
Abstract
Mucopolysaccharidosis type IIIA (MPSIIIA) is a rare lysosomal storage disorder caused by mutations in the sulfamidase gene. Accumulation of glycosaminoglycan (GAG) inside the lysosomes is associated with severe neurodegeneration as well as peripheral organ pathological changes leading to death of affected individuals during adolescence. There is no cure for MPSIIIA. Due to the limitation of the blood–brain barrier, enzyme replacement therapy and gene therapy strategies attempted thus far have not achieved whole-body correction of the disease. After the systemic administration of an adeno-associated virus 9 (AAV9) vector encoding for sulfamidase under the control of a ubiquitous promoter, we were able to obtain widespread expression of the therapeutic transgene in brain and in peripheral organs, and sulfamidase activity in serum of both male and female MPSIIIA mice. This was accompanied by the normalization of GAG storage levels in most peripheral organs. In brain, decrease in GAG tissue content following AAV9 gene transfer of sulfamidase was associated with the resolution of neuroinflammation. Finally, correction of disease phenotype resulted in a remarkable prolongation of survival of both male and female AAV-treated MPSIIIA mice. This proof-of-concept study will be relevant to the future development of therapies for MPSIIIA.
Introduction
A mouse model of MPSIIIA, resulting from a spontaneous missense mutation in the sulfamidase gene, has been identified (Bhattacharyya et al., 2001). MPSIIIA mice show similar pathological alterations to those observed in MPSIIIA patients, such as severe neurodegeneration, neuroinflammation, increased liver and spleen size, and shortened lifespan (Bhaumik et al., 1999; Crawley et al., 2006).
There is no cure for MPSIIIA; existing treatments merely try to alleviate the symptoms of the disease without treating the cause. Several strategies are currently being pursued to treat MPSIIIA (Savas et al., 2004; Crawley et al., 2006; Roberts et al., 2006; Fraldi et al., 2007b; Hemsley et al., 2008; McIntyre et al., 2008; Hemsley et al., 2009a; Hemsley et al., 2009b; McIntyre et al., 2010; Crawley et al., 2011; Ruzo et al., 2012) (Shire Human Genetic Therapies, Inc., 2010), most of them relying on the fact that lysosomal enzymes can be taken up by cells from the extracellular medium through mannose-6-phosphate receptor (M6PR)-mediated endocytosis (Enns et al., 2008). Consistently, in MPSIIIA mice, enzyme replacement therapy (ERT) was able to markedly decrease the HS accumulation in non-neurological tissues, but the inability of sulfamidase to cross the blood–brain barrier (BBB) at normal doses (Gliddon and Hopwood, 2004; Urayama et al., 2008; Vogler et al., 2005) prevented disease correction in the central nervous system (CNS).
Direct administration of recombinant sulfamidase to the brain parenchyma or into the cerebrospinal fluid (CSF) has been tested in MPSIIIA mice and dogs ( Savas et al., 2004; Hemsley et al., 2008; Hemsley et al., 2009a; Hemsley et al., 2009b; Crawley et al., 2011). However, clinical translation of these approaches may be challenging as the enzyme has a short half-life (Crawley et al., 2011), requiring repeated administrations and the widespread distribution of the neurological alterations, which would require injections in multiple locations in the brain.
Adeno-associated vectors (AAV) have proven highly efficient in transducing post-mitotic cells in a wide range of tissues, including CNS and liver, resulting in long-lasting transgene expression (Mingozzi et al., 2011b; Nathwani et al., 2011). We previously showed that a single intravenous administration of an AAV8 vector expressing sulfamidase was driving high levels of transgene expression from the liver, efficiently correcting non-neurological alterations of MPSIIIA mice, and mediating a partial reversal of the neurological pathology. The only limited disease correction in the brain was due to the inability of most AAV serotypes, including AAV8, to cross the BBB (Ruzo et al., 2012). Similarly, it has been documented that the direct intracranial administration of AAV vectors to MPSIIIA mice results in correction of the pathological alterations in the brain but not in peripheral organs (Fraldi et al., 2007b).
Adeno-associated virus 9 (AAV9), a recently isolated AAV serotype (Gao et al., 2004), can cross the BBB from the bloodstream and transduce neurons and astrocytes in multiple brain areas, while also transducing the liver and heart very efficiently (Duque et al., 2009; Foust et al., 2009; Gray et al., 2011). This strategy has been successfully used to revert the neurological pathology in a mouse model of MPSIIIB (Fu et al., 2011).
Here, we tested the ability of an AAV9 vector expressing the sulfamidase transgene delivered systemically to mediate widespread gene delivery to the brain and to peripheral organs. This approach resulted in correction of GAG accumulation in CNS and peripheral organs of male and female MPSIIIA mice; this was associated with a significant prolongation of the lifespan of treated animals, regardless of sex. These results demonstrate that systemic gene transfer with AAV9 vectors efficiently targets the brain and several peripheral tissues and provides a potential treatment strategy for MPSIIIA.
Materials and Methods
Animals
Congenic C57Bl/6 mice carrying an inactivating missense mutation (G91A) in the sulfamidase gene were used (Crawley et al., 2006). Affected MPSIIIA and unaffected littermates were bred from heterozygous founders (The Jackson Laboratory, Bar Harbor, MA). Genotyping was performed by polymerase chain reaction (PCR) as previously described (Bhattacharyya et al., 2001; Ruzo et al., 2012). All experimental procedures were approved by the Ethics Committee in Animal and Human Experimentation of the Universitat Autònoma de Barcelona.
Vector administration to mice
The AAV expression cassette used in the study was generated by cloning the cDNA of murine sulfamidase (Clone ID: D330015N16; RIKEN, Japan) or green fluorescent protein (GFP) under the control of the ubiquitous promoter CAG (chicken β-actin promoter and CMV enhancer) into AAV backbones. AAV9 vectors were produced by a triple transfection of HEK293 cells and purified as described (Ayuso et al., 2010), resulting in a prep titer of 3.5×1013 vg/ml. Vectors were delivered intravenously in a total volume of 200 μl of PBS.
