
Abstract
Human papillomavirus (HPV) is involved in the development of anogenital tumors and also in the development of oropharyngeal head and neck carcinomas, where HPV-16, expressing the E6 and E7 oncoproteins, is the most frequent serotype. Although vaccines encoding L1 and L2 capsid HPV proteins are efficient for the prevention of HPV infection, they are inadequate for treating established tumors. Hence, development of innovative vaccine therapies targeting E6/E7 is important for controlling HPV-induced cancers. We have engineered a nononcogenic mutated E7-specific plasmo-retroVLP vaccine (pVLP-E7), consisting of plasmid DNA, that is able to form recombinant retrovirus-based virus-like particles (VLPs) that display E7 antigen into murine leukemia virus Gag proteins pseudotyped with vesicular stomatitis virus envelope glycoprotein (VSV-G). pVLP-E7 vaccinations were studied for their ability to generate specific immune responses and for induction of protective immunity against tumor cell challenge in preventive and therapeutic models. The produced VLPs induce the maturation of human dendritic cells in vitro and mount specific E7 T cell responses. Intradermic vaccinations of mice with pVLP-E7 show their efficacy to generate antigen-specific T cell responses, to prevent and protect animals from early TC-1 tumor development compared with standard DNA or VLP immunizations. The vaccine efficacy was also evaluated for advanced tumors in mice vaccinated at various time after the injection of TC-1 cells. Data show that pVLP-E7 vaccination can cure mice with already established tumors only when combined with Toll-like receptor-7 (TLR7) and TLR9 agonists. Our findings provide evidence that pVLPs, combining the advantages of DNA and VLP vaccines, appear to be a promising strategy for the treatment of HPV-induced cancers.
Introduction
Therefore, strategies to develop innovative vaccine therapies targeting E6/E7 should be further explored. One potential strategy to enhance the immunogenicity of vaccine antigens is to display E7 into/onto virus-like particles (VLPs). VLPs consist of viral structural proteins that assemble into particles structurally similar to infectious viruses. However, because they lack viral nucleic acid, VLPs are absolutely noninfectious. Thus, they represent a safer alternative to attenuated viruses or viral vectors (Jennings and Bachmann, 2008; Buonaguro et al., 2011). On the basis of their particulate nature, VLPs provide an inherent advantage over soluble antigens (Link et al., 2012), which have been shown to fail in several vaccine approaches owing to weak immunogenicity or instability (Bachmann and Jennings, 2010). Indeed, particulate antigens can be taken up by antigen-presenting cells (APCs) and processed by the class II presentation pathway (Xiang et al., 2008), but also into the alternative class I presentation pathway (cross-presentation), in contrast to soluble antigen (Agnandji et al., 2011). Altogether, VLPs are commonly more immunogenic than recombinant protein immunogens, and are able to stimulate both the humoral and cellular arms of the immune system (Jennings and Bachmann, 2008). Depending on their nature, these stable and versatile pseudo-particles may possess excellent adjuvant properties capable of inducing innate and adaptive immune responses. As a consequence, VLPs can also be exploited as vaccine platforms for antigen presentation (Chackerian, 2007). The genetic insertion of target sequences into viral structural proteins to generate chimeric particles has been the most commonly used method for displaying heterologous epitopes on VLPs. Many different VLP types have been adapted for this purpose (Jennings and Bachmann, 2008), and there have been notable successes in developing vaccines that protect animal models from infection by malaria (Agnandji et al., 2011) and influenza A virus (Galarza et al., 2005). We developed a vaccine platform specifically aimed at inducing such responses against multiple antigens displayed by recombinant retrovirus-based VLPs based on Gag of Moloney murine leukemia virus (MuLV) (Bellier et al., 2006; Dalba et al., 2007) and demonstrated their efficacy for vaccination against hepatitis C (Garrone et al., 2011). Recombinant retrovirus-based VLPs can be produced either ex vivo after cell transfection with plasmid DNA encoding wild-type or chimeric Gag proteins and envelope glycoproteins, or in vivo after injection of the same plasmid DNA, named plasmo-retroVLPs (pVLPs). We previously demonstrated that pVLPs induce higher cellular (Bellier et al., 2006) and humoral (Bellier et al., 2009) immune responses against vaccine antigens in comparison with standard DNA vaccines that do not form VLPs. Furthermore, this strategy combines the straightforwardness, the stability, the large-scale and low-cost production of DNA vaccines, and the immunostimulatory properties of VLP vaccines.
In this study, we engineered a pVLP DNA vaccine that produces VLPs harboring a nononcogenic mutated E7 antigen into MuLV Gag proteins and that are pseudotyped with vesicular stomatitis virus glycoprotein (VSV-G). The latter is known to improve the uptake of VLPs by APCs and also to favor immune T cell responses (Temchura et al., 2008). Therefore, we designed a sequence encoding chimeric Gag–E7 fusion proteins that can self-assemble into E7-recombinant retrovirus-based VLPs (VLP-E7) and be released from transfected cells. We studied the ability of pVLPs to generate specific immune responses as well as in vivo antitumor effects, using TC-1 epithelial cells that overexpress HPV-16 E6/E7 oncoproteins.
Materials and Methods
Plasmid constructs
The Gag coding sequence for all constructs was derived from MuLV. pBL36 is a plasmid that encodes Gag under the control of the human cytomegalovirus (CMV) promoter. pBL211, a plasmid carrying a Gag–GFP fusion gene under the control of the human CMV promoter, was obtained from the EPX145-68 plasmid (Garrone et al., 2011) by insertion of a PCR fragment with an MluI restriction site between the NruI and NheI sites. A PCR-synthesized DNA fragment encoding green fluorescent protein (GFP) with an MluI site introduced at the 5′ end was then inserted at the MluI site to produce the CMV-Gag-GFP plasmid. pBL36ΔE7, a plasmid carrying a gag-ΔE7 cassette under the control of the human CMV promoter, was obtained from pBL36 and from the pET-15bE7 plasmid that encodes a nononcogenic deleted 21–26 HPV-16 E7 protein inactivated at the retinoblastoma (Rb)-binding motif (ΔE7). The ΔE7 gene was amplified by PCR with primers containing MluI and XbaI restriction sites. The PCR product was purified, sequentially digested with MluI and XbaI, and inserted by ligation into pBL36, giving rise to pBL36ΔE7. pBL36, pBL211, and pBL36ΔE7, presented in Fig. 1A, are referred to as pGag, pGag-GFP, and pGag-E7, respectively. pG2AGag-GFP, encoding a G2A-mutated form of Gag-GFP, was previously described (Bellier et al., 2009). pcDNA3-Δ7, referred to as pE7, is a plasmid in which the ΔE7 gene, amplified by PCR from the pET-15bE7 plasmid with primers containing BamHI and XbaI restriction sites, was inserted by ligation into the pcDNA3 plasmid. The pMΔ2G plasmid, referred to as pVSV-G and kindly provided by D. Trono (Swiss Federal Institute of Technology, Lausanne, Switzerland), encodes the vesicular stomatitis virus G (VSV-G) envelope protein.

