
Abstract
Gene-therapy medicinal products are currently applied to patients enrolled in authorized clinical trials to demonstrate safety and efficacy. Given a positive outcome, marketing authorization can subsequently be achieved via the centralized procedure coordinated by the European Medicines Agency. With Regulation (EC) No. 1394/2007 in force, advanced therapy medicinal products, including gene- and cell-therapy products, can be excepted from the obligation of obtaining a marketing authorization via the centralized procedure under specific conditions (so-called “hospital exemption”). This hospital exemption allows the application of gene-therapy medicinal products prepared on a non-routine basis for an individual patient and used under the exclusive professional responsibility of a medical practitioner. Here, we explain the requirements to be fulfilled in order to fall under this exemption, the implementation of this regulation into the German national legislation, and its impact on gene-therapy product development in the future.
Introduction
Despite this progress, GTMPs have so far only reached the Asian market, where three different products are commercially available (Guo and Xin, 2006). In neither Europe nor the United States has any GTMP obtained marketing authorization so far. Reasons for this limited success of GTMPs are plentiful. Some are listed here: (i) The manufacturing process is often complex and costly. Especially, the transition from laboratory scale to a standardized and highly controlled industrial production process can be particularly challenging, e.g., for products containing autologous cells, which can show an intrinsically broad batch-to-batch variability. (ii) The demonstration of clinical efficacy can often be hampered by too few eligible patients, especially for rare inherited diseases. (iii) To date, GTMPs are developed mainly by science-driven small- and medium-sized enterprises (SMEs) or university hospitals, both lacking sufficient personnel and financial resources in order to comply with state-of the-art pharmaceutical standards. Their limited experience in regulatory processes turned out to be an additional burden when the European Commission decided to regulate GTMPs under the centralized European pharmaceutical legislation.
Here we illustrate the recent revision of the European GTMP regulation, especially focusing on the so-called hospital exemption clause, which was introduced by the Regulation (EC) No. 1394/2007 for advanced therapy medicinal products (ATMPs). Due to the high impact of this clause for the future development of GTMPs, we explain its legal basis, the requirements to be fulfilled to fall under this regulation, and its implications for gene therapy.
Regulation of GTMPs Under the European Pharmaceutical Legislation
In 2007, Regulation (EC) No. 1394/2007 integrated GTMPs together with somatic cell-therapy medicinal products (sCTMPs) and tissue engineered products (TEPs) in a new overarching pharmaceutical group named ATMPs (Sanzenbacher et al., 2007). Whereas GTMPs (see Fig. 1 for definition) and sCTMPs have been covered by the European pharmaceutical legislation before, TEPs—such as biotechnologically manufactured cell-based skin substitutes or chondrocytes for autologous chondrocyte implantation—were not regulated at the Community level. Nevertheless, at that time several TEPs were legally on the market in many Member States complying with highly divergent national legislation, if existent at all. Therefore, one major goal of Regulation (EC) No. 1394/2007 was to provide a harmonized legal definition and regulatory approach for this product group.

Revised legal definition of GTMPs. The definitions for GTMPs and sCTMPs were introduced in Directive 2009/120/EC, amending Directive 2001/83/EC, respectively. The TEPs and the combined products are defined in Regulation (EC) No. 1394/2007.
Regulation (EC) No. 1394/2007 has become applicable as of December 2008 and forms now, together with Directive 2001/83/EC and the respective amendments, the main legal framework for the regulation of ATMPs. Products that have been legally on the market (mainly TEPs) must be compliant with these laws and get an approval via the centralized procedure by December 30, 2011 (sCTMPs) or December 30, 2012 (TEPs) in order to maintain market access.
This overall legislative setup is clearly focused on centralized marketing authorization and stipulates a mandatory approval procedure for all ATMPs at the European level: To place GTMPs on the market, the product has to go through clinical trials to demonstrate its safety and efficacy before an application for marketing authorization can be filed at the European Medicines Agency (EMA) (Schüle et al., 2010). Only on the basis of a positive opinion of the Committee for Advanced Therapies (CAT) and the Committee for Human Medicinal Products (CHMP) and finally marketing authorization by the European Commission can the ATMP be marketed in all European Union Member States. For GTMP-specific issues, these legal provisions are further supplemented by a number of guidelines (see list of relevant documents: EMA ATMP-guidelines). Other options for making pharmaceuticals, including ATMPs, available for patients include clinical trials and compassionate use. However, data from confirmatory phase clinical trials demonstrating safety and efficacy are usually required to make the compassionate-use regulations applicable [see the specific requirements in article 83 to Regulation (EC) No. 726/2004]. This fits rarely to the stage of development of many GTMPs, which often have undergone only early-phase clinical studies until today.