Sample collection
At the time of sacrifice, mice were anesthetized by intraperitoneal (IP) injection of ketamine/xylacine (100/10 mg/kg). Blood was extracted by cardiac puncture, and animals were perfused via cardiac puncture with 10 ml of phosphate buffered saline (PBS). The entire brain and all other tissues were collected and either snap-frozen or formalin-fixed until analyzed.
Sulfamidase activity and GAG quantification
Samples were sonicated in water and sulfamidase activity was assayed in supernatants with a 4-methylumbelliferone-derived fluorogenic substrate (Moscerdam Substrates, Oegstgeest, The Netherlands), as previously described (Karpova et al., 1996). Sulfamidase activity was normalized against the total protein content quantified by Bradford assay (Bio-Rad, Hercules, CA). For glycosaminoglycans (GAG) quantification, tissues were weighted and digested with proteinase K and extracts were clarified by centrifugation and filtration (Ultrafree MC, Millipore, Billerica, MA). GAG levels were determined with the Blyscan sulfated glycosaminoglycan kit (Biocolor, Carrickfergus, United Kingdom) using chondroitin 4-sulfate as standard. Results were normalized to wet tissue weight. All samples were processed and analyzed in parallel.
Histology
Tissues were fixed in 4% formaldehyde, embedded in paraffin, and sectioned. For immunohistochemical detection, paraffin sections were incubated overnight at 4°C with rat anti-LAMP1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-LIMP2 (Novus Biologicals, Littleton, CO), rabbit anti-GFAP (Dako Cytomation, Glostrup, Denmark), lectin BSI-B4 (Sigma, St Louis, MO) and goat anti-GFP (Abcam, Cambridge, MA). Secondary antibodies were biotinylated rabbit anti-rat IgG (Dako Cytomation) or biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA). Brightfield sections were stained with 3,3-diaminobenzidine (Sigma, St Louis, MO), counterstained with hematoxilin, and images were obtained with an Eclipse E800 optical microscope (Nikon, Tokyo, Japan). LIMP-2, GFAP, and BSI-B4 signal were quantified in 3–5 images/animal of each brain region, using NIS-Elements Advanced Research 2.20 software. For fluorescence sections, streptavidin-conjugated Alexa 488 (Molecular Probes, Eugene, OR) or streptavidin-Alexa 568 (Molecular Probes) were used for visualization. Nuclei were stained with TOPRO-3, and images were obtained with a confocal microscope (Leica Microsystems, Wetzlar, Germany).
Vector genome copy number
Tissues were digested overnight at 56°C in 400 μl of Proteinase K solution (0.2 mg/ml). Total DNA was isolated from supernatants by extraction using standard techniques. DNA was resuspended in distilled water and quantified using a NanoDrop ND-1000 (NanoDrop, Wilmington, DE). Vector genome copy number in 20 ng of total DNA was determined by quantitative real-time PCR with primers and probe specifics for the woodchuck hepatitis virus posttranscriptional regulatory (WPRE) element in the AAV-GFP vector. Forward primer: 5′-CGG CTG TTG GGC ACT GA-3 ′; Reverse primer: 5′-GGA AGG TCC GTC GAA TTG A-3; Probe: 5′-ATG GCT GCT CGC CTG TGT TGC C-3′. The final values were determined by comparing to a reference standard curve, built from serial dilutions of the linearized plasmids used for AAV vector production spiked into 20 ng of non-transduced genomic DNA.
Statistical analysis
Results are expressed as the mean±SEM. Statistical comparisons were made by either t-test or one-way ANOVA. Statistical significance was considered if p<0.05. Survival analysis was done by the Kaplan–Meier method and the log-rank test was used for comparisons.
Results
Systemic administration of an AAV9 vector encoding for sulfamidase reduces lysosomal GAG storage in CNS of male and female MPSIIIA mice
Recent studies suggest that AAV serotype 9 vectors have the ability to cross the blood-brain barrier (BBB) when delivered systemically. We confirmed these results by injecting 2-month-old wild-type (WT) and MPSIIIA mice intravenously with 1×1012 vector genomes (vg) of an AAV9 vector encoding GFP. Two weeks after vector administration, widespread brain transduction was observed in different parts of the CNS (Fig. 1), regardless of genotype, along with the efficient targeting of liver and heart, typical of this vector serotype (Supplementary Fig. S1; Supplementary Material available online at

Widespread CNS transduction after intravenous delivery of AAV9. Immunosfluorescent detection of GFP in different brain sections was performed 2 weeks after intravenous delivery of 1x1012 vg to 2-month-old wild-type (WT, left panels) and MPSIIIA (middle panels) male mice (n=3/group). Similar patter of transduction was observed for both genotypes. GFP immunostaining of sections obtained from uninjected MPSIIIA mice (right panels) were used as control. Scale bars: 10 μm, except cerebellum 5 μm. CNS, central nervous system; AAV9, adeno-associated virus 9; GFP, green fluorescent protein; MPSIIIA, mucopolysaccharidosis type IIIA.
Two-month-old WT and MPSIIIA males and females (n=3–4) were intravenously injected with 1012 vg of an AAV9-GFP vector and sacrificed 15 days later. Vector genome copies were determined through Taqman-quantitative real-time polymerase chain reaction (PCR) assay with primers and probe described in the Materials and Methods section. The five brain regions analyzed correspond to the same five sections analyzed and depicted in Figure 3. CNS, central nervous system; AAV-9, adeno-associated virus 9; GFP, green fluorescent protein; WT, wild-type; MPIIIA, mucopolysaccharidosis type IIIA.
The efficient target of both central nervous system (CNS) and peripheral tissues with AAV9 vectors prompted us to test the therapeutic efficacy of systemic gene transfer of the sulfamidase transgene in MPSIIIA mice. Two-month-old mice MPSIIIA were used in this study. At this age, animals already show pathological accumulation of glycosaminoglycans (GAGs) in the brain (Supplementary Fig. S3a) and other tissues (Ruzo et al., 2012). Lysosomes appear enlarged, and activation of astroglial and microglial cells denotes the presence of neuroinflammation (Supplementary Fig. S3b–d).