Validation of plasmo-retroVLP (pVLP) DNA constructs.
Cell lines and primary human cells
293T cells (CRL-1573; American Type Culture Collection [ATCC], Manassas, VA) were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 2 mM
Cell transfection, in vitro production, and characterization of virus-like particles
293T cells, seeded in 175-cm2 culture flasks (15×106 cells per plate), were cotransfected 24 hr later according to a calcium phosphate transfection protocol with 50 μg of plasmid DNA including either pGag, pGag-GFP, or pGag-E7 with or without pVSV-G at a 3:1 ratio. Sixteen to 18 hr later, medium was replaced with FCS–free DMEM, and then supernatants were harvested 48 hr later, filtered through 0.45-μm (pore size) membranes, concentrated (Centricon; Millipore, Molsheim, France), and purified by ultracentrifugation through a 2-ml 20% sucrose cushion (Sigma-Aldrich, Saint-Quentin-Fallavier, France) in an SW41 Beckman rotor (107.170×g, 2 hr at 4°C). Pellets were resuspended in phosphate-buffered saline (PBS). The production of VLPs was quantified by protein measurement according to the Bradford method (Quick Start Bradford dye reagent; Bio-Rad Laboratories, Marnes la Coquette, France). Samples corresponding to 1 μg of protein were analyzed by Western blot as described elsewhere (Bellier et al., 2006). VLPs were detected with rat anti-mouse mAbs (clone R187, CRL-1912 cells; ATCC) that recognize the MuLV p30 Gag capsid protein, and polyclonal rabbit anti-rat immunoglobulins conjugated with horseradish peroxidase (HRP; Dako, Trappes, France). Signals were detected with an ECL Plus Western blotting detection system (GE Healthcare, Saclay, France). For electron microscopy (EM) analysis, purified particles were first diluted 1:5 in cacodylate buffer. Two microliters of the particle solution was placed on formvar-coated grids, fixed in osmium vapor for 5 min, and then incubated with uranyl acetate (2% in water) for 8 min. The samples were washed three times in water, dried, and observed at magnifications of ×20,000–×60,000 by transmission electron microscopy (CM120 TEM; Philips).
In vitro capture of VLPs by iDCs
iDCs (106 cells/ml) were seeded in flat-bottomed 12-well tissue culture plates in the presence of VLPs (3 μg/ml) produced by 293T cells transfected with pGag-GFP plus pVSV-G. Medium alone was used as negative control. The capture of VLPs by DCs was performed according to a modified protocol described elsewhere (Dupuy et al., 2005). Briefly, cells were centrifuged (1000×g) at 30°C for 3 hr and then incubated at 37°C overnight until use. Cell phenotypes were analyzed by flow cytometry, using the following mAbs: allophycocyanin (APC)-conjugated anti-CD11c (clone B-ly6), phycoerythrin–cyanine 5 (PE-Cy5)-conjugated anti-CD83 (clone HB15e), biotin-conjugated anti-CD80 (clone L307.4) detected with peridinin chlorophyll protein (PerCP)–cyanine 5.5–streptavidin, all purchased from BD Biosciences (Pont-de-Claix, France); and phycoerythrin (PE)-conjugated anti-CD40 (clone MAB89) and phycoerythrin–cyanine 7 (PE-Cy7)-conjugated anti-HLA-DR (clone Immu-357) from Beckman Coulter (Villepinte, France). Data were acquired with an LSR II flow cytometer (BD Biosciences) and flow cytometry analyses were performed with FloJo software (Tree Star, San Carlos, CA). For microscopy observation, 5×105 cells were plated on a poly-
In Transwell experiments, day 5 iDCs were plated in the lower chamber at 5×105 cells per well in 500 μl of RPMI supplemented with 10% FCS, IL-4, and GM-CSF, and cocultured with 106 transfected 293T cells per well seeded in 200 μl of DMEM–10% FCS in the upper chamber of 24-well Transwell plates (Costar Transwell permeable support, product 3413; Corning, Lowell, MA). On day 7, DCs were harvested for further analysis.
In vitro priming of human T cells with DCs infected with VLP-E7
VLP-E7 (3 μg/ml)-loaded iDCs (105) from HLA-A2 healthy donors were cocultured with 106 autologous T cells in 200 μl of RPMI medium supplemented with 10% hABS in a flat-bottomed 96-well tissue culture plate (Corning). On day 3, primed T cells were transferred in fresh medium supplemented with recombinant human (rh)IL-15 (10 ng/ml; Miltenyi Biotec) and rhIL-7 (10 ng/ml; Miltenyi Biotec) into flat-bottomed 48-well tissue culture plates. On day 10, cells were counted and restimulated at a 10:1 ratio with VLP-E7-loaded iDCs for an additional 10 days. DCs loaded with wild-type VLPs were used as negative control. After two rounds of stimulation, T cells were harvested, counted, and assayed for their antigen specificity by interferon (IFN)-γ enzyme-linked immunospot (ELISPOT) assay after restimulation for 24 hr with DCs loaded with E7 peptide (see below).
Mice
Female C57BL/6J (H-2b ) mice were purchased from Elevage Janvier (Le Genest-Saint-Isle, France) and were 7 weeks old when experiments were initiated. Mice were maintained under specific pathogen-free conditions, and manipulations were performed according to European Economic Community guidelines and approved by the local ethics committee.
Immunizations and tumor challenges
Mice were anesthetized by intraperitoneal injection of xylazine (Rompun 2%, 10 mg/kg; Bayer Pharma, Puteaux, France) and ketamine (Imalgene 1000, 150 mg/kg; Merial, Lyon, France). Before DNA immunization and injection of tumor cells, mice were shaved on the lower back.
