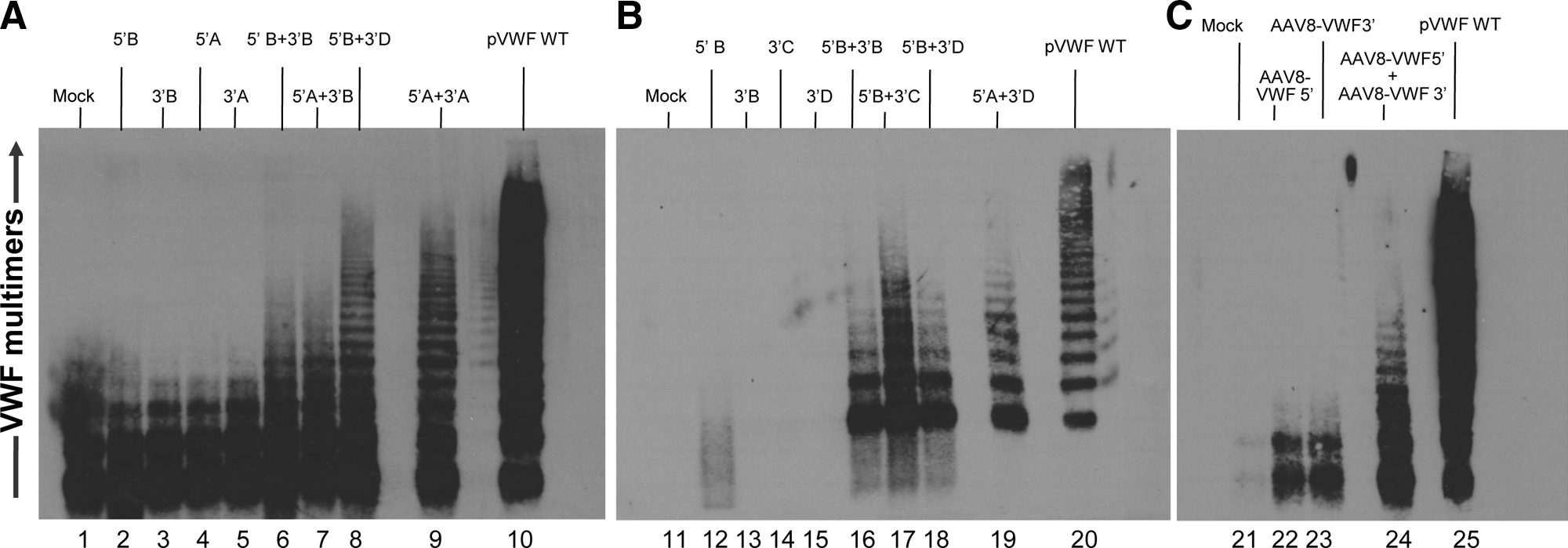
Evaluation of the ability of combinations of the 5′ and 3′ VWF segmental pre-mRNA trans-splicing constructs to generate VWF multimers in vitro.
To assess AAV8-mediated VWF segmental pre-mRNA trans-splicing in vitro, 293orf6 cells were infected with either AAV8-VWF 5′ alone, AAV8-VWF 3′ alone, or AAV8-VWF 5′ plus AAV8-VWF 3′. VWF protein expression was evaluated by Western analysis. Multimers were detected from cells infected with the combination of AAV8-VWF 5′ + AAV8-VWF 3′ (Fig. 2C, lane 24), but not in mock-infected cells or cells infected with either AAV8-VWF 5′ or AAV8-VWF 3′ alone (Fig. 2C, lanes 21–23).
Evaluation of mVWF segmental pre-mRNA trans-splicing mediated by AAV8 SPTS vectors in vivo
Our previous study demonstrated that mouse liver hepatocytes could express VWF production short term after high-volume injection of VWF-expressing plasmids into the tail vein (Pergolizzi et al., 2006). In a strategy to express VWF cDNA on a persistent basis, the AAV8 serotype was chosen to express VWF by segmental pre-mRNA trans-splicing in vivo, based on studies showing that AAV8 capsid has high tropism to hepatocytes and thus mediates high-efficiency gene transfer to liver in rodents and humans (Gao et al., 2002; Sarkar et al., 2004; Thomas et al., 2004; Nathwani et al., 2006, 2011). AAV8-VWF 5′ and AAV8-VWF 3′ vectors were premixed, each 2×1011 GC, and administered to VWF−/− mice via the tail vein. At 2, 4, and 8 weeks postinjection the mice were monitored by Western analysis for plasma VWF multimers. However, despite the function of the vectors in vitro to generate VWF multimers (Fig. 2C), there was inefficient expression from the segmental trans-spicing to generate enough VWF to create visible multimers. Although there were some very low molecular weight bands from the combined vectors at 2 weeks (Fig. 3A, lanes 6–10), the mice receiving only AAV8-VWF 3′ also gave a similar pattern (Fig. 3A, lane 5), and thus these are likely nonspecific. No visible multimers were detected from the combined vectors at either the 4- or 8-week time point (Fig. 3B and C).