Male and female MPSIIIA mice received 1×1012 vg of an AAV9 vector encoding for the sulfamidase transgene under the control of a constitutive promoter (AAV9-Sgsh) and were followed for 8 months after gene transfer. As controls, untreated MPSIIIA mice or unaffected littermates were followed for an equal amount of time.
At 10 months of age, untreated MPSIIIA mice had significantly higher levels of GAG compared to unaffected littermates in all areas of the brain that were tested (Fig. 2a and b). Conversely, AAV9-Sgsh treated animals showed complete correction of pathological accumulation of GAG throughout the brain, with the exception of the portion that encompasses the more caudal part of the occipital cortex and the more rostral part of the brainstem (Section IV) (Fig. 2a and b), where the correction of the disease phenotype was only partial.
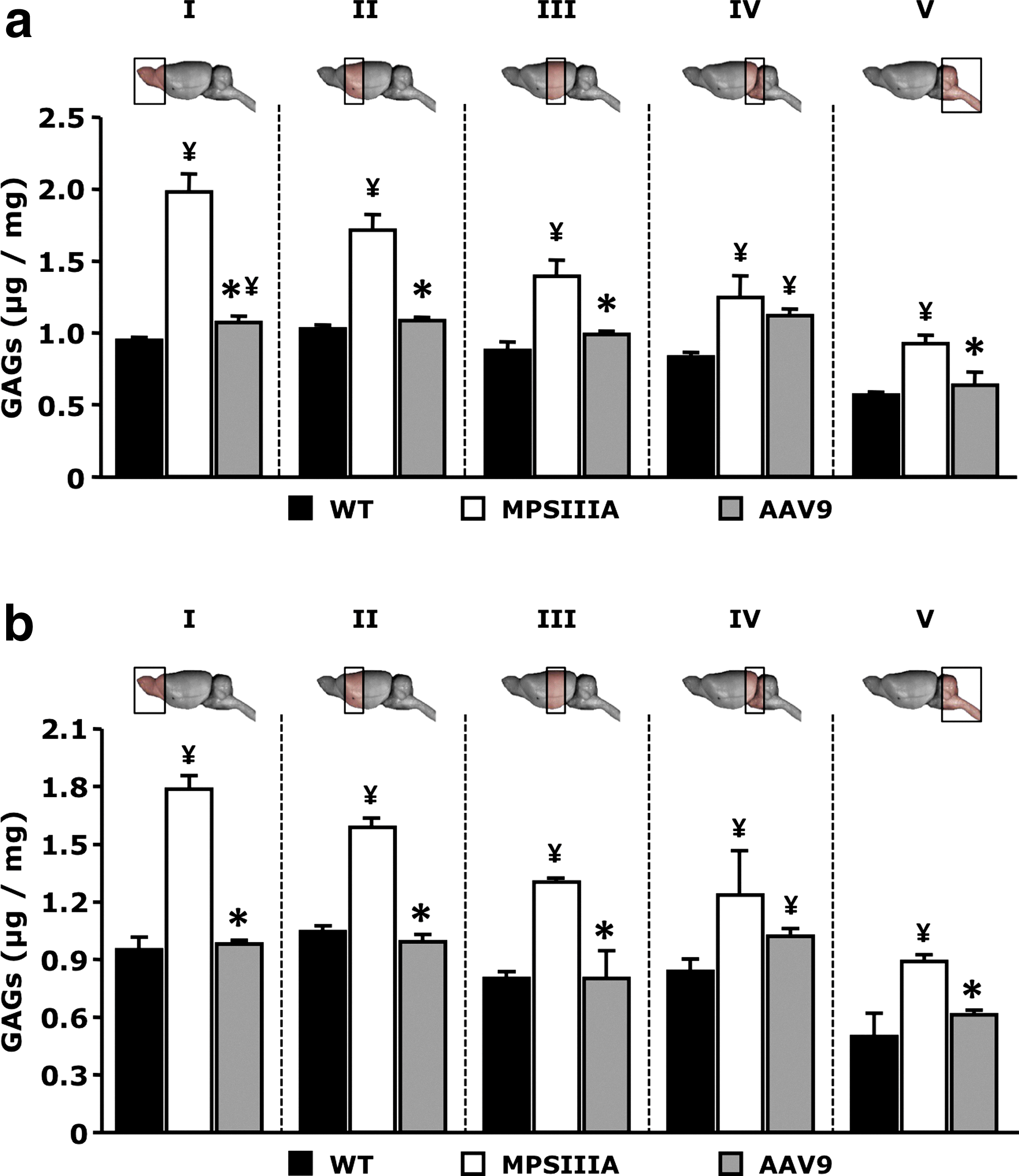
Glycosaminoglycan (GAG) levels in the CNS after intravenous delivery of AAV9-Sgsh vector. GAG content in different areas of the brain from unaffected littermates (WT), untreated MPSIIIA mice (MPSIIIA), or AAV9-Sgsh treated male (AAV9)
In agreement with these results, sulfamidase transgene product activity was detectable throughout the brain at levels between about 6 and 13% of normal (Table 2). Female animals showed slightly lower levels of sulfamidase expression in the brain, which did not seem to result in decreased clearance of GAG from brain tissue. Interestingly, section IV in treated animals contained notably less sulfamidase activity than any of the other sections, which was consistent with the only partial correction of GAG storage observed in that same area of the brain (Fig. 2).
Percentage of WT sulfamidase activity±SEM achieved in the brain of MPSIIIA mice after the intravenous AAV9-Sgsh treatment (n=3–4). The five brain regions analyzed correspond to the same five sections analyzed and depicted in Figure 3. Sgsh, sulfamidase.
Clearance of GAG from CNS is associated with marked histopathological improvements and reduction of neuroinflammation
Histological analysis of brain tissue was performed in male animals 8 months after AAV9-Sgsh systemic gene transfer. A marked decrease in LIMP-2 staining was observed in vector-treated animals, to levels undistinguishable from unaffected littermates (Fig. 3). Conversely, intense LIMP-2 staining was detectable in untreated MPSIIIA animals, indicating lysosomal compartment enlargement consequent to GAG accumulation.