Immunization
Mice (five or six animals per group) were intradermally vaccinated three times at 2-day or 2-week intervals with 10 μg of plasmid DNA (7.5 μg of pGag or pGag-E7 plus 2.5 μg of pVSV-G, or 10 μg of pE7 alone) in 40 μl of 0.9% NaCl to analyze primary and protective memory immune responses, respectively. The skin was immediately electroporated with tweezer-type electrodes (CUY650 P3 electrodes; Sonidel, Dublin, Ireland), using a BTX ECM830 generator (Harvard Apparatus, Les Ulis, France). Eight pulses at 60 V were given at a duration of 20 msec with a 200-msec interval. Alternatively, mice were subcutaneously injected three times with 25 μg of VSV-G-pseudotyped E7-recombinant VLPs (VLP-E7). To evaluate the induced immune responses, mice were killed 10 days after the last vaccination. Splenocytes were collected and the T cell immune response was monitored by IFN-γ ELISPOT assay (Mabtech, Sophia-Antipolis, France). For in vivo tumor protection, mice were vaccinated three times at 2-week intervals with 10 μg of plasmid DNA or 25 μg of VLP-E7, using the same procedure of vaccination described previously. Two weeks after the last immunization, mice were anesthetized and challenged, per mouse, with 5×104 TC-1 cells suspended in 0.15 ml of PBS and injected subcutaneously into the flank. For in vivo tumor treatment, mice were first subcutaneously injected with 5×104 TC-1 cells and then immunized 3, 7, or 11 days later as described previously by three injections at 2-day intervals. In some groups, vaccinations were combined with the use of adjuvants added locally to the tumors: imiquimod (Aldara 5%, 5 mg/mouse; Meda Pharmaceuticals, Somerset, NJ) was used as a topical treatment and CpG oligodeoxynucleotides (ODN 1668; Coley Pharmaceutical, Wellesley, MA) were directly injected (50 μg of CpG in 50 μl of 0.9% NaCl) into the periphery of tumors.
Monitoring of tumor growth
Mice were monitored every 2 days for tumor progression. Tumor growth was determined by direct palpation and tumor volumes were estimated by measuring the largest diameter (L) and the smallest diameter (l). Tumor volumes were calculated according to the formula: 0.5×(L×l 2). When tumors reached volumes of 1500 mm3, mice were killed. Survival curves were determined by Kaplan–Meyer analysis.
Systemic and local effects of adjuvants
Imiquimod and CpG adjuvants were locally administered in unvaccinated mice that had received 5×104 TC-1 cells, 11 days previously. Two days later, mice were killed and cell suspensions from spleen, draining lymph nodes, and tumors digested with collagenase (Liberase, 0.75 mg/ml; Roche Diagnostics, Meylan, France) and DNase (Pulmozyme, 30 IU/ml; Roche Laboratories) were analyzed by flow cytometry with the following mAbs: PE-conjugated anti-CD3, BD Horizon V500-conjugated anti-CD4, Alexa Fluor 700-conjugated anti-CD8, fluorescein isothiocyanate (FITC)-conjugated anti-B220, FITC-conjugated anti-CD11c, BD Horizon PE-CF594-conjugated anti-CD45, and BD Horizon V500-conjugated anti-MHC class II (IAe) (all from BD Biosciences); Alexa Fluor 700-conjugated anti-CD11b, eFluor (eF) 450-conjugated anti-F4/80, APC–eF780-conjugated anti-GR-1, PE–Cy7-conjugated anti-CD25, PerCP-eF710-conjugated anti-NKp46, and Pacific Blue-conjugated anti-FoxP3, all from eBioscience (Paris, France). Biotin-conjugated anti-CD45 (BD Biosciences) was detected with APC–eF780-conjugated streptavidin (eBioscience).
ELISPOT assay
Specific IFN-γ production by human T cells primed with VLP-E7-loaded DCs for 20 days was determined in a standard human ELISPOT assay (Mabtech). Briefly, 5×105 primed T cells were restimulated for 24 hr at 37°C in 5% CO2 with 5×104 DCs loaded for 4 hr with an equal mixture (50 μg/ml) of HLA-A2-restricted immunodominant E7 peptides (11–19 and 86–93; tebu-bio, Le Perray-en-Yvelines, France). Medium alone and concanavalin A (ConA; Sigma-Aldrich) at 2 μg/ml were used as negative and positive controls, respectively. E7-specific IFN-γ production by splenocytes of immunized mice was also determined in a standard murine ELISPOT assay. Briefly, splenocytes (5×105 cells per well) were stimulated for 48 hr with a 5-μg/ml concentration of H-2b-restricted immunodominant HPV-16 E7 peptides (49–57; AnaSpec, Fremont, CA). Spots were counted with an AID ELISPOT reader (ELR03; AID Autoimmun Diagnostika, Strassberg, Germany). Results, expressed as spot-forming units (SFU) per 106 cells, represent the mean±SEM of triplicates. Background levels were ≤12 SFU/106 T cells and ≤46 SFU/106 splenocytes for human and murine experiments, respectively.
Results
pVLP-transfected cells produce recombinant retrovirus-based VLPs
We designed the constructs pGag, pGag-GFP, and pGag-E7 to induce the formation of control wild-type, fluorescent, and E7-recombinant VLPs, respectively (Fig. 1A). For validation, the various constructs were transfected into 293T cells and cell supernatants were analyzed after viral particle purification by Western blot, using anti-Gag mAbs. Wild-type Gag, Gag–GFP, and Gag–E7 fusion proteins of the expected sizes were detected in purified supernatants (Fig. 1B), revealing VLP formation. The shape and size of the recombinant VLPs formed from 293T cells after their transfection with the various pVLPs (pGag, pGag-GFP, or pGag-E7 with or without pVSV-G) were studied by EM. Thus, Gag–E7-based VLPs were similar to native MuLV particles with diameters ranging from 70 to 90 nm (Fig. 1C). We then examined whether purified VLPs can be taken up by immature human monocyte-derived dendritic cells (iDCs). For this purpose, fluorescent VLPs were purified and concentrated from the supernatant of 293T cells cotransfected with pGag-GFP and pVSV-G, and incubated with iDCs for 24 hr. Figure 1D shows a representative image obtained by fluorescence microscopy of DCs that have captured fluorescent Gag–GFP VLPs. To mimic the in vivo observation and to visualize the VLP formation after pVLP administration, we developed an in vitro model using Transwell plates seeded with Gag–GFP-transfected 293T cells and iDCs in the upper and bottom chambers, respectively. In addition, we investigated the influence of adding VSV-G envelope protein to VLPs on the uptake efficacy by iDCs. Thus, 293T cells were transfected with pGag-GFP in the presence or absence of pVSV-G and, 24 hr later, DCs were analyzed by fluorescence-activated cell sorting (FACS) in order to quantify the percentage of GFP-positive DCs. Alternatively, 293T cells were transfected with pGag-GFP alone or with pG2AGag-GFP plus pVSV-G as a negative control because the G2A mutation in Gag prevents the assemblage of capsid proteins and thus the release of VLPs. As shown in Fig. 2A, more than 15% of DCs were GFP positive when cocultured with 293T cells transfected with pVLPs composed of Gag–GFP- and VSV-G-encoding sequences. In contrast, low levels of GFP were detected in DCs cocultured with 293T cells transfected with either pGag-GFP alone or pG2AGag-GFP plus pVSV-G. Because VSV-G expression does not change the amount of VLPs produced (data not shown), these data reveal that VSV-G pseudotyping significantly favors VLP uptake by iDCs. Altogether, we demonstrate that administration of pVLPs induces the production of VLPs that can be subsequently taken up by human DCs.