In article 28 to Regulation (EC) No. 1394/2007—the hospital exemption clause—the European Commission introduced a possibility to annul the aforementioned obligation for a mandatory centralized marketing authorization under restricted conditions (Fig. 2). Thus, the clause is applicable for those ATMPs (including GTMPs) that are prepared on a non-routine basis according to specific quality standards for an individual patient, are used within the same Member State in a hospital under the exclusive professional responsibility of a medical practitioner, and are individually prescribed. The wording already indicates that the hospital exemption concept targets individual patient-centered care rather than the approval for placing a medicinal product with a high commercial interest on the market.

The hospital exemption clause (Article 28, subsection 2 of Regulation No. 1394/2007). The exemption of certain ATMPs from the centralized procedure has been introduced by amending article 3 of Directive 2001/83/EC.
The hospital exemption clause explicitly states that the competent authorities of the Member States shall authorize the manufacturing of these products and ensure the compliance of traceability and pharmacovigilance with European requirements. With the intention to provide flexibility and to cover all the different products, article 28 includes legally undefined terms as well as potential inconsistencies. For example, the requirement that the product should be “prepared on a non-routine basis” seems to be contradicted by the second paragraph of article 28 subsection 2, stating that “quality standards referred to in this paragraph are equivalent to those provided for at Community level with respect to advanced therapy medicinal products.” Thus, the hospital exemption allows no major deviations from the principles of GMP-compliant manufacturing. Outsourcing of the manufacturing process, shipment, or export of the concerned ATMP to other countries, even within the European Union, is explicitly excluded by the exemption.
In contrast to Regulations, which become law in all Member States at the moment they come into force, Directives have to be implemented into the law of each Member State within a defined time frame before they become legally effective. As article 28 to Regulation (EC) No. 1394/2007 is amending Directive 2001/83/EC, the specific legal regulation and supervision of ATMPs applied to patients under the hospital exemption is left to EU Member States. Each Member State is obliged to implement the provisions of article 28, but is also enabled to respect and incorporate its specific national circumstances. Germany is one of the first Member States where implementation of the hospital exemption clause has been completed and national provisions have been put in place.
Implementation of the Hospital Exemption in Germany
In Germany, the hospital exemption has been implemented by the 15th amendment of the “German Medicinal Products Act” [AMG (Arzneimittelgesetz); see list of relevant documents for German and English versions] by introducing article 4b (Sondervorschriften für Arzneimittel für neuartige Therapien). If the ATMP is intended to be distributed to other users, article 4b stipulates a mandatory authorization by the competent national regulatory authority. Hence, an applicant has to submit an application to the Paul-Ehrlich-Institut (PEI) as the responsible German competent authority for vaccines and biomedicines. TEPs and sCTMPs that have been placed on the German market according to former national law may fall under the transitional period of the ATMP regulation and, on the national level, under the transitional period for the hospital exemption specified in article 144 AMG. In this case, an application for the hospital exemption had to be submitted by August 1, 2010 (sCTMP, GTMP) or January 1, 2011 (TEP), to keep the product legally on the German market until a decision on the application could be taken by PEI. Whereas this transitional phase was highly relevant for quite a number of TEPs, no GTMP was concerned. To fall under the German hospital exemption, the product must be manufactured, prescribed, and used in Germany and should comply with the following prerequisites, as specified in article 4b AMG: (i) it must be prescribed by a physician as an individual preparation for an individual patient; (ii) it must be prepared on a non-routine basis according to specific quality standards; and (iii) it must be used in a specialized facility for health care under the professional responsibility of a medical practitioner.