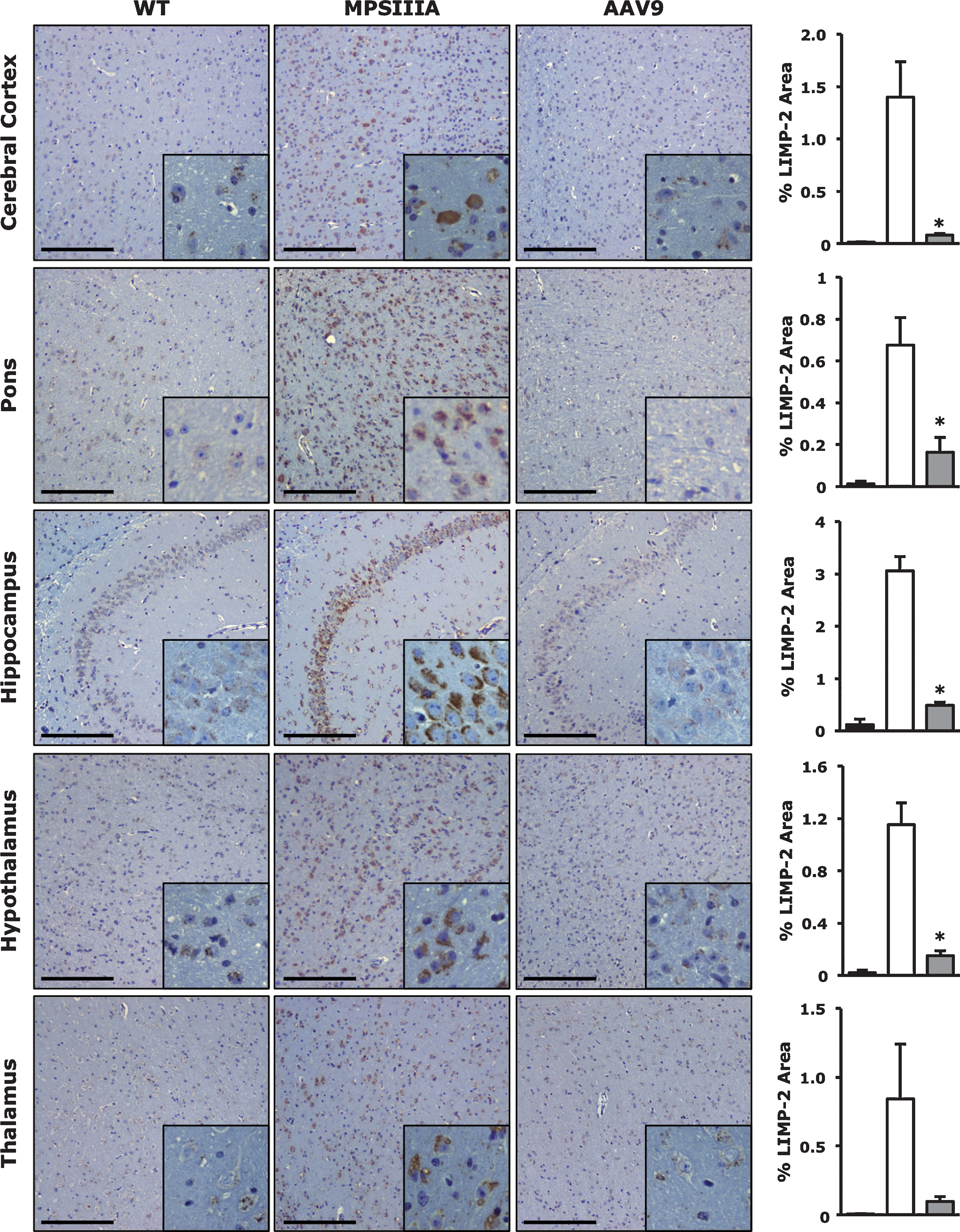
Correction of lysosomal distention in the CNS of MPSIIIA mice following systemic AAV9-Sgsh gene transfer. Tissues from unaffected littermates (WT), untreated MPSIIIA mice (MPSIIIA), or AAV9-Sgsh treated male mice (AAV9) were harvested 8 months after gene transfer and immunostaining for LIMP2 was performed in cerebral cortex, pons, cerebellum, hypothalamus, and thalamus. Histograms depict the corresponding signal quantification in all treatment groups. Results are shown as mean±SEM. n=3 to 4 per group. *, p<0.05 vs. untreated MPSIIIA. Scale bars, 200 μm.
Specific staining of brain tissue for astrogliosis and microgliosis was used to evaluate the neuroinflammatory response to neurodegeneration and the disease-induced deleterious effects on glial cells (Hemsley et al., 2008; Hemsley et al., 2009a). Staining for GFAP, a marker upregulated in activated glial cells, demonstrated complete reversal of astrogliosis in AAV9-Sgsh treated MPSIIIA mice compared to untreated affected animals (Fig. 4a). Similarly, immunostaining of activated microglia with BSI-B4 lectin revealed the complete absence of reactive microglia from all brain areas tested from treated animals compared with untreated controls (Fig. 4b).