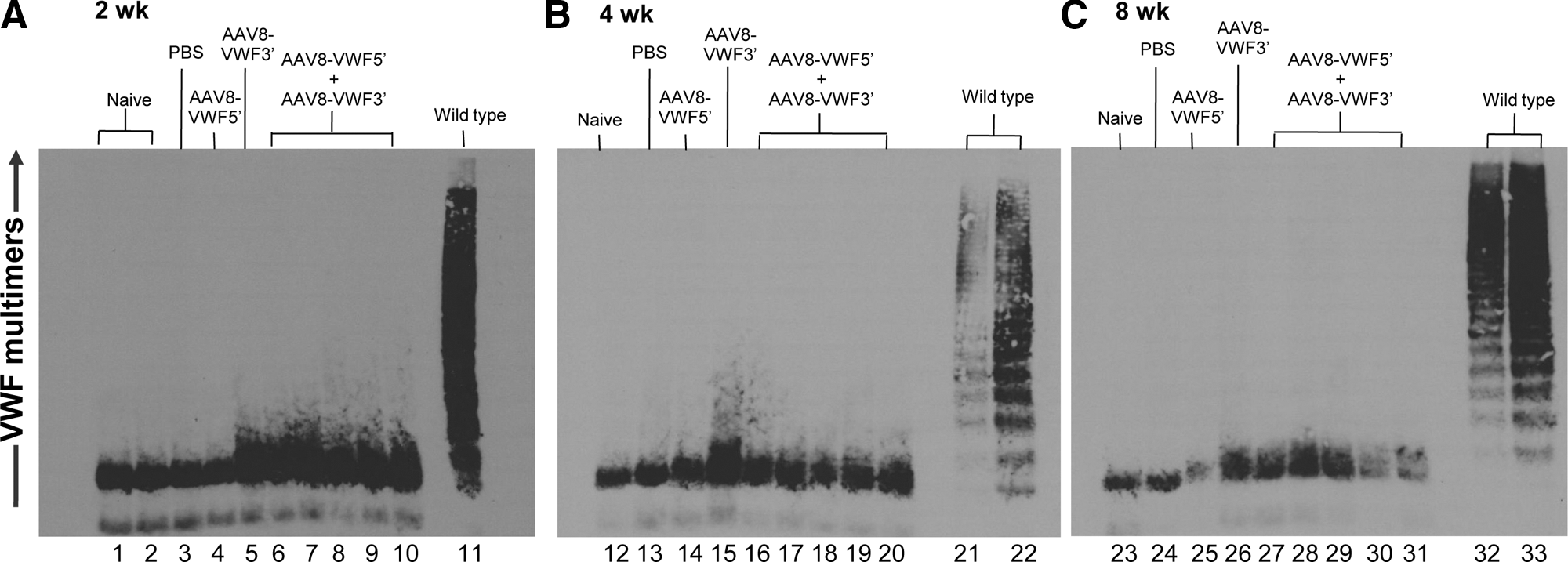
Assessment of VWF multimers generated by segmental pre-mRNA trans-splicing in vivo after intravenous administration of AAV8-VWF segmental pre-mRNA trans-splicing (SPTS) vectors to VWF knockout mice. AAV8-VWF 5′ and/or AAV8-VWF 3′ (each 2×1011 genome copies) or PBS was administered intravenously to VWF−/− mice. At 2, 4, and 8 weeks postinjection, blood was collected and evaluated for VWF multimers by Western analysis.