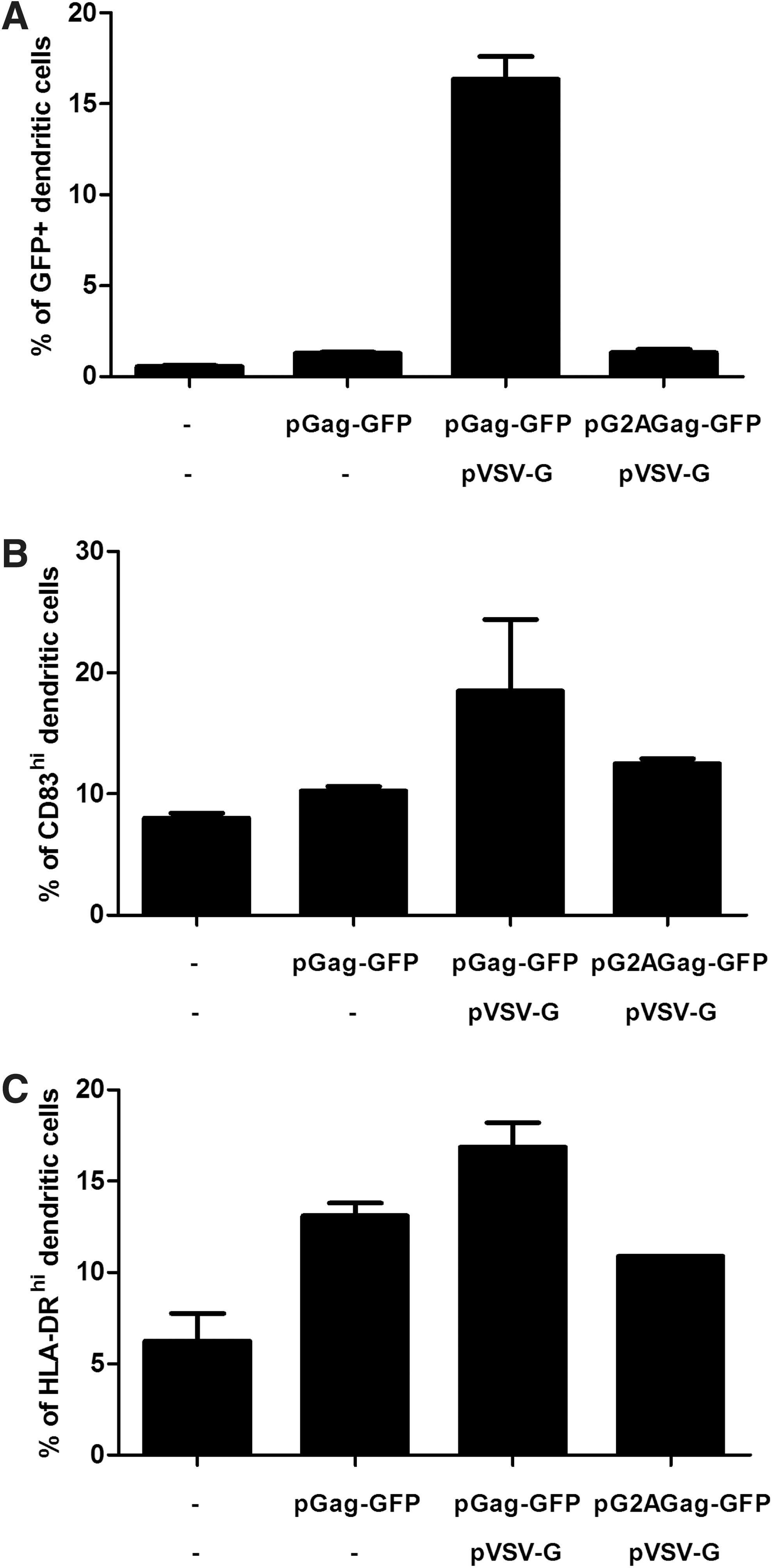
VLP release from pVLP-transfected cells and their uptake by iDCs. 293T cells untransfected or transfected with pGag-GFP in the presence or absence of vesicular stomatitis virus envelope glycoprotein (VSV-G)-encoding plasmids were seeded on the upper chamber of Transwell plates and iDCs were added in the bottom chamber. Alternatively, a sequence of Gag harboring the G2A mutation that prevents VLP release was used as control (pG2AGag-GFP). After 24 hr, percentages of fluorescent DCs were measured by flow cytometry
Retrovirus-based VLPs induce the maturation of human monocyte-derived dendritic cells
Because released VLPs did bind to DCs, we investigated whether the binding of VLPs to iDCs could induce cell activation. We analyzed the global expression of maturation markers such as CD83 and HLA-DR by iDCs cocultured in Transwell plates with 293T cells that were transfected as previously described. Figure 2B and C shows that VSV-G-pseudotyped VLPs produced by pGag-GFP plus pVSV-G-transfected cells increased the expression of activation markers. In contrast, lower maturation states of DCs were observed when 293T cells were transfected with pGag-GFP alone or with pG2AGag-GFP plus pVSV-G. Whether such an effect was due only to simple contact between VLPs and DCs or more specifically to their capture by DCs was further addressed. Thus, iDCs were incubated with a 3-μg/ml concentration of purified fluorescent VLPs or RPMI medium alone as a negative control. After 24 hr, 3% of DCs appeared to be GFP+ (data not shown) and expression levels of CD40, CD80, CD83, and HLA-DR markers were analyzed by FACS analysis gated on GFP status (positive or negative) and compared with the negative control. Figure 3A reveals that CD40, CD80, CD83, and HLA-DR markers were considerably upregulated in GFP+ DCs, but not in GFP– DCs as compared with the negative control. Mean fluorescence intensities from GFP+ cells demonstrated 44-, 22-, 6-, and 23-fold increases in CD40, CD83, CD80, and HLA-DR expression levels, respectively. When analyses were performed on the total DC population (GFP+ and GFP– cells), we still detected VLP-induced activation (data not shown). In addition, we observed that boiled VLPs lost the ability to induce DC activation (data not shown). Altogether, the results indicate that DC activation is exclusively linked to VLP uptake favoring their maturation, and may subsequently enhance their ability to better present antigens to T cells.