What is meant by “prepared on a non-routine basis” is further defined in section 2 of article 4b AMG. Accordingly, a product should be manufactured in small quantities and, if based on a routine manufacturing process, variations in the process, medically justified for an individual patient, are carried out. Alternatively, the product has not yet been manufactured in sufficient quantities so that the necessary data to enable a comprehensive assessment are not yet available. This means that data with respect to product quality, clinical efficacy, and safety are available, but not to the extent required for marketing authorization via the centralized procedure. Thus, manufacturing on a non-routine basis is not (solely) linked to a specific maximal quantity. In fact, all above-mentioned aspects will have to be considered to decide if a product will be valid for an application according to article 4b. However, GTMPs based on autologous cells will obviously more readily comply with these requirements than products that are used off-the-shelf, such as oncolytic viruses.
The manufacturing of the ATMP should be conducted according to specific quality standards. In Germany, the issuing of the required manufacturing authorization is the responsibility of the federal state authorities (Länderbehörden). One of the prerequisites is that the applicant will have to hold a manufacturing license covering the concerned ATMP. Compared with conventional medicinal products, concerns about serious adverse events are usually more pronounced when applying GTMPs. These concerns may be derived from particular knowledge (e.g., insertional mutagenesis) or from uncertainties about the mode of action and potential toxicities. Thus, safety standards on traceability and pharmacovigilance have to be implemented at the national level according to articles 14 and 15 of Regulation (EC) No. 1394/2007 and must be equivalent to those provided at Community level for centrally authorized ATMPs. The requirements of the German pharmacovigilance system ensure that PEI is notified about any suspected adverse reactions to an ATMP used under the hospital exemption (article 63 AMG). According to article 15 of Regulation (EC) No. 1394/2007, the authorization holder and the respective specialized health-care facility in which the ATMP is used will have to establish and maintain a system for patient and product traceability. This system should ensure that the individual product and its starting and ancillary materials can be traced through sourcing, manufacturing, packaging, storage, transport, and delivery to the applying hospital. The requirements for traceability of the human donor cells and tissues are laid down in Directives 2004/23/EC and 2002/98/EC for human blood cells, respectively. In Germany, these requirements have been implemented into the “TPG-Gewebeverordnung” (TPG-GewV) and the ordinance “Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung” (AMWHVÄndVO), respectively.
The holder of the hospital exemption authorization is legally obligated to report regularly to PEI about the scale of manufacture and about the data for the comprehensive assessment of the medicinal product.
Finally, as many GTMPs consist of, or contain, genetically modified organisms (GMOs), the potential risk of the GMO for the environment needs to be evaluated (Anliker et al., 2010). Therefore, an application under hospital exemption for GMO-containing GTMPs must be accompanied by an environmental risk assessment (ERA) specifically performed on the basis of the information specified in Annexes III and IV of Directive 2001/18/EC and in accordance with the principles of Annex II to the Directive and its supplementing Commission Decision 2002/623/EC on the deliberate release of GMOs.
How to Apply at PEI—Practical Issues
For authorizing an ATMP under article 4b AMG in Germany, an application must be filed with the PEI. Depending on the responsibilities, a manufacturer, a clinician, or a pharmaceutical enterprise may apply. To support and guide the applicant, PEI developed several documents available on its Web site (see relevant documents: PEI documents article 4b AMG). As application under the hospital exemption scheme is only possible for applicants located in Germany, most information and modules are currently provided in German. However, PEI will accept applications in English as well.
Besides general information on the application procedure and fees, several modules are available that can be used for preparation of the application dossier. These modules are based on the internationally used application dossier format CTD (common technical document), but have been specifically adjusted and simplified according to the needs of the hospital exemption application procedure. Moreover, they contain additional information and decision trees to facilitate the application (Fig. 3). The applicant is asked to give his own estimation on classification and applicability of the hospital exemption for his product. A checklist specifically designed for GTMPs requests basic description about the used vector, its structure, replication potential, and application route. As explained above, high-quality standards are expected to be in place. Module M-3 provides guidance on which information on product quality, e.g., particle numbers, titer, tropism, specificity, potency of transgene expression, and qualification of master and working cell bank, is required. Finally, information on the nonclinical and clinical development has to be provided and should focus primarily on the actual product, but may be supported by information or data achieved with comparable products.