Expression of mVWF lentivirus in vitro
PCR analysis of cells transduced in vitro with lentiviral DNA showed the presence of both viral genes and VWF exons (data not shown). Lentiviral titering of these vectors by limiting dilutions and by p24 antigen ELISA indicated high infectivity (109 PU/ml) and yields of 18–30 ng of p24 particles/ml. No apparent loss of infectivity or low yields were observed with the full-length VWF lentiviruses. Transduction of the VWF lentiviral vectors (Fig. 4A) into human lung carcinoma line A549 mediated robust expression of full-length VWF multimers by either promoter (PGK or CMV; Fig. 4B, lanes 2 and 3).

Lentivirus-directed expression of the murine VWF cDNA in vitro.
Lentivirus-mediated gene transfer in vivo
As a comparison of the ability of the PGK versus CMV promoter VWF lentiviral vectors to function in hepatic gene transfer in vivo, lentiviruses expressing hAAT under the control of either of the two promoters were administered by direct hepatic administration to neonatal mice. Using AAT as a surrogate secreted plasma protein for VWF, we observed that the expression levels with both promoters remained stable for at least 12 weeks, at about 1 μg of hAAT per milliliter (Fig. 4C).
To assess lentivirus-mediated correction of the VWF−/− mouse, newborn VWF−/− mice were injected intrahepatically with lentiviruses expressing the VWF cDNA (1 or 4 μg of p24 or 1–2×109 PU), plus recombinant human VWF protein (5 μg, to prevent internal bleeding). Almost all of the injected pups survived without any bleeding or complications and developed into adult mice. Immunohistochemistry was performed on serial liver sections from lentivirus-transduced and negative control VWF−/− mice, using anti-VWF or irrelevant IgG antibodies (Fig. 5). Deposits of the antibody (brown to red color) were observed within most hepatocytes in the liver sections from Lenti-VWF-administered mice, especially in the hepatocytes around portal triads, indicative of positive antibody detection of VWF proteins (Fig. 5B and D compared with Fig. 5A and C, antibody control). The LentiPGK.mVWF-treated mouse livers also had abundant areas of positive staining in the endothelial cells lining portal veins (compare Fig. 5F with 5E). No staining was observed in nontreated control VWF−/− livers (data not shown).

Immunohistochemical localization of VWF in the liver of VWF−/− mice after intrahepatic administration of LentiPGK.mVWF to neonates. The lentiviral gene transfer vector expressing mVWF under the control of the PGK promoter (1 μg of p24) was administered to 2-day-old VWF−/− neonates via the intrahepatic route. After 3 weeks, the mice were killed and liver sections were stained with an anti-VWF antibody
On the basis of the positive immunohistochemistry findings of VWF expression in liver, the neonatal injections were repeated in newborn mice and assessed over time for detectable levels of plasma VWF and phenotypic correction of bleeding times. Increasing doses of Lenti-VWF (1 to 4 μg) were administered by direct intrahepatic injection in neonates. The Lenti-VWF-injected mice were monitored by Western analysis once every 4 weeks for plasma VWF multimers and by bleeding time assays once at 12 weeks. No significant differences were observed with the CMV and PGK promoters; only the CMV data are shown (Fig. 6).

VWF multimers, bleeding time, and blood loss after intrahepatic administration of lentivirus expressing murine VWF.
The results were variable regardless of the dose administered. Several mice of each neonatal litter developed long-term expression of VWF multimers (e.g., see Fig. 6A, lanes 3, 8, and 9, and Fig. 6B, lanes 15, 16, 18, 19, and 20); whereas other mice failed to develop detectable VWF multimers. The highest number of positive VWF responders occurred with the use of higher lentivirus dose (4 μg; Fig. 6B). Densitometry showed that the lower molecular weight multimers from the LentiCMV.mVWF-treated mice were distinct from the background of the VWF−/− mice (see, e.g., Fig. 6C, lane 13 [negative] vs. lanes 15, 18, and 19 [treated]). In the wild-type VWF+/+ plasma from C57BL/6 mice, medium weight multimers were more prevalent, but the lower molecular weight multimers match up with the lower molecular weight multimers from the Lenti-VWF-treated mouse plasma (Fig. 6C, lanes 18 and 19 [treated] vs. lane 21 [positive]).
