
Abstract

Proximal spinal muscular atrophy (SMA) is one of the leading genetic causes of infant death in the world. SMA is an autosomal recessive degenerative disease characterized by selective loss of α motor neurons of the anterior horn of the spinal cord, which leads to atrophy of limb and trunk muscles (Crawford and Pardo, 1996). SMA results from the loss or mutation of the SMN (survival motor neuron) gene (Lefebvre et al., 1995). In humans, the SMN gene is duplicated to yield two SMN genes (SMN1 and SMN2). SMA results from the loss of or mutation in SMN1 but—in most cases—retention of SMN2. SMN1 and SMN2 differ by a single nucleotide (C-to-T) transition within an exon splice enhancer of exon 7 (Lorson et al., 1999; Monani et al., 1999). mRNA transcripts from SMN1 contain exon 7, which encodes full-length SMN (FL-SMN), whereas most (80–90%) of the transcripts from SMN2, on the other hand, lack exon 7 and produce a truncated SMN protein lacking exon 7 (SMNΔ7) (Fig. 1; reviewed in Burghes and Beattie, 2009).
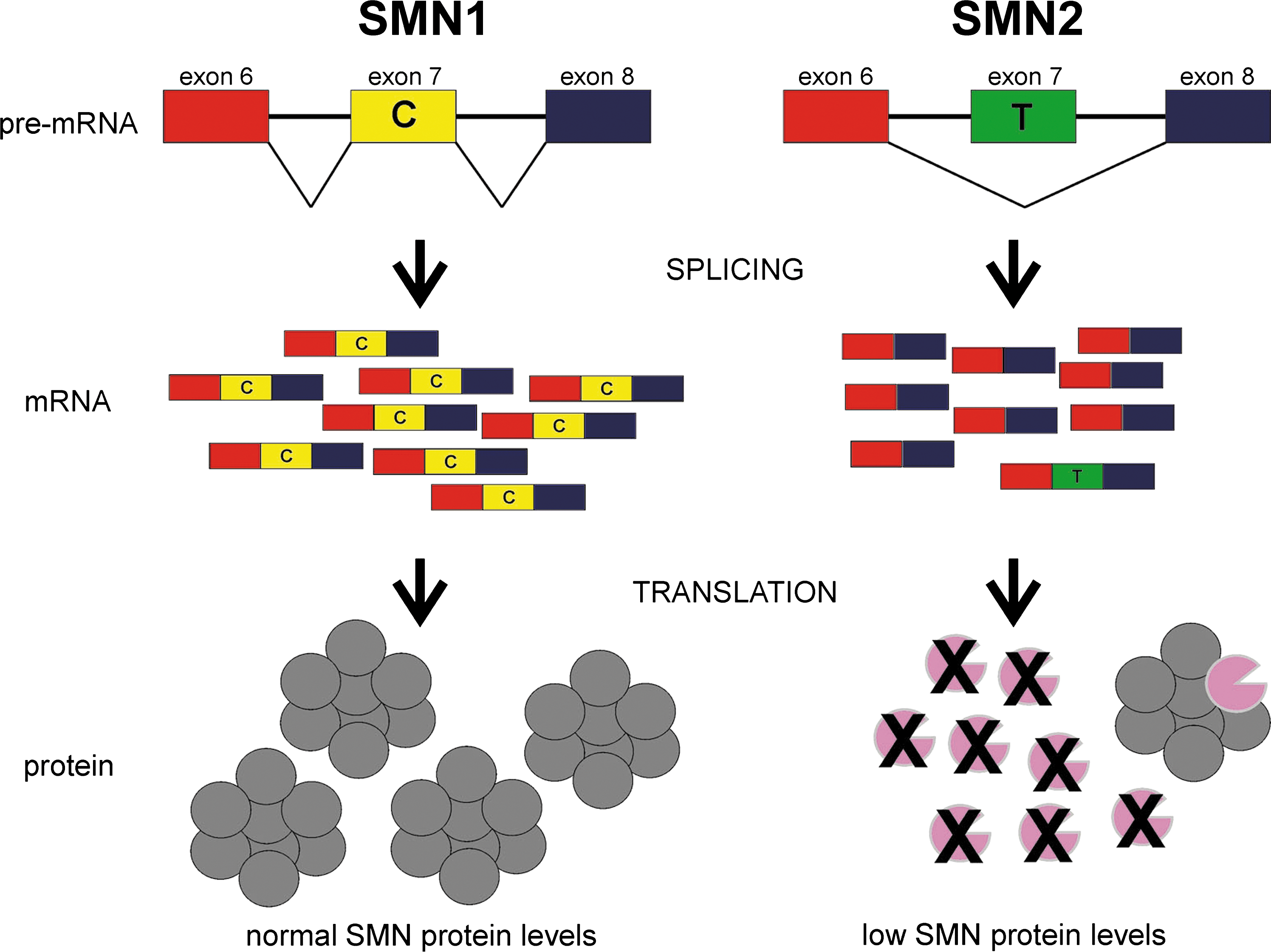
The effect of a single-nucleotide transition from C to T in SMN exon 7 on splicing and translation. The SMN gene is duplicated in humans. The primary difference between SMN1 and SMN2 in humans is a C→T transition within exon 7. This transition disrupts an exonic splicing enhancer element within exon 7 leading to the majority (80–90%) of SMN2 mRNA transcripts lacking exon 7 (SMNΔ7). The resultant SMNΔ7 protein is unstable and rapidly degraded; however, this truncated protein is able to form heteromeric complexes with full-length SMN (FL-SMN) protein. SMA results from the loss of or mutation in SMN1 but retention of SMN2. SMN2 can produce some FL-SMN protein but it is much less than that produced from SMN1. Reprinted from Butchbach and Burghes (2004) with permission from Elsevier.