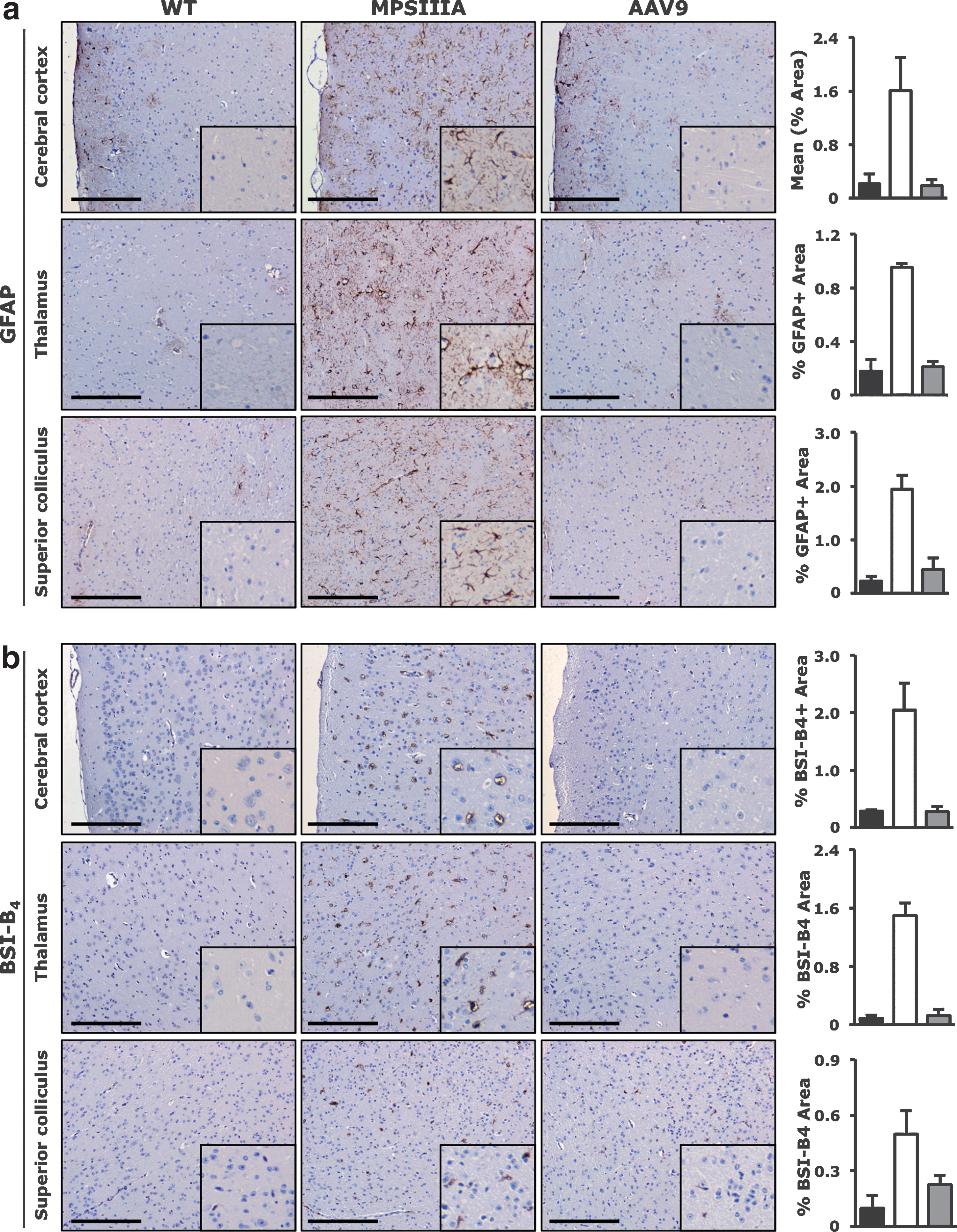
Correction of astrogliosis and microgliosis in the CNS of MPSIIIA mice following systemic AAV9-Sgsh gene transfer. Tissues from unaffected littermates (WT), untreated MPSIIIA mice (MPSIIIA), or AAV9-Sgsh treated male mice (AAV9) were harvested 8 months after gene transfer.
Intravenous AAV9-Sgsh vector administration results in the detection of supraphysiological levels of sulfamidase transgene product in liver and serum of male mice
Following AAV9-Sgsh gene transfer, MPSIIIA male mice displayed levels of sulfamidase transgene activity 6-fold and 5-fold higher in liver lysates and serum, respectively, compared with unaffected littermates (Fig. 5a and b). Female MPSIIIA animals injected at the same dose had sulfamidase transgene product levels in liver and serum equivalent to 100% of normal (Fig. 5a and b). Gender differences in liver transduction efficacy have previously been reported for AAV vectors (Davidoff et al., 2003).
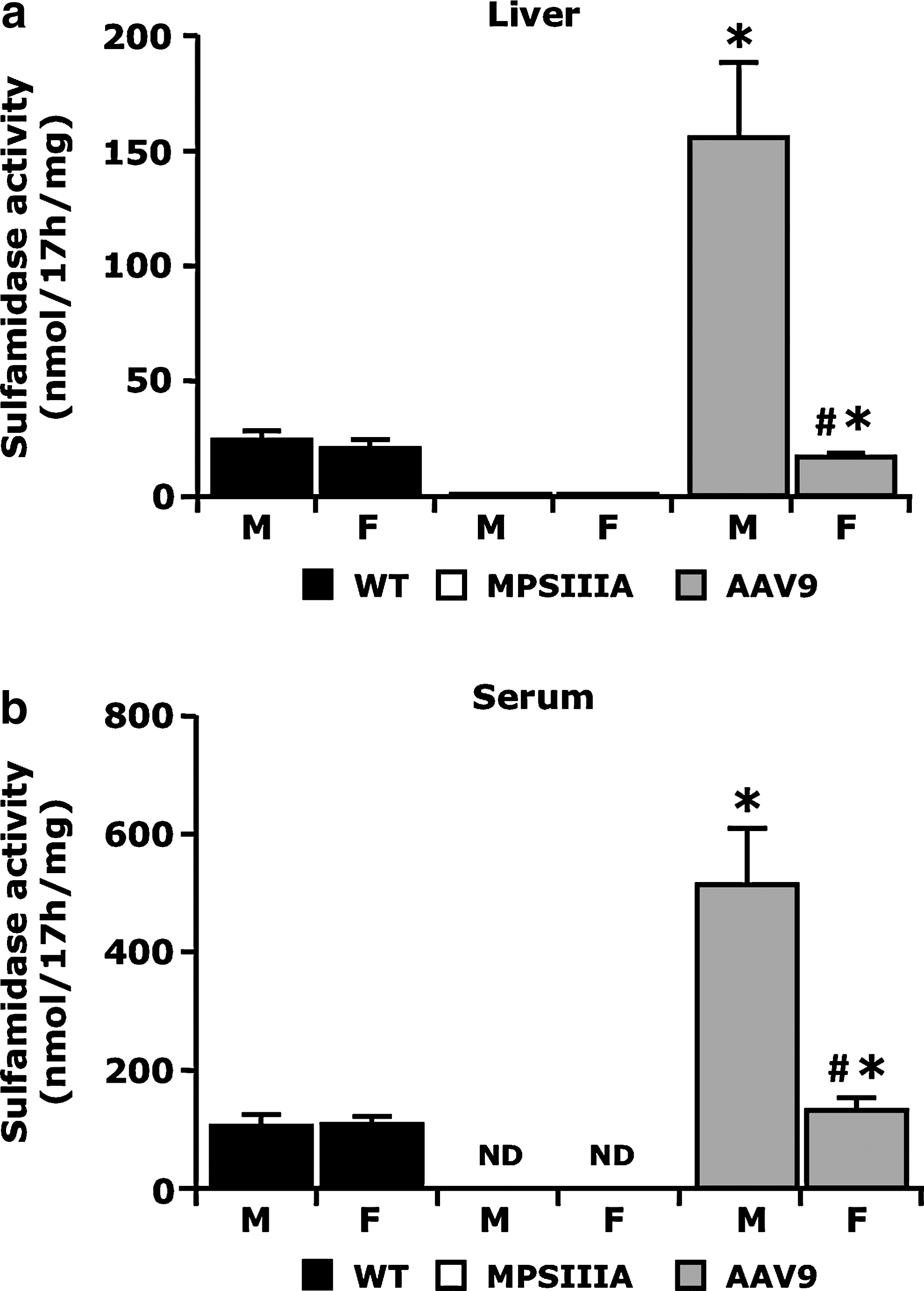
Sulfamidase activity in liver and serum following AAV9-Sgsh vector gene transfer.