By week 12 posttreatment, 9 of 27 mice from 4 separate litters displayed complete bleeding cessation during the tail snip bleeding time assays. An additional 3 of the 27 treated mice displaying bleeding cessation that was reduced but not normalized compared with age-matched wild-type C57BL/6 control mice (Fig. 6D). When these LentiCMV.mVWF-treated neonate litters were compared for overall blood loss during the bleeding assay, 11 of the 27 treated adult mice showed reductions (RBC volume, <100 μl) far below that observed with knockout negative VWF−/− controls, with 5 of these treated mice demonstrating near wild-type mouse blood loss levels (Fig. 6E). Thus, approximately one-third of the VWF−/− mice demonstrated partial or complete reversal of the severe VWF coagulation defect.
Discussion
Because von Willebrand disease is a monogenetic disorder with a phenotype easily assessed in blood, it could be an ideal candidate for gene therapy (Federici, 2004; Mannucci, 2004; Sadler, 2006; Mikhail and Kouides, 2010). The challenge in achieving this is the large size of the VWF cDNA (∼8.4 kb) which, together with an active promoter, conflicts with packing constraints in most viral gene transfer vectors (Kumar et al., 2001; Dong et al., 2010; Lai et al., 2010; Wu et al., 2010). In the present study, we examined two different approaches to viral vector delivery of the VWF cDNA to correct the coagulation-related phenotype of the VWF−/− mouse. First, in order to take advantage of the efficiency of AAV8 vectors to transfer genes to hepatocytes in vivo (Thomas et al., 2004; Nathwani et al., 2006, 2011), but to circumvent the packaging constraints of AAV vectors, we split the VWF cDNA and expressed the 5′ and 3′ fragments in two AAV8 vectors using segmental pre-mRNA trans-splicing, using a hybridization domain and splice donor and acceptor site to fuse the two fragments at the pre-mRNA level. However, although effective in vitro in generating VWF multimers, the SPTS strategy in vivo was not efficient enough to generate detectable VWF multimers or to correct the coagulation defects of the VWF−/− mice. As a second strategy, we expressed the full-length murine VWF cDNA in a lentiviral vector. Not only did the lentivirus-VWF vector generate VWF multimers in vitro, but direct hepatic administration to neonatal VWF−/− mice resulted in the generation of VWF protein in hepatocytes and liver endothelium, and approximately 33% of the treated mice generated VWF multimers in serum and partial or full correction of the coagulation defect. Although improvements in efficiency will have to be made, all together the data demonstrate the potential of lentiviral delivery of VWF cDNA as a candidate for gene therapy to at least partially treat severe VWD.
Prior attempts of cell and gene therapy for von Willebrand disease
Bone marrow transplants of wild-type marrow into pigs with VWD have been attempted with limited success (Bowie et al., 1986; Nichols et al., 1995). Lin and colleagues (2002) established that cultured autologous “blood outgrowth endothelial cells” (BOECs) isolated from blood maintained expression of VWF through multiple passages, and De Meyer and colleagues (2006) used lentiviral transduction of the canine-VWF cDNA to correct VWF-deficient cultured canine BOECs in vitro. Although the ex vivo approach to treat VWD with genetically corrected BOECs is possible, large numbers of these cells will be needed as there is no selective advantage to the transfected cell progeny, and successful therapy will therefore require repeated infusions of large numbers of genetically modified cells to produce therapeutic VWF plasma levels.
Transient in vivo correction of the mouse model of VWD has been achieved by high-volume tail vein administration to deliver to the liver plasmids expressing the VWF cDNA under the control of an active promoter. Our group (Pergolizzi et al., 2006), Marx and colleagues (2008), and De Meyer and colleagues (2008) demonstrated correction of VWD in VWF-deficient mice by this strategy. However, in all of these studies, there was only transient expression of VWF and correction of the VWF−/− mouse was observed for less than 2 weeks.