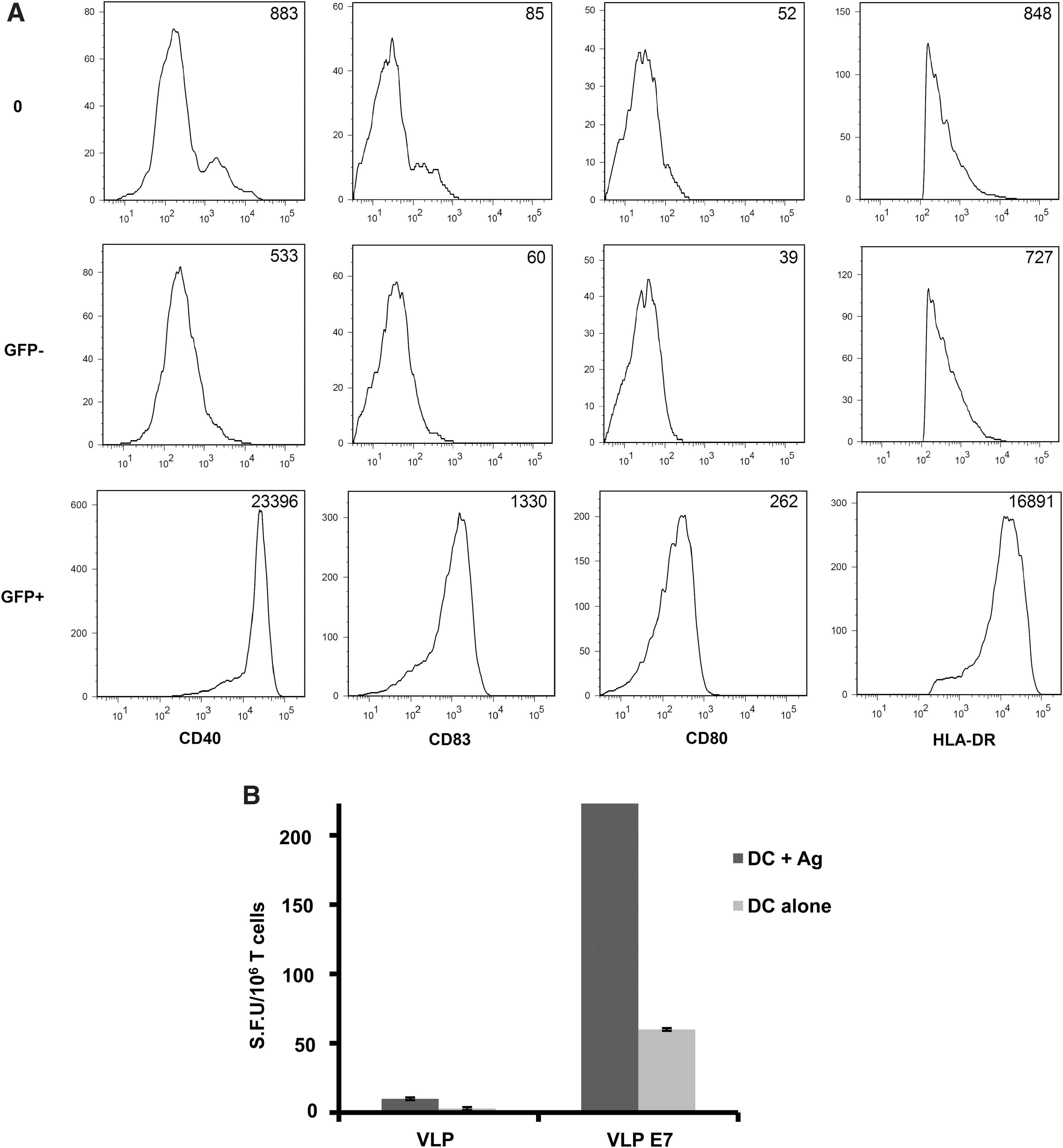
In vitro effects of VLPs on maturation of DCs and on T cell priming. iDCs were incubated
DCs loaded with VLP-E7 can prime T cells to mount in vitro an anti-E7 immune response
Whether DCs that take up VLP-E7 can elicit an antigen-specific immune response was further addressed. For this purpose, iDCs (from HLA-A2 donors) incubated with VLP-E7 were cocultured with autologous T cells as described in Materials and Methods. After two rounds of stimulation, primed T cells were restimulated with HLA-A2 DCs loaded with HLA-A2-restricted E7 peptides, and the anti-E7-specific T cell response was assessed by IFN-γ ELISPOT. Primed T cell restimulation with unloaded DCs and T cells primed with control VLPs were used as negative controls. As shown in Fig. 3B, a specific anti-E7 MHC class I response could be observed when T cells were primed with DCs incubated with VLP-E7. These data indicate that VLP-E7 is efficiently captured by human DCs, which then induce in vitro a specific E7 response.
Preventive vaccinations with pVLP-E7 fully protect mice from HPV-induced tumors
To evaluate whether vaccination with pVLP DNA vaccine can induce a specific immune response and can also protect mice from tumor challenge, mice were vaccinated three times with pVLP-E7 and compared with mice undergoing VLP vaccination with VLP-E7 or DNA vaccination with pE7, which does not form VLPs. On the basis of the results from Fig. 2, the VSV-G glycoprotein was systematically incorporated in pVLP or VLP vaccines for immunization. Ten days after the last vaccination, the cellular immune response was measured by IFN-γ ELISPOT assay. Results showed that pE7 and VLP-E7 vaccines induced a slight, albeit not significant, IFN-γ response (Fig. 4A). In contrast, pVLP-E7 yielded a significantly (p<0.01) stronger specific T cell response than the other conditions. Although one vaccination with pVLP-E7 was sufficient to generate a specific E7 response (data not shown), it turned out that three vaccinations resulted in a better immune response. In a second set of experiments, mice were vaccinated three times at 2-week intervals and then challenged with TC-1 cells. Data showed that mice vaccinated with pVLP-E7 were fully protected from TC-1 tumor growth (Fig. 4B and C). Although TC-1 tumor growth was slightly delayed in mice vaccinated with pE7 or VLP-E7, tumor volumes were not significantly diminished (Fig. 4B), and all mice died or were euthanized at times similar to those at which nonvaccinated mice died (Fig. 4C).