The detection of sulfamidase transgene product at similar levels in liver lysate and serum in both males and females suggest the enzyme activity detected in the bloodstream following gene transfer is mainly deriving from the transduced liver as previously shown with AAV8 vectors (Ruzo et al., 2012).
AAV9-Sgsh systemic administration efficiently corrects GAG accumulation in peripheral tissues of MPSIIIA animals
Systemic administration of the AAV9-Sgsh vector resulted in correction of the pathological accumulation of GAG in all organs tested. Eight months after gene transfer, liver, heart, lung, spleen, and kidneys of AAV-treated MPSIIIA mice, healthy littermate controls, and untreated MPSIIIA controls were harvested and analyzed. Gene transfer completely corrected GAG accumulation in all tissues tested, with the exception of kidneys in female mice, which showed a marked amelioration of the phenotype but not complete normalization of GAG content in tissue lysates (Fig. 6a). The lower levels of serum sulfamidase transgene product found in treated females (Fig. 5b), and the fact that kidneys are known to be refractory to correction by enzyme replacement therapies (Garcia et al., 2007), may explain this finding.
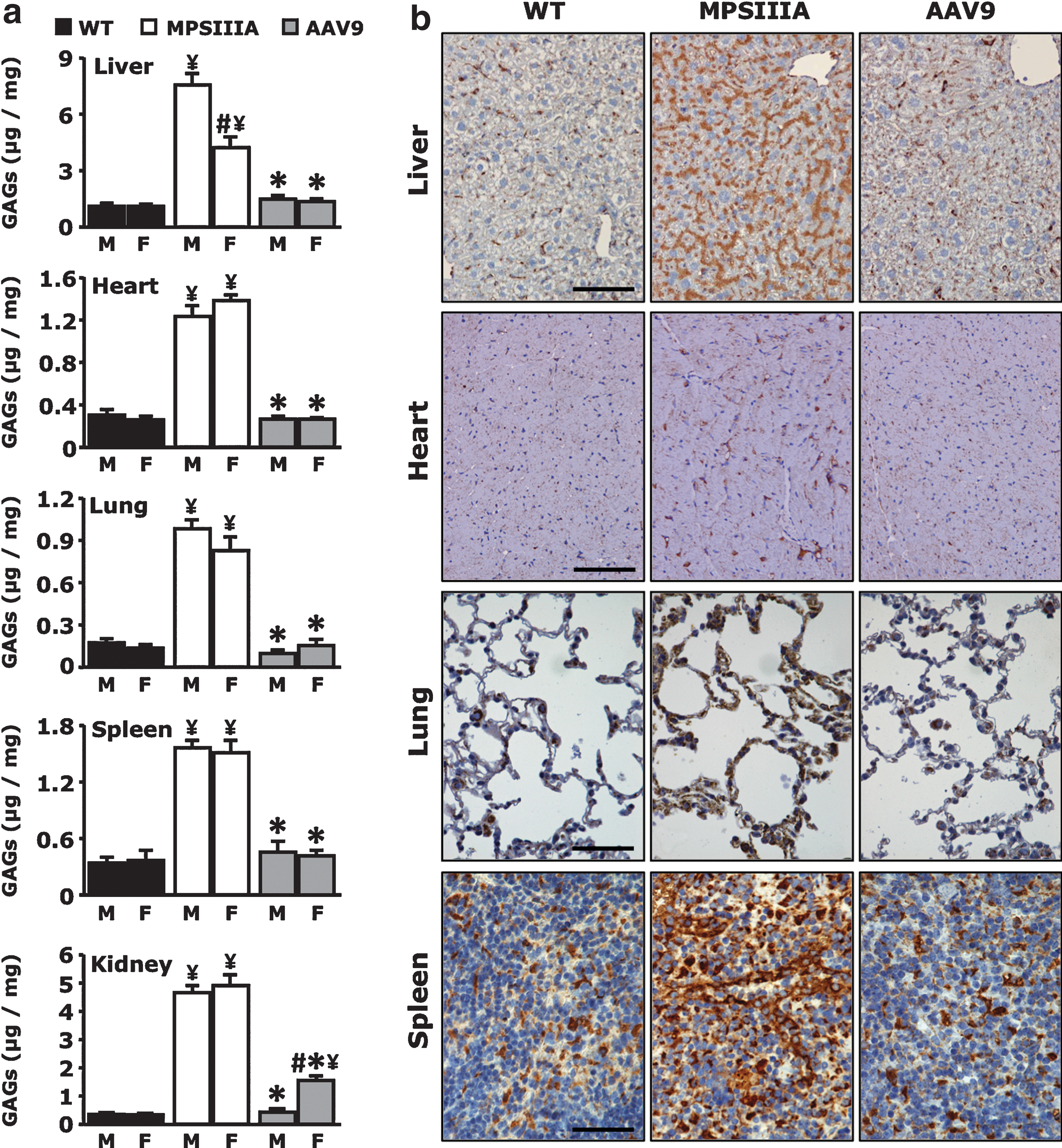
Correction of lysosomal pathology in peripheral tissues following AAV9-Sgsh vector administration.
