
Abstract

Although the double-blinded, placebo-controlled, randomized trial design is used in the evaluation for almost all new therapeutics, the rigor of this design has rarely been used in clinical trials assessing the safety and effectiveness of gene therapy. For some gene-therapy applications, such as the recent trial for hemophilia B (Nathwani et al., 2011), the double-blinded, placebo-controlled, randomized trial is not critical, because the phenotype (plasma factor IX activity) is simple to measure, the result is definitive, and there is no biologic reason why the phenotype would be altered by a placebo. However, for many of the disorders that gene therapy is designed to treat, the phenotype is complex, the outcome measurements have variability, and how the patient and physicians perceive the effect of the therapy may cloud the evaluation of safety and efficacy (Finniss et al., 2010).
Why has the gene-therapy field been slow to adopt the double-blinded, placebo-controlled, randomized trial design? In part, because investigators, subjects, and regulators have viewed gene therapy as different from traditional therapeutics, with theoretical concerns of germ-line gene transfer, insertional mutagenesis, recombination of viral vectors with other viruses, and, for some applications, a therapy that cannot be reversed. As a result, gene-therapy trials rarely have a placebo control, particularly a viral vector control. Further, many of the diseases to which gene therapy has been applied are untreatable, lethal disorders. When there are convincing experimental animal studies demonstrating efficacy, there is reluctance to not offer to subjects a possible curative (albeit unproven) therapy. Finally, many in vivo gene therapies require an invasive surgical procedure to administer the vector to the affected organ, and there is the ethical issue of carrying out a surgical procedure to administer a placebo.
One model of this tension among safety, ethics, and rigorous trial design is the application of gene therapy intended to induce therapeutic angiogenesis in the myocardium for individuals with diffuse coronary artery disease. For example, a decade ago, we carried out a cardiac gene-therapy trial using an E1−E3− serotype 5 adenovirus (Ad) vector coding for the vascular endothelial growth factor (VEGF) 121 isoform (Rosengart et al., 1999). The AdVEGF121 vector was administered directly to the myocardium. After a safety study where the gene therapy was used as an adjunct to coronary artery bypass surgery, we carried out a trial where the AdVEGF121 vector was the sole therapy, delivered to the heart through a minithoracotomy. Using the subjects as their own control, most showed improvement in exercise tolerance testing after therapy. A subsequent trial with the same vector in a larger group of subjects (the REVASC trial) also showed efficacy (Stewart et al., 2006). But critics (appropriately) pointed out that using the patient as his or her own control or using a randomized contemporaneous untreated control group was not the same as having a rigorous blinded, placebo-controlled, randomized design. Surgery itself can be a “placebo,” as observed in the Parkinson's disease trial of embryonic dopamine neurons to the brain versus sham neurosurgery [burr holes in the skull without administration of fetal cells (Freed et al., 2001)] and the knee osteoarthritis trial of arthroscopic surgery and lavage/debridement versus sham arthroscopic surgery (Moseley et al., 2002). Further, needle sticks to the myocardium induce angiogenesis (Pelletier et al., 1998), as may the Ad per se (Harvey et al., 1999; Moldovan et al., 2000; Squadrito and De Palma, 2011). Finally, the primary variable for this type of study is exercise tolerance, a test where the performance may be influenced by what the subjects believe about their cardiac health (Zachary and Morgan, 2011).
In the decade since our AdVEGF121 trial, we have designed a VEGF-based angiogenic cassette (VEGF-All6A+) that expresses all three isoforms of VEGF (121, 165, and 189), mimicking the endogenous expression of the VEGF gene (Fig. 1). Not only is the resulting Ad serotype 5 vector (AdVEGF-All6A+) efficacious at inducing angiogenesis in experimental animals, it does so at doses 1 to 2 logs lower than any of the VEGF individual isoforms delivered by the same Ad vector, with the same promoter (Whitlock et al., 2004; Amano et al., 2005).