In patients with SMA, the copy number of SMN2 correlates with disease severity (Coovert et al., 1997; Lefebvre et al., 1997; McAndrew et al., 1997). Ablation of the mouse SMN1 gene (mSmn; the SMN gene is not duplicated in mice) by gene targeting leads to embryonic lethality (Schrank et al., 1997). Insertion of SMN2 into mSmn null mice by transgenesis rescues the embryonic lethality phenotype (Monani et al., 2000); however, mice with low copy numbers (i.e., one or two) of SMN2 develop severe (type I-like) SMA and die at 6–8 days (Hsieh-Li et al., 2000; Monani et al., 2000). mSmn nullizygous mice with three or four copies of SMN2 develop a milder SMA phenotype than those mice with two copies of SMN2 (Hsieh-Li et al., 2000; Michaud et al., 2010). Mice with very high copy numbers (i.e., eight) of SMN2, on the other hand, are phenotypically normal when compared with nontransgenic littermates (Monani et al., 2000). On the basis of these observations, SMN2 is a phenotypic modifier of SMA both in humans and in mice.
Increasing SMN protein levels in neurons (using the prion protein promoter as a driver) but not in mature skeletal muscle (using the skeletal muscle actin promoter as a driver) ameliorates the phenotype of mice with very severe SMA (SMN2+/+ ; mSmn–/– ) (Gavrilina et al., 2008). SMN gene replacement by either lentiviral (Azzouz et al., 2004) or adeno-associated viral (AAV) (Foust et al., 2010; Passini et al., 2010; Valori et al., 2010; Dominguez et al., 2011) vectors significantly improves the phenotype of SMA mice and prolongs their survival. Severe SMA mice that also contain the SMNΔ7 transgene (SMN2+/+ , SMNΔ7+/+ , mSmn–/– ) develop a less severe SMA phenotype and these mice die at 13–15 days (Le et al., 2005). Introduction of SMA-linked intragenic SMN mutations [SMN(A2G) and SMN(A111G)] onto a severe SMA mouse background significantly ameliorates the severe SMA phenotype depending on the copy number of the mutant SMN transgenes (Monani et al., 2003; Workman et al., 2009). These transgenic and gene therapy mouse studies show that increasing SMN expression, whether it is FL-SMN, SMNΔ7, or mutant SMN, can be beneficial to SMA mouse models.
SMN2 is a strong target for the development of therapeutic agents for SMA. Numerous studies have identified many compounds that increase SMN expression by activating the SMN2 promoter in cultured cells. Histone deacetylase (HDAC) inhibitors such as trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA) have been shown to increase SMN expression and improve the survival and phenotype of SMA mouse models (Avila et al., 2007; Narver et al., 2008; Riessland et al., 2010). One problem with using TSA and SAHA as therapeutics for SMA is that it is not known whether these compounds specifically increase SMN2 promoter activity, as these HDAC inhibitors can modulate the expression of multiple genes. An ultrahigh-throughput screen of compounds that induce SMN2 promoter activity in motor neuron-like cells identified 2,4-diaminoquinazoline as a chemical scaffold on which derivatives with stronger potency and favorable pharmacological properties can be synthesized (Jarecki et al., 2005). One such derivative, D156844, increases SMN protein expression in cultured SMA fibroblasts as well as in—albeit modestly—spinal cord extracts from SMNΔ7 SMA mice (Thurmond et al., 2008; Butchbach et al., 2010). Oral administration of this compound significantly increases (by ∼21–30%) the mean life span and motor function of SMNΔ7 SMA mice when given before motor neuron loss (Butchbach et al., 2010). Although the change in SMN expression in vivo is small and the effect on survival of SMA mice is small when compared with the AAV-mediated gene replacement experiments (Foust et al., 2010; Passini et al., 2010; Valori et al., 2010; Dominguez et al., 2011), this study demonstrates the feasibility of drug discovery and optimization for the development of clinical therapeutic candidates for SMA.