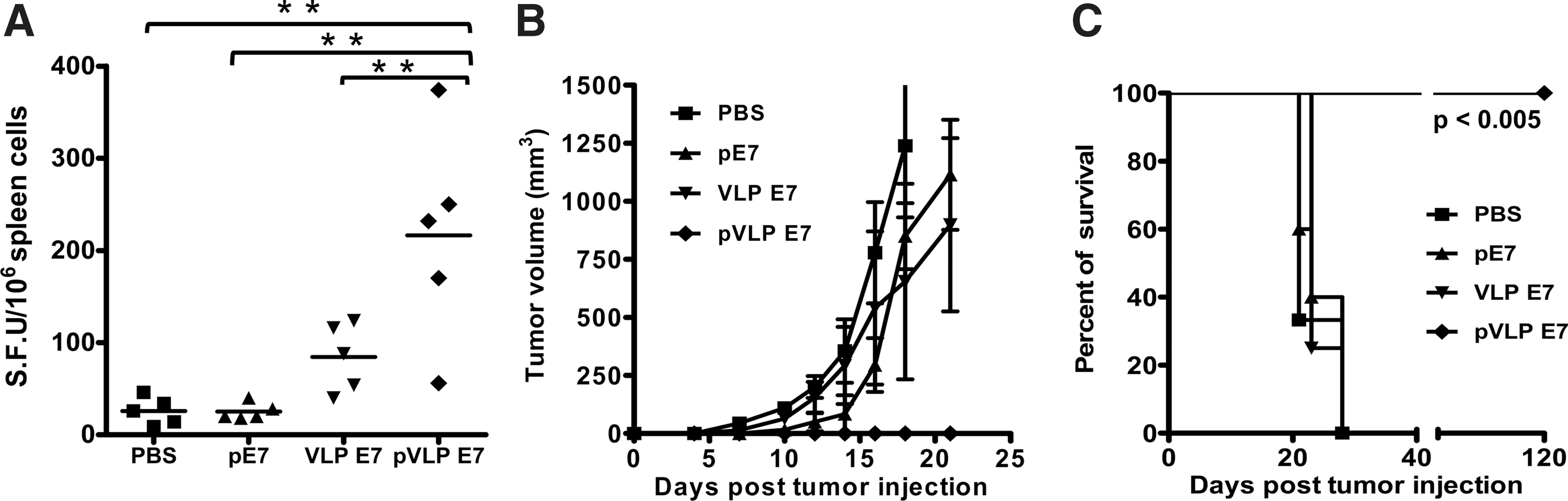
Advantage of pVLP-E7 vaccinations compared with pE7 DNA and VLP-E7 vaccines for antigen-specific T cell response induction and tumor protection. C57BL/6 mice (five animals per group) were immunized three times with 10 μg of pE7 or pVLP-E7, injected intradermally in association with electroporation. As controls, mice were immunized subcutaneously with 25 μg of VLP-E7 or PBS.
Efficacy of therapeutic vaccinations in mice bearing TC-1 tumors depends on tumor burden
It is well known that patients with advanced solid tumors are less responsive to treatment than patients with small tumors. Whether pVLPs can cure mice bearing tumors at various stages was further addressed. For this purpose, mice were infused with TC-1 cells and then underwent pVLP-E7 vaccination 3, 7, or 11 days after the administration of tumor cells. When treated as soon as 3 days after tumor injection, tumor regression was observed in most cases and more than 80% of the mice survived without any detectable tumors for at least 120 days (Fig. 5A and B). When mice were treated 7 and 11 days after tumor injection, a decrease in tumor growth was observed compared with untreated mice, resulting in significantly (p<0.01) prolonged survival (Fig. 5A and B). However, no tumor regression was observed and all mice died or were killed (Fig. 5B). These findings indicate that the therapeutic antitumor effect of pVLP-E7 depends on the tumor burden.
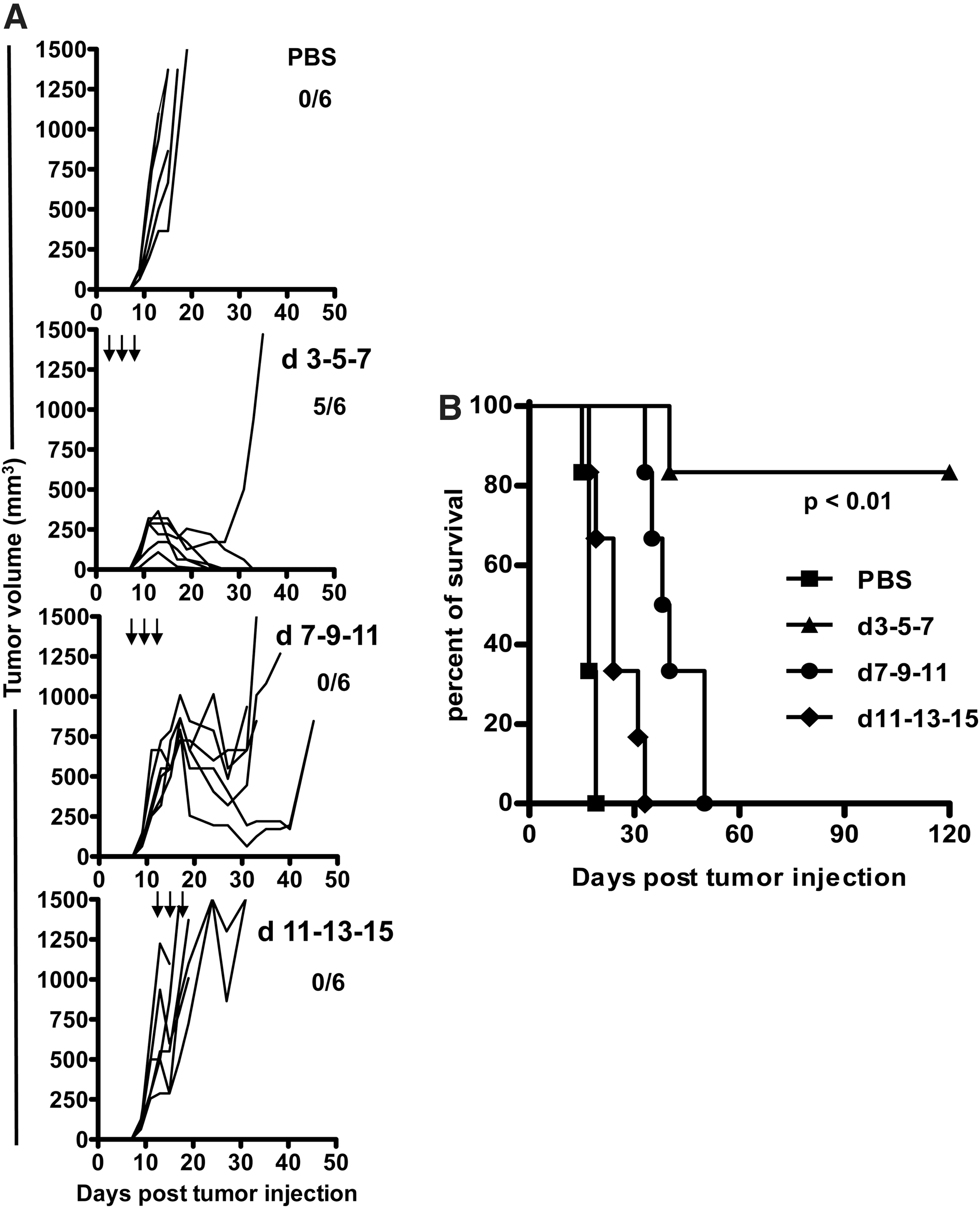
Efficiency of therapeutic vaccinations with pVLP-E7 at various times after E7-positive tumor inoculation.