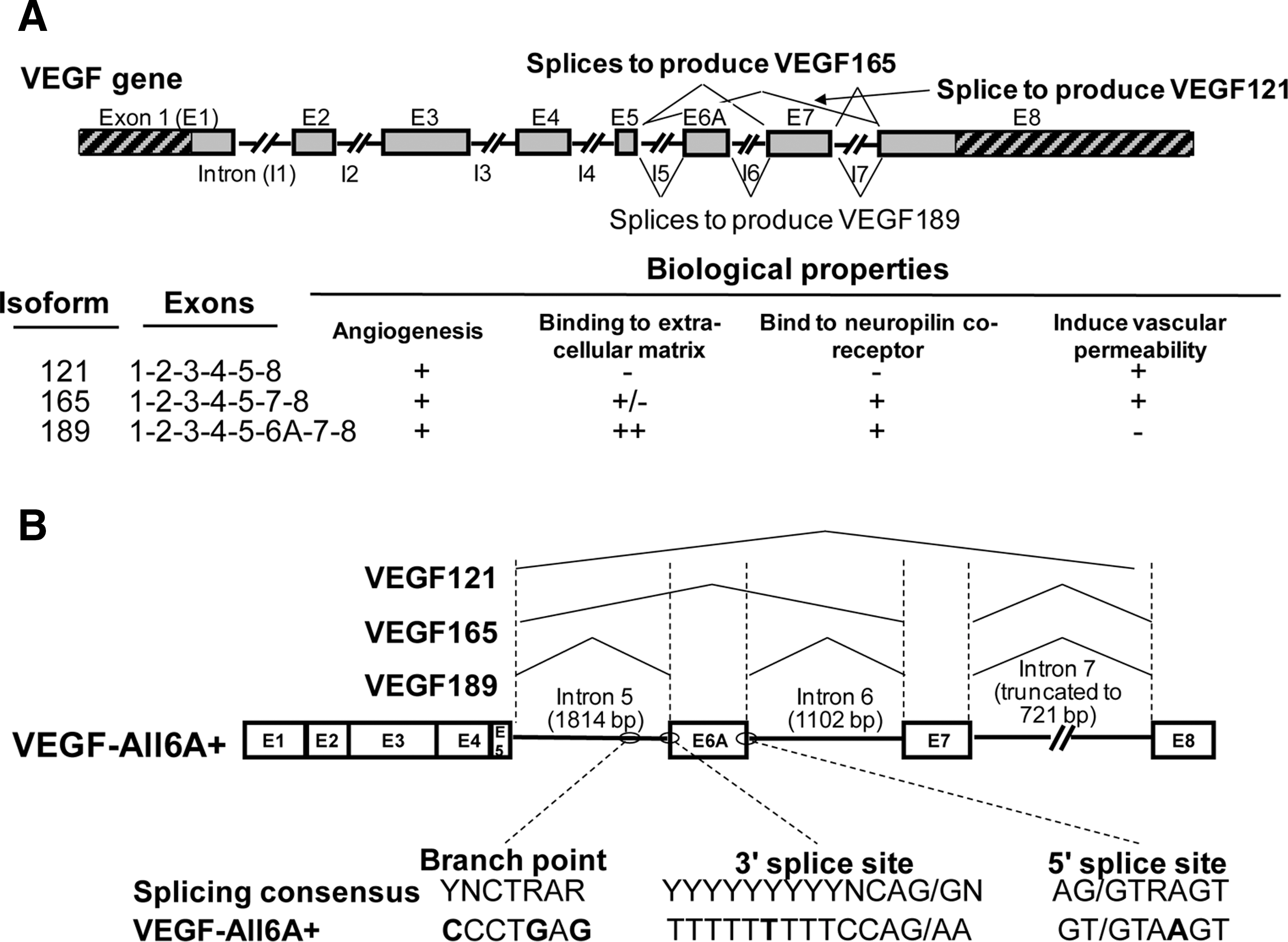
The normal and alternate splicing of the VEGF transcript and the design of the AdVEGF-All6A+ expression cassette.
Our plan is to assess the AdVEGF-All6A+ vector in a patient population similar to that in our original AdVEGF121 trial (individuals with moderate to severe coronary artery disease that are on maximal medical therapy, but are not candidates for coronary artery bypass surgery or coronary artery stents) (Rosengart et al., 1999). Fifty-two percent of these patients will die within 5 years, a survival rate similar to that of cancer of the oral cavity, pharynx, colon, and rectum (Allen et al., 2004; NCI, 2011). Further, their quality of life is poor: they have angina class III (symptoms with everyday living activity) or class IV (inability to perform any activity without angina, or even angina at rest) (Campeau, 1976).
However, in contrast to our AdVEGF121 trial, where the patient served as his or her own control, and the REVASC study, where there was a contemporary randomized untreated control group, our plan is to use the classic design of a double-blind, placebo-controlled, randomized trial (Fig. 2). After a dose-ranging safety study to determine the maximum tolerated dose, the subjects will be randomized (in a 3-to-1 ratio) to AdVEGF-All6A+ (the active drug) or AdNull (the placebo, an identical vector but without the VEGF-All6A+ transgene). The active drug or placebo will be administered directly to the myocardium through a minithoracotomy in a fashion similar to our AdVEGF121 trial (Rosengart et al., 1999).

Design of the AdVEGF-All6A+ trial. pu, particle units.