Another way to increase SMN2-derived SMN expression is to modulate the splicing of SMN2 pre-mRNAs such that exon 7 is included in a greater proportion of SMN2 mRNAs. Tetracycline derivatives such as PTK-SMA1 have been shown to increase FL-SMN protein levels by promoting the inclusion of exon 7 in SMN2 mRNAs (Hastings et al., 2009). Although PTK-SMA1 increases SMN protein expression in vivo in mouse liver, it is unable to cross the blood–brain barrier (BBB) or the blood–spinal cord barrier (BSCB), thus limiting its use as a therapeutic for SMA. Another approach to modulate SMN2 splicing involves the use of antisense oligonucleotides (ASOs). Blockade of an intronic splicing silencing (ISS) element at the 5′ end of the intron between exons 7 and 8 with an ASO promotes the inclusion of exon 7 in SMN2 transcripts (Singh et al., 2006; Hua et al., 2008). Repeated intracerebroventricular (ICV) administration of an 2′-O-methyl ASO increased FL-SMN protein expression and improved motor function in SMNΔ7 SMA mice (Williams et al., 2009). The effect of this ASO on the survival of SMNΔ7 SMA mice was not examined in this study. ICV administration of SMN2 ISS ASOs with a 2′-O-(2-methoxyethyl) phosphorothionate increases SMN expression in the CNS (Hua et al., 2010); intravenous administration of these ASOs, however, does not increase FL-SMN protein levels in the CNS (Hua et al., 2008). Embryonic ICV treatment of mice with mild SMA (four copies of SMN2 on an mSmn nullizygous background) with this ASO rescues tissue necrosis observed in these mice (Hua et al., 2010). ASOs may be valid therapeutics for SMA except that most ASOs examined to date are unable to penetrate the physiological barriers around the CNS, that is, the BBB and BSCB.
Bifunctional RNAs have two functional domains: a moiety that is complementary to a specific RNA (the ASO region) and a splicing factor (such as A2F, heterogeneous nuclear ribonucleoprotein [hnRNP] A1, and Tra2β) binding site. One of the first examples of using bifunctional RNAs to modulate SMN2 splicing contained an ASO region that bound to exon 7 and an A2F exonic splicing enhancer (ESE) element (Skordis et al., 2003). This tailed bifunctional RNA promotes inclusion of exon 7 in SMN2 transcripts and increases FL-SMN protein levels in SMA fibroblasts. Placing this bifunctional RNA in a U7 small nuclear RNA vector (U7-ESE-B) markedly promoted exon 7 inclusion and increased FL-SMN protein in SMA fibroblasts (Marquis et al., 2007). Transgenic expression of U7-ESE-B into mice with very severe SMA led to an increase in FL-SMN protein in the CNS and markedly improved survival (Meyer et al., 2009). AAV2-mediated delivery of a bifunctional RNA containing an intron 6 ASO and tandem ESE sites increased the inclusion of exon 7 in SMN2 transcripts and SMN protein levels in SMA fibroblasts (Baughan et al., 2006). A bifunctional RNA that contains an ASO moiety directed against a negative splicing regulatory element (E1) in intron 6 coupled to a moiety of tandem Tra2-binding elements (Tra2-E1) increased the expression of FL-SMN protein in SMA fibroblasts (Baughan et al., 2009). ICV injection of Tra2-E1 2′-O-methyl RNAs into mice with very severe SMA increased FL-SMN protein in the CNS and modestly increased the average life span of these mice. Introduction of 2′-O-methyl bifunctional RNAs containing an ASO against exon 8 and tandem hnRNP A1-binding elements (exon8-hnRNPA1) by ICV injection into neonatal SMNΔ7 SMA mice increased FL-SMN protein levels in the brain and spinal cord (Dickson et al., 2008).