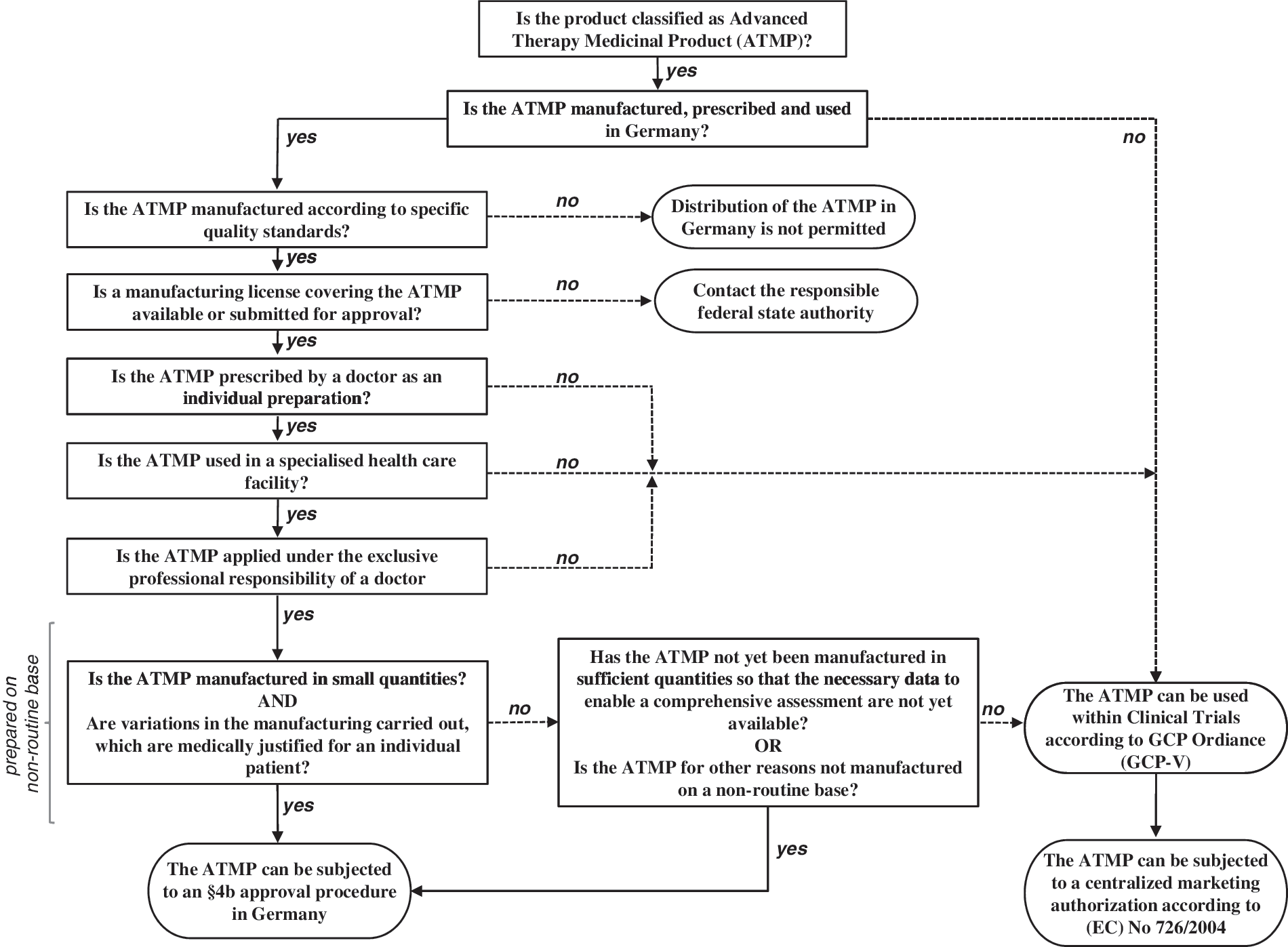
Decision tree depicting the requirements to be fulfilled by an ATMP to fall under the hospital exemption in Germany.
Despite the described available support, questions and uncertainties regarding a specific product application may remain. Thus, PEI encourages applicants to arrange for a preceding informal advice meeting. Although not legally binding, such face-to-face meetings are usually extremely helpful and informative for both the applicant and the authority.
The evaluation procedure may take up to 5 months. Due to the novelty of the application procedure and possible deficiencies of the dossier, a clock stop may be introduced to clarify outstanding issues. The authorization shall be revoked, once the requirements of section 4b AMG are no longer fulfilled.