Gene therapy for von Willebrand disease using segmental pre-mRNA trans-splicing vectors
SPTS is a strategy to get around the insert size limitations of AAV vectors, typically 4.7 to 5.3 kb (Grieger and Samulski, 2005; Dong et al., 2010; Lai et al., 2010; Wu et al., 2010) and significantly smaller than the cDNA for VWF (8.4 kb). In an attempt to adapt the SPTS strategy to treat VWD, we inserted 5′ and 3′ fragments of the murine VWF cDNA into two AAV8 vectors as a segmental pre-mRNA trans-splicing delivery system. This successfully produced VWF monomers in vitro, but failed to efficiently express detectable levels of VWF multimers in VWF knockout mice. This result is likely due to an insufficient level of cells coinfected with both the 5′ and 3′ coding AAV8 vectors. In contrast, trans-splicing of large genes using dual AAV vectors has been achieved in muscle by using AAV9 vectors encoding the 5′ and 3′ segments of alkaline phosphatase (Ghosh et al., 2007, 2009) and AAV6 vectors encoding dystrophin (Zhang and Duan, 2012). Whether the greater success of these studies compared with our AAV8-mediated segmental pre-mRNA trans-splicing to liver was due to the relative ease of detection of the transgenes used (intracellular vs. secreted for VWF), to the AAV serotype, and/or to the differences in the organs (liver vs. muscle) being treated is unclear. For segmental pre-mRNA trans-splicing of the VWF cDNA with AAV8, the inefficiency observed could theoretically be overcome by increasing the dose of the two vectors, but such high doses may be inappropriate for human clinical use for safety reasons. Thus, we have abandoned the SPTS AAV8 strategy for in vivo treatment of VWD.
In vivo gene therapy for von Willebrand disease with lentiviral vectors
A promising approach to in vivo gene therapy for large genes is to use replication-defective lentiviruses, with a packaging constraint of 8.5 to 9.7 kb (Kumar et al., 2001). Using a third-generation lentivirus, our studies focused on the use of in vivo lentivirus-mediated expression of the mouse VWF transgene in the liver of neonatal VWF−/− mice. Experimental trials in mice and rabbits have demonstrated that in vivo lentiviral gene therapy can be successful for targeting adult and neonatal livers, including in vivo gene transfer of marker genes such as GFP and eGFP, as well as correcting factor VIII and IX deficiency and Fabry's disease (α-galactosidase) (Kafri et al., 1997; Kootstra et al., 2003; Chen et al., 2004; Waddington et al., 2004; Yoshimitsu et al., 2004; Salani et al., 2005; Moreno et al., 2008). Salani and colleagues (2005) demonstrated that lentivirus could mediate transgene expression in the liver of neonatal mice, and Moreno and colleagues (2008) found that lentivirus could be used in utero to transfer genes to the fetus. Intrahepatic administration to the neonatal liver has been successful in rabbits (Moreno et al., 2008). Relevant to treatment of VWD, Niiya and colleagues (2009) used lentiviruses to deliver the VWF-cleaving enzyme ADAMSTS13 by the intravenous route in utero to fetal ADAMSTS13-deficient mice, which led to liver transduction by the lentiviral vector with generation of normal VWF multimeric status.
We directly administered lentivirus into neonatal liver to deliver the lentiviral mVWF transgene to hepatocytes and liver sinusoidal endothelium. Direct administration of the lentiviral-VWF into 2-day neonatal liver demonstrated production of VWF in liver hepatocytes and endothelial cells, with consequent partial or complete correction of the coagulation defect in approximately one-third of the mice. The variability of the response in the inbred VWF−/− mice to therapy with the lentiviral vector is an interesting observation, but it has been observed in other studies (Kootstra et al., 2003; Waddington et al., 2004). Given that only 5% of the wild-type VWF level is necessary to correct the defect, it is likely that what we are observing is a critical threshold of correction, where the two-thirds of treated VWF−/− mice are not expressing adequate VWF protein levels to observe a corrected phenotype. Thus, a higher “cure rate” may be achievable by increasing the dose, modifying elements in the vectors, and/or repeat dosing.
VWD is an inherited disorder associated with a lifelong predisposition to mucocutaneous bleeds (Federici, 2004; Mannucci, 2004; Sadler, 2006; Mikhail and Kouides, 2010). Depending on the severity of the disease, the patient may require daily to weekly administration of VWF concentrates or plasma cryoprecipitates, with the associated prohibitive costs (Mannucci, 2004; Mirchandani et al., 2011). Even small improvements in local production of VWF could alleviate at least some of the consequences of the coagulation defect, suggesting it is worthwhile to continue to evaluate in vivo lentivirus VWF gene transfer as a possible therapy for severe VWD.
Footnotes
Acknowledgments
The authors thank N. Mohamed for help in preparing this manuscript. These studies were supported, in part, by NIH U01 HL66952; and the Malcom Hewitt Wiener Foundation (Greenwich, CT). J.R. is supported, in part, by the National Foundation for Cancer Research. R.W. is supported, in part, by T32 HL094284; and S.R. is supported, in part, by NIH R01 HL102449.
Author Disclosure Statement
The authors have no conflicts to disclose.