Trans-splicing is a process by which two separately transcribed mRNAs are involved in a splicing event wherein a composite transcript is produced (reviewed in Wood et al., 2007). A trans-splicing approach promotes the inclusion of exon 7 in SMN2 mRNA transcripts in vitro and, to a lesser extent, increases SMN protein expression and function in SMA fibroblasts (Coady et al., 2007). By adding to the SMN2 trans-splicing vector a cassette that produces a small RNA that is complementary to the intron 7:exon 8 splice junction (the trans-splicing:antisense combination pMU3 vector), treatment with pMU3 resulted in a more marked increase in FL-SMN protein expression and small nuclear ribonucleoprotein (snRNP) assembly (an assayable function of SMN) than from the trans-splicing vector (Coady et al., 2008). Furthermore, ICV administration of pMU3 into newborn SMA mice led to a significant increase in FL-SMN protein levels in the spinal cord and a 60% increase in the average life span (from postnatal day 5 [PND5] to PND7, with the day of birth defined as PND1) of mice with very severe SMA (two copies of SMN2 and nullizygous for mSmn) (Coady et al., 2008; Coady and Lorson, 2010). As shown by these studies, trans-splicing may be a viable strategy for increasing SMN protein levels in patients with SMA.
In this issue of Human Gene Therapy, Shababi and colleagues (2011) show the combined effects of a trans-splicing RNA directed at SMN2 and the neurotrophic factor insulin-like growth factor-1 (IGF-1) on the expression of SMN protein and on the life span of mice with very severe SMA. The novelty of this study lies in the multifaceted approach the nonviral vector uses to ameliorate the phenotype of SMA mice, that is, increasing SMN expression by trans-splicing and protecting from motor neuron loss with IGF-1. IGF-1 was selected as a neuroprotective factor in this study because this neurotrophin has been shown to ameliorate the survival and phenotype of the SOD1(G93A) transgenic mouse model for the motor neuron disease amyotrophic lateral sclerosis (ALS) (Kaspar et al., 2003; Lepore et al., 2007). As with the pMU3 vector, the trans-splicing:antisense:IGF-1 combination vector (pMU4) increases FL-SMN protein levels and snRNP in the CNS of SMNΔ7 SMA mice (Shababi et al., 2011). A single ICV injection of pMU4 into newborn mice with very severe SMA led to a significant increase (∼80%; i.e., PND9 from PND5) in the average life span (Shababi et al., 2011).
One limitation with using plasmid DNAs—or any other oligonucleotide-based biopharmaceutical, for that matter—revolves around the delivery of these compounds to target cells. Most plasmid DNAs and ASOs are unable to penetrate the BBB or BSCB, and therefore direct injection into the CNS is required for delivery. Repeated ICV injections can be problematic for neonatal mice and can lead to neurodevelopmental abnormalities and premature death (from hydrocephalus; M.E.R. Butchbach, personal observations). This risk of complications can be somewhat alleviated by using a different route of administration, such as intrathecal injection, or by using an implantable long-term infusion system such as a microosmotic pump. Unfortunately, microosmotic pumps cannot be used for therapeutics delivery in neonatal and preweanling mice, but this chronic infusion approach has been successfully used by Hua and colleagues (2010) to deliver ASOs to the CNS of mice with mild SMA. Another option would be to modify the plasmid DNA (or oligonucleotide) itself to improve delivery. One approach in which delivery of therapeutic plasmid DNAs could be improved is through the use of a facilitated delivery system (Viola et al., 2010). Systemic delivery of plasmid DNAs to target cells—in this case, spinal motor neurons—could be enhanced by physical means such as electroporation (De Vry et al., 2010) or hydrodynamic delivery (Herweijer and Wolff, 2007). Therapeutic plasmid DNAs can also be formulated with nanocarriers such as cationic liposomes, polymers such as polyethylenimine (PEI), and RGD polypeptides to form functionalized nanoparticles (reviewed in Pathak et al., 2009; Viola et al., 2010). Another way to circumvent the problem of delivery requires the use of a different mode of gene delivery. Viral gene vectors such as those based on AAV—especially the AAV8 and AAV9 serotypes—have been shown to efficiently deliver their target gene (SMN) to the neonatal spinal cord to ameliorate the motor neuron disease in SMA mice (Foust et al., 2010; Passini et al., 2010; Valori et al., 2010; Dominguez et al., 2011).
Multiple ways to increase SMN expression, ranging from chemical activators of the SMN2 promoter to viral vector-based SMN replacement to oligonucleotide-based manipulation of SMN2 pre-mRNA splicing, are currently being investigated. Multifunctional approaches such as bifunctional RNAs as well as multifaceted approaches wherein two different processes are being targeted (such as promoting exon 7 inclusion in SMN2 transcripts and IGF-1-mediated neuroprotection as shown in this issue of Human Gene Therapy) are also showing promise as therapeutics for SMA. Although each of these approaches has its respective limitations, optimization of each of these approaches so as to maximize increased SMN expression and to minimize unfavorable traits such as poor CNS penetration and off-target toxicity is ongoing. At the same time, combination therapies, for example, increasing SMN promoter activity with D156844 and increasing SMN2 exon 7 inclusion with bifunctional RNAs, are being considered as therapeutics for SMA.