Adjuvants improve the efficacy of pVLP-E7 therapeutic vaccinations in TC-1 advanced tumors
Immunomodulatory agents such as adjuvants are widely used to potentiate vaccine-induced responses by acting on sensors of innate immunity such as Toll-like receptors (TLRs). Because we observed that pVLP-E7 was not efficient enough when used as therapeutic vaccine for TC-1 tumors established on day 11, we evaluated whether a combination of adjuvants such as imiquimod and CpG oligodeoxynucleotides (ODNs), which act on TLR7 and TLR9, respectively, may enhance the antitumor response. The use of such adjuvants was justified by the fact that, when imiquimod was applied topically and CpG ODN was injected into the tumor periphery, they were able by themselves to recruit both in the spleen and locally (draining lymph nodes and tumor environment) innate cells such as granulocytes, macrophages, and dendritic cells (Supplementary Fig. S1; supplementary data are available online at
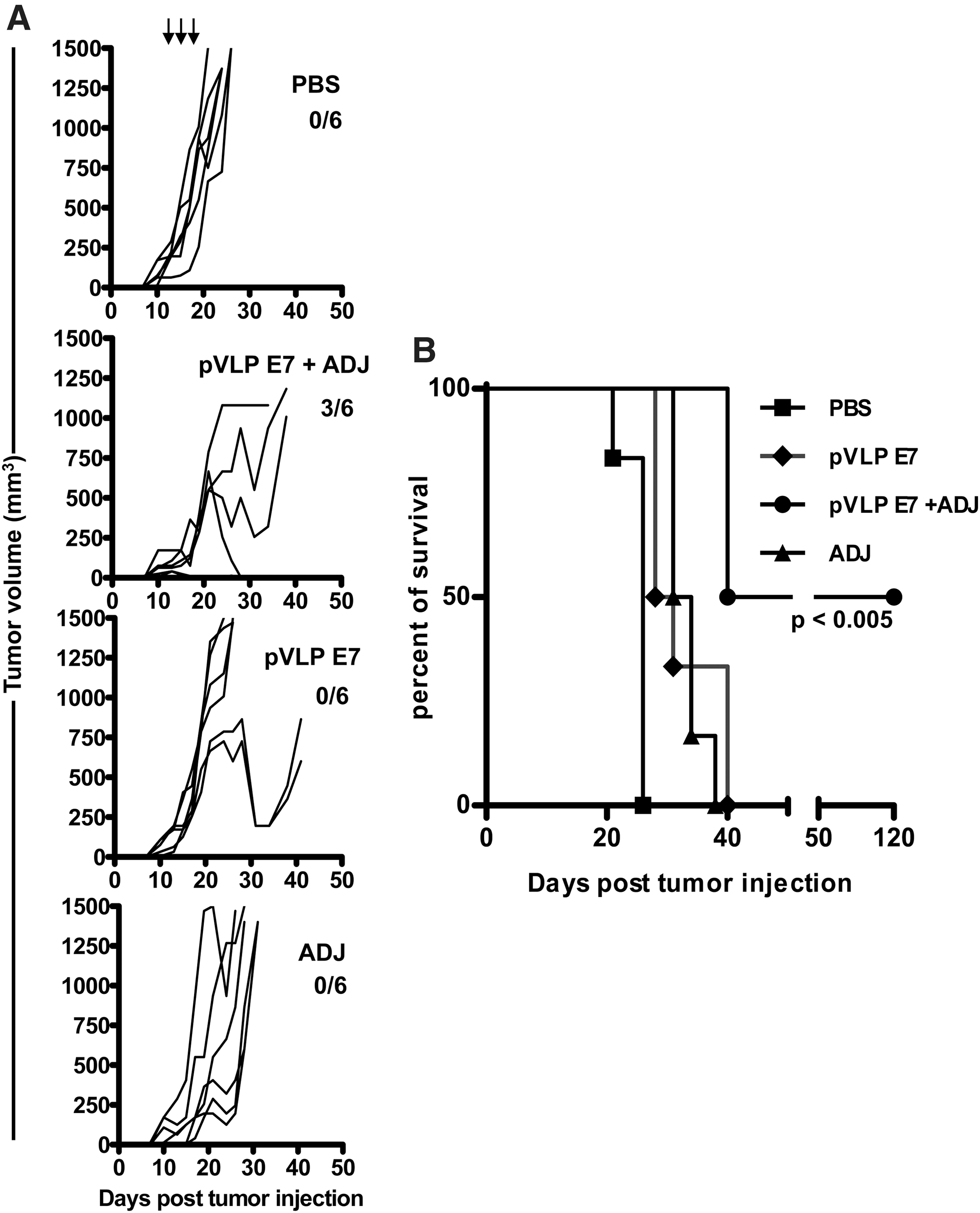
Efficacy of late therapeutic vaccinations with pVLP-E7 in combination with adjuvant therapy.
Discussion
In this paper, we have described a strategy of vaccination for HPV-induced cancer that is based on the use of pVLP DNA vaccines producing HPV-16 E7 recombinant retrovirus-based VLPs. Data showed that the VLPs produced can induce the maturation of human DCs and elicit a specific E7 T cell response. Using an in vivo tumor model, we demonstrated the efficiency of pVLP vaccination to prevent and protect animals from E7-positive tumor development compared with standard DNA or VLP immunization. To evaluate this strategy for advanced and well-established tumors, a situation more closely related to clinical settings, we addressed this question by vaccinating the mice at various times after the injection of tumorous cells. Thus, we found that pVLP vaccination can cure mice with already established TC-1 tumors only when associated with adjuvants.