Immunohistochemistry for the lysosome marker LAMP-1 in male mice confirmed tissue GAG measurement results. Intense staining of lysosomes was detectable in untreated MPSIIIA mice, while AAV9-Sgsh treated animals showed complete reversion of the storage phenotype to levels similar to unaffected littermates (Fig. 6b).
Widespread correction of GAG accumulation in CNS and in peripheral tissues results in prolonged lifespan of animals
Systemic delivery of AAV9-Sgsh resulted in a remarkable extension of the lifespan on MPSIIIA mice, both males and females (Fig. 7b and c). While all untreated MPSIIIA animals died by 17 months of age, all AAV-treated animals survived past that time point. The mean survival of male MPSIIIA mice was extended from 15.7±0.5 (n=12) to 22.3±1.2 (n=9) months following AAV9-Sgsh treatment (p=0.0003); similarly mean survival of female MPSIIIA animals was extended from 13.0±0.5 (n=12) to 21.1±1.3 (n=7) months (p=0.001). The improved survival of AAV treated mice constitutes additional evidence of the therapeutic potential of systemic AAV9 gene transfer for sulfamidase.
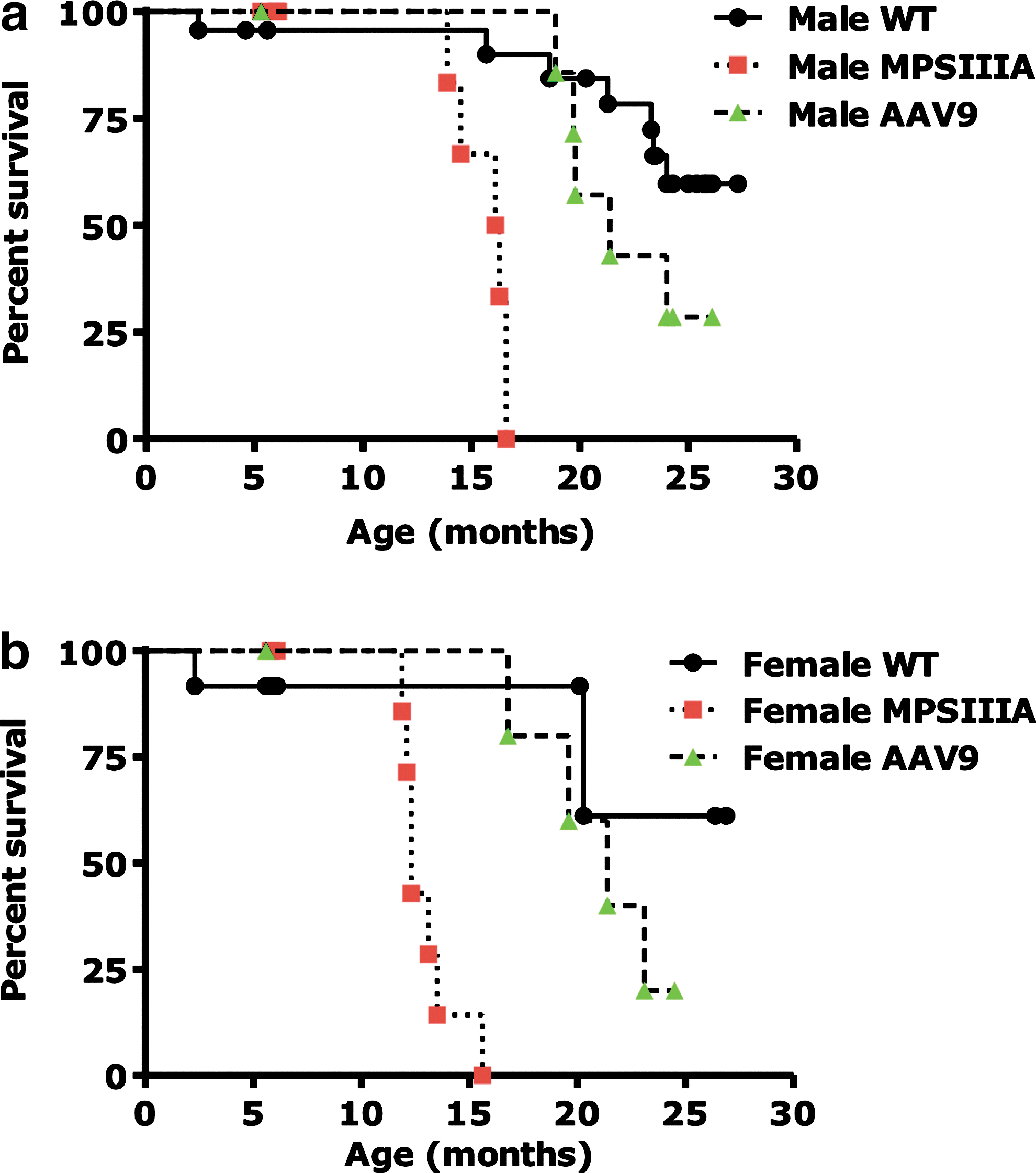
Prolonged survival of MPSIIIA animals following AAV9-Sgsh gene transfer.
Discussion
The ultimate goal of treatments for disorders affecting the CNS, as well as other peripheral organs, is to achieve efficient delivery of the therapeutic agent to the entire body. This could be obtained by delivering a therapeutic agent systemically through a peripheral vein, particularly for those diseases, such as MPSIIIA, amenable for enzymatic cross-correction (Enns et al., 2008). However, the BBB constitutes a major obstacle to the achievement of this goal, as protein therapeutics and the majority of gene transfer vectors fail to reach the brain when introduced into the bloodstream (Scherrmann, 2002). Possible solutions to this problem include the direct CNS administration of the missing enzyme (Shire Human Genetic Therapies, Inc., 2010) or the gene therapy vector (Worgall et al., 2008), the use of hematopoietic stem cells modified to produce the missing enzyme (Biffi et al., 2006), or the use of modified gene therapy vectors that target the brain endothelium, which then secretes the therapeutic protein across the BBB (Chen et al., 2009).