Why use a double-blind, placebo-controlled randomized design? Because this is the only way to begin to develop confidence that the AdVEGF-All6A+ vector may be efficacious. Without a rigorous design, no matter how “efficacious” the AdVEGF-All6A+ vector appears to be relative to the subjects as their own control or to a historic or contemporaneous untreated control group, there will always be the questions: was it the VEGF-All6A+ transgene, or was it the Ad per se, the surgery, or a combination of these factors?
Is it ethical to carry out a double-blind, placebo-controlled, randomized trial where some of the subjects will have a minithoracotomy but will receive myocardial injection of the AdNull placebo, not the AdVEGF-All6A+ vector? The issues relate to the respect for autonomy (voluntary informed consent) and the balance of beneficence and nonmalfeasance (whether there is direct, collateral, or aspirational benefit) (Dekkers and Boer, 2001).
Although there is postoperative discomfort, the risks of the minithoracotomy in this population are not high. The procedure requires general anesthesia and takes about 1 hr, and the total hospital stay is 2 to 3 days. In our prior study in the identical population, using the same surgical procedure to administer AdVEGF121 to the heart, there were no unanticipated serious adverse events and no deaths associated with the procedure (Rosengart et al., 1999; Crystal et al., 2002).
Could we do only a skin incision as “sham surgery”? No, because without the same postoperative course, the subjects and the treating physicians would know who received placebo. Importantly, sham surgery would not allow testing of the central hypothesis—that the VEGF-All6A+ expression cassette is more efficacious than simple needle sticks to the heart and/or the Ad per se induced angiogenesis (Pelletier et al., 1998; Chu et al., 1999; Harvey et al., 1999; Moldovan et al., 2000; Squadrito and De Palma, 2011).
Why use a minithoracotomy and epicardial administration of the vector? Why not use the intracoronary or intraventricular routes, both of which can be done by arterial catheterization, a less invasive procedure? In our view, the trials using intracoronary administration of angiogenic gene-transfer vectors have not been successful, because the intracoronary route is 1 to 2 logs less effective than the epicardial route in delivering the vector to the myocardium (Hackett et al., 2000; Lee et al., 2000). Catheter-based cardiac navigation systems have been used in angiogenic gene-transfer trials to deliver genes to the myocardium via the intraventricular route. But the investigators using this approach concluded that they could not be certain the vector was always injected into the myocardium, and only a fraction of the free wall of the left ventricle could be treated with confidence (Gyongyosi et al., 2005; Stewart et al., 2009). Importantly, with the intraventricular route, there is the significant risk that the vector will be injected intravascularly, with the attendant high risk of antivector innate immunity and a serious adverse event (Schnell et al., 2001; Crystal et al., 2002; Raper et al., 2003). In contrast, the minithoracotomy/epicardial route provides absolute control of the sites of myocardial injection, limits inadvertent intravascular administration, and has been shown to be safe in our prior AdVEGF121 trial and in the REVASC trial (Rosengart et al., 1999; Stewart et al., 2006).
Finally, there are parallels in what is being proposed and the randomized, placebo-controlled trials of new chemotherapeutic agents for fatal cancers: the therapy being tested is superimposed on standard optimal medical therapy, and there is discomfort and risk, including the risk of death from the therapy. The potential participants are adults, and with proper informed consent process taking care to avoid therapeutic misconception (Arkin et al., 2005), they can make the decision as to whether their quality of life versus the potential benefit is worth the risk.
Because of the ethical issues involved, it is important that this protocol and others like it are reviewed in a public forum not only with experts in gene therapy and cardiac disease, but also with ethicists and the lay public. In the gene-therapy field, we have such a forum in the National Institutes of Health DNA Recombinant Advisory Committee (RAC;
In summary, this is research; no matter how rational and no matter how many positive experimental animal studies, we need to evaluate gene therapy in humans, and we need to do it in a rigorous fashion. Gene therapy has reached the crossroads at which one path paves the way for real drug development and the opportunity to finally deliver the high expectations for this technology. The alternate route is to continue with the usual gene-therapy trial design, with studies leading to publications of academic interest but dead ends in terms of drug development toward regulatory approval. We suggest that gene therapy has matured as a clinical development field where we can now adopt the gold standard of the double-blinded, placebo-controlled, randomized trial, including, if necessary, the use of surgery for vector delivery.
Footnotes
Acknowledgments
We thank N. Mohamed and D.N. McCarthy for help in preparing the manuscript. These studies were supported, in part, by The Qatar Foundation and Weill Cornell Medical College in Qatar; and Lisa and James Cohen.
Author Disclosure Statement
No competing financial interests exist.