Various vaccine strategies directed against E6 and E7 oncoproteins have already been developed for HPV-induced cancer. Most of them are based on the use of long peptides (Zwaveling et al., 2002), E7 proteins coupled to bacterially derived targeting molecules (Vingert et al., 2006; Berraondo et al., 2007), recombinant VLPs (Greenstone et al., 1998; Di Bonito et al., 2009) and defective viral vectors (Daemen et al., 2004; Riezebos-Brilman et al., 2005; Gomez-Gutierrez et al., 2007), and DNA vaccines (Monie et al., 2009; Wu et al., 2011), and some of them have already led to clinical trials (Kenter et al., 2008; Trimble et al., 2009). However, pVLPs for such an application have not been reported yet. For this application, pVLPs are formed of sequences encoding both Gag–E7 fusion protein and VSV-G envelope glycoproteins that subsequently form in vivo recombinant VLPs. Thus, this vaccine, which combines both DNA and VLP strategies, strongly enhanced antigen-specific T cell responses whereas standard DNA vaccination as well as VLPs alone turned out to be inefficient in our study (Fig. 4). In addition, the intradermal injection of pVLPs associated with electroporation reinforces the attraction of pVLP vaccination because this DNA delivery method has been shown to elicit stronger CD8+ T cell responses than intramuscular injection and intradermal gene gun delivery (Best et al., 2009). Altogether, these results highlight the potential of this vaccine strategy that combines advantages of VLP and DNA-based vaccines to elicit T cell responses.
Moreover, pseudotyping of VLPs with VSV-G is expected to improve their efficiency of antigen delivery into APCs as well as to favor specific CD8+ T cell immune responses through antigen cross-presentation and cross-priming mechanisms. It has been reported that VSV-G facilitates VLP uptake by APCs through endocytosis, and by promoting fusion between VLP envelope and endosome membranes (Temchura et al., 2008). This guarantees efficient delivery of VLP contents into cytoplasm, thus facilitating their interaction with proteasomes, protein degradation, and association with class I MHC molecules (Marsac et al., 2002). Along this line, we observed that nonpseudotyped Gag–E7 VLPs failed to efficiently prime T cells (data not shown). In addition, VSV-G glycoproteins are known to activate the TLR4-dependent pathway (Georgel et al., 2007), which could thus increase DC activation. Indeed, it has been shown that TLR4 activation in DCs and macrophages enhances the interferon-dependent pathways and their ability to mature and secrete IL-12 (Re and Strominger, 2001). Altogether, VSV-G glycoproteins play a critical role as adjuvant and must be included in our strategy to favor the immunogenicity of antigens and the vaccine efficacy.
More accurate models to investigate cancer vaccine efficacy are based mainly on preventive vaccination or therapeutic vaccination in which tumors are at an early stage. Numerous studies with a wide variety of transplantable tumors revealed that many vaccine preparations are extraordinarily effective in preventing tumor development but are largely ineffective in treating established malignancies (Zwaveling et al., 2002; Daemen et al., 2004; Riezebos-Brilman et al., 2005; Gomez-Gutierrez et al., 2007; Monie et al., 2009; Wu et al., 2011). Moreover, among therapeutic tumor vaccines that present some efficacy in animal models with small tumor inoculum, most of them lose their therapeutic value when the tumor burden is increased (Mansilla et al., 2012). A similar observation has been reported for HPV-16-specific immunotherapeutic trials (Welters et al., 2010). Here, we reveal that our strategy efficiently protects animals from tumor development and controls tumor growth at an early stage. However, tumor growth was not controlled when vaccinations alone were performed in mice with tumors whose diameter was greater than 5 mm. This observation may be explained by cancer cell escape during tumor development from innate and adaptive immune responses by selection of nonimmunogenic tumor cell variants and/or by active suppression of the immune response (Chaput et al., 2008; Pages and Kroemer, 2011). In the tumor environment after TC-1 injection, more than 10 and 20% of CD4+CD25+FoxP3+ regulatory T cells (Tregs) within total CD4+ cells were detected on days 13 and 18, respectively (our unpublished data). Such Treg infiltration during TC-1 tumor development may account for the fact that late vaccinations may be less efficient. It has already been published that rapid emergence of Tregs at the early steps of tumor development results in a tolerogenic environment (Darrasse-Jeze et al., 2009). We can speculate that adjuvants added to the vaccines will, by recruiting and stimulating innate immune cells (see Supplementary Fig. S1), counterbalance the Treg infiltration and subsequently boost the effector immune responses.
Interestingly, we showed that the combination of pVLP vaccination with adjuvants improved inhibition of tumor growth and gave rise to 50% tumor-free survival. The immunoadjuvants imiquimod and CpG ODN, as TLR7 and TLR9 agonists, have already been reported to enhance therapeutic HPV DNA vaccination, allowing the eradication even of large tumors in the TC-1 model (Chuang et al., 2010; Mansilla et al., 2012). Nevertheless, it might be important to take into account the route of administration of CpG ODN. Indeed, it has been reported that in contrast to local application of CpG ODN, which stimulates immune responses, systemic injection of CpG ODN suppresses adaptive T cell immunity (Wingender et al., 2006) via induction of indoleamine 2,3-dioxygenase (IDO), a potent activator of regulatory T cells (Baban et al., 2009). In our study, CpG ODN was injected into the periphery of the tumors, but similar results were obtained when CpG ODN was directly added to the vaccine formulation (data not shown). Because one paper indicates that a TLR9 adjuvant combined with electroporation-mediated delivery of HPV-16 E7-encoding DNA enhances antitumor responses (Ohlschlager et al., 2011), further experiments using imiquimod or CpG ODN alone or in combination may be necessary to better evaluate their effects on the tumor microenvironment in the context of pVLP vaccination associated with electroporation. Indeed, better deciphering the tumor–host interactions both in animal models as well as in patients may be useful to design innovative immunotherapeutic strategies for head and neck cancer (Badoual et al., 2010; Allen et al., 2012).
Thus, this paper justifies interest in pVLP vaccination in oncology and highlights for the first time the efficiency of pVLPs in controlling HPV-induced established tumors in combination with TLR7 and TLR9 agonists.
Footnotes
Acknowledgments
The authors thank Dominique Langui (Plateforme d'Imagerie Cellulaire Pitié-Salpêtrière, PICPS, INSERM UMRS 975/CNRS UMR 7225/UPMC, Paris, France) for expert technical assistance in electron microscopy. This work was supported by grants to F.M. Lemoine from Cancéropôle Île de France, Association pour la Recherche contre le Cancer (ARC), and La Ligue Nationale contre le Cancer. Author contributions: G.L., F.P., D.K., F.M.L., and B.B. conceived and designed the experiments; G.L., F.P., R.M., C.B., and C.H. performed the experiments; G.L., F.P., F.M.L., and B.B. analyzed the data; G.L., F.P., E.T., F.M.L., and B.B. wrote and corrected the paper; G.L. and F.P. contributed equally to this work as first authors; and F.M.L. and B.B. contributed equally to this work as last authors.
Author Disclosure Statement
The authors declare no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.