AAV9 is a recently isolated AAV serotype (Gao et al., 2004), which has been shown to cross the BBB of mice, cats, and macaques (Duque et al., 2009; Foust et al., 2009; Bevan et al., 2011; Gray et al., 2011), driving efficient brain gene transfer in mouse models of diseases (Fu et al., 2011). Here we tested the efficacy of systemic AAV9 gene transfer for sulfamidase in achieving whole-body correction of the pathological accumulation of GAG in MPSIIIA mice.
For the first time, we showed that the levels of CNS transduction attained with an AAV9 vector encoding for sulfamidase are sufficient to fully reverse the brain disease phenotype of MPSIIIA in both male and female animals. This included the reduction of GAG tissue accumulation throughout the brain, the reduction of lysosome size, and a remarkable reduction in neuroinflammation. Similarly, the natural high tropism of AAV9 for liver and heart (Inagaki et al., 2006) resulted in widespread correction of the disease in peripheral organs. Gender differences in efficiency of liver transduction by AAV vectors were evident in our study, as previously reported (Davidoff et al., 2003; Ruzo et al., 2012), resulting in considerably lower liver and serum sulfamidase activity in female MPSIIIA mice compared to male animals. However, albeit lower, the values achieved in MPSIIIA females were still considerably high, within the range of healthy littermates, which explains the normalization of GAG accumulation observed in most somatic tissues analyzed from animals of both genders.
We previously showed that it is possible to ameliorate the MPSIIIA CNS phenotype in affected mice by expressing sulfamidase in liver, thus achieving supraphysiological levels of enzyme activity in serum (Ruzo et al., 2012). This result was obtained using an AAV8 vector encoding for sulfamidase injected systemically, resulting in complete correction of pathological GAG accumulation in peripheral organs and a partial reduction of GAG content in the brain. However, the failure to completely correct GAG accumulation in the brain resulted in only partially improved survival of male animals and no improvement of survival of female animals, which expressed the sulfamidase transgene at lower levels than males. Thus, these results suggest that sex differences in levels of AAV liver transduction (Davidoff et al., 2003) prevent reaching therapeutic levels of transgene product activity in the female brain. Additionally, they highlight the importance of correcting the CNS alterations typical of MPSIIIA in prolonging animal survival. Here, the higher degree of phenotype reversal at the CNS level, achieved by crossing the BBB with the AAV9 vector and expressing the transgene throughout the brain, resulted in a remarkable prolonged survival of animals, both males and females.
Intracranial delivery of AAV5 vectors encoding for the sulfamidase transgene and the Sulfatase Modifying Factor 1 (Fraldi et al., 2007a) in MPSIIIA mice resulted in amelioration of the CNS disease phenotype with functional improvement in cognitive functions (Fraldi et al., 2007b). No peripheral organ disease correction was documented in this study. Similarly, systemic lentiviral gene transfer in MPSIIIA mouse models did not achieve the same levels of disease correction in CNS described here (McIntyre et al., 2008; McIntyre et al., 2010).
Finally, clinical translation of the results described here will take advantage of data generated in the context of intravenous delivery of AAV vectors, in which the safety and efficacy of the approach was tested in the context of gene transfer studies for hemophilia B (Manno et al., 2006; Nathwani et al., 2011). Potential drawbacks of the systemic administration of AAV vectors to target the CNS are related to the high doses of vector required to achieve therapeutic efficacy. First, the production of such amounts of GMP-quality vectors needed for human administration may represent a challenge. Second, systemic exposure to high vector doses would result in a more widespread biodistribution of vector genomes, which may pose safety concerns. Systemic exposure to high doses of AAV vectors may also trigger the activation of capsid-specific CD8+ T cell responses directed against the viral capsid in a dose-dependent manner (Manno et al., 2006; Mingozzi et al., 2007; Mingozzi et al., 2009; Nathwani et al., 2011), which may or may not affect the CNS (Lowenstein et al., 2007). Due to the lack of reliable experimental animal models to study the effect of activation of capsid T cells on AAV vector transduction (Li et al., 2007a; Li et al., 2007b; Wang et al., 2007), careful immunomonitoring for capsid-triggered T cell responses in future clinical trials will be essential to define the immunogenicity profile of systemic delivery of AAV9 vectors encoding for sulfamidase in humans (Mingozzi et al., 2011a). Finally, AAV vectors delivered systemically may be neutralized by anti-AAV NAb (Jiang et al., 2006; Manno et al., 2006; Scallan et al., 2006), which are highly prevalent in humans (Boutin et al., 2010).
In summary, we demonstrated that it is possible to obtain reversal of GAG accumulation in brain and peripheral organs of MPSIIIA mice with the intravenous administration of an AAV9 vector encoding for sulfamidase. Disease correction in both male and female animals resulted in a remarkable improvement of survival in both sexes. These results are important for the development of therapies for MPSIIIA and other diseases in which targeting of both CNS and peripheral organs is necessary.
Footnotes
Acknowledgments
We thank Xavier Leon and Marta Moya for technical assistance. A. Ruzo and A. Ribera were recipients of predoctoral fellowships from Ministerio de Ciencia e Innovación (Spain), S.M. from Generalitat de Catalunya, and P.V. from Fundación Ramón Areces. This work was supported by grants from MPS España Foundation, Ministerio Ciencia e Innovación, Plan Nacional I+D+I (SAF2008-00962), and from Laboratorios Esteve, S.A., Spain, and from European Community (CLINIGENE LSHB-CT-2006-018933).
Author Disclosure Statement
A. Ruzo, E. Ayuso, and F. Bosch are co-inventors on a patent application for the use of AAV vectors for the treatment of MPSIIIA.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.