Outlook and Situation in the European Union
The option to apply for a hospital exemption comes at a point in time where several GTMPs have demonstrated nonclinical or even clinical benefits. Some member states, such as Germany and the United Kingdom, have defined and published the requirements and authorization procedure, and first applications have been filed. Detailed information is available on the Web sites of these competent authorities. Other EU Member States are still in the process of converting the hospital exemption regulation into national law. In any case, applications can now be filed to obtain a hospital exemption for GTMPs. It must be clearly emphasized, however, that for a systematic assessment of safety and efficacy, clinical trials are irreplaceable. Consequently, data gained from GTMPs used under the hospital exemption scheme cannot substitute for systematic product development and controlled clinical trials.
According to the authors' view, the idea of the hospital exemption is to preserve and facilitate promising patient-directed therapy options and to provide a certain degree of flexibility in the regulation of ATMPs. Accordingly, there are many reasons why going for a hospital exemption may be a good choice for certain types of GTMPs. Advantages are already evident, e.g., the option to treat patients who then benefit from receiving otherwise unavailable treatments within an authorized setting, but under adapted requirements and a less cost-intensive application procedure. Others may become clear once practical experience with GTMPs under hospital exemption has been collected. However, it must be clearly emphasized that for a systematic assessment of safety and efficacy clinical trials are irreplaceable. Consequently, data gained from GTMPs used under the hospital exemption scheme cannot substitute for a systematic product development and investigation under a controlled clinical trial regime.
Gene therapy, whether by in vivo or ex vivo vector application, is a complex therapeutic approach that requires experienced physicians with a strong background in molecular medicine. Moreover, patients suffering from genetic disease are usually rare. Both facts argue for a few or even single centers specialized for certain types of genetic disease and treatment strategies in Europe. In such a setting, applying a GTMP under the hospital exemption seems more appropriate than under the centralized marketing authorization. Moreover, the GTMP is then prescribed by the medical practitioner. This may allow financial compensation by the health insurance systems. Especially for SMEs which often have to rely on long-term financial support by investors, this may be an important option toward reaching financial balance.
Footnotes
Acknowledgments
This work was supported by grants of the 7th European Community programme (agreement 222878, PERSIST) and the LOEWE Center for Cell and Gene Therapy Frankfurt funded by Hessisches Ministerium für Wissenschaft und Kunst (III L 4- 518/17.004 (2010)) to C.J.B. The authors wish to thank Jürgen Scherer and Marion Frech for critical reading of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
Relevant Documents
726/2004. The European Parliament and the Council of the European Union. Off. J. Eur. Comm. L136, 1–33 (2004). <
1394/2007. Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004 (Note: shall apply from 30 December 2008) (Official Journal L 324, 10/12/2007, pp. 121–137) <
2001/18/EC. Directive 2001/18/EC of the European Parliament and of the Council of 12 March 2001 on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC (Official Journal L 106, 17/4/2001, pp. 1–39). <
2001/83/EC. The European Parliament and the Council of the European Union. Off. J. Eur. Comm. L311, 67–126 (2001). <
2002/98/EC. The European Parliament and the Council of the European Union. Off. J. Eur. Comm. L33, 30–40 (2003). <
2002/623/EC. Commission Decision 2002/623/EC establishing guidance notes supplementing Annex II to Directive 2001/18/EC of the European Parliament and of the Council on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC.24 July 2002. Official Journal L 200, 30 July 2002, pp. 22–33. <
2004/23/EC. The European Parliament and the Council of the European Union. Off. J. Eur. Comm. L102, 48–58 (2004). <
2009/120/EC. The Commission of the European Communities. Off. J. Eur. Comm. L242, 15(9) (2009), pp. 3–12. <
AMG. Gesetz über den Verkehr mit Arzneimitteln [Arzneimittelgesetz (AMG)]. Arzneimittelgesetz in der Fassung der Bekanntmachung vom 12. Dezember 2005 (BGBl. I S. 3394), das zuletzt durch Artikel 1 der Verordnung vom 19. Juli 2011 (BGBl. I S. 1398) geändert worden ist. <
AMWHVÄndVO. Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHVÄndV): V. v. 26.03.2008 BGBl. I S. 521 (Nr. 12); Geltung ab 05.04.2008.
EMA ATMP-guidelines. <
Medicinal Products Act [Arzneimittegesetz (AMG)] of the Federal Republic of Germany (Nonofficial translation). <
PEI documents article 4b AMG. <
TPG-GewV. TPG-Gewebeverordnung vom 26. März 2008 (BGBl. I S. 512). <