
Abstract
Dystroglycanopathies are a group of congenital muscular dystrophies (CMD) often caused by mutations in genes encoding glycosyltransferases that lead to hypoglycosylation of α-dystroglycan (α-DG) and reduce its extracellular matrix-binding activity. Overexpressing LARGE (formerly known as like-glycosyltransferase) generates an extracellular matrix-binding carbohydrate epitope in cells with CMD-causing mutations in not only LARGE but also other glycosyltransferases, including POMT1, POMGnT1, and fukutin, creating the possibilities of a one-for-all gene therapy. To determine the feasibility of LARGE gene therapy, a serotype 9 adeno-associated viral vector for overexpressing LARGE (AAV9-LARGE) was injected intracardially into newborns of two mouse models of CMD: the natural LARGE mutant Largemyd mice and protein O-mannose N-acetylglucosaminyltransferase 1 (POMGnT1) knockout mice. AAV9-LARGE virus treatment yielded partial restoration of α-DG glycosylation and ligand-binding activity. The muscular dystrophy phenotype in skeletal muscles was ameliorated as revealed by significantly reduced fibrosis, necrosis, and numbers of centrally located nuclei with improved motor function. These results indicate that LARGE overexpression in vivo by AAV9-mediated gene therapy is effective at restoring functional glycosylation of α-DG and rescuing the muscular dystrophy phenotype in deficiency of not only LARGE but also POMGnT1, providing evidence that in vivo LARGE gene therapy may be broadly useful in dystroglycanopathies.
Introduction
Many of these patients have mutations in genes encoding glycosyltransferases (or putative glycosyltransferases), including POMT1 (encoding protein O-mannosyltransferase 1) (Beltran-Valero de Bernabé et al., 2002); POMT2 (van Reeuwijk et al., 2005); POMGnT1 (encoding protein O-mannose N-acetylglucosaminyltransferase 1) (Yoshida et al., 2001); LARGE (encoding a protein formerly known as like-glycosyltransferase) (Longman et al., 2003); FKTN (encoding fukutin) (Kobayashi et al., 1998); and FKRP (encoding fukutin-related protein) (Brockington et al., 2001; Beltran-Valero de Bernabé et al., 2004). POMT1 and POMT2 form an enzyme complex that transfers mannose to serine or threonine residues to initiate O-mannosyl glycosylation (Manya et al., 2004; Akasaka-Manya et al., 2006). POMGnT1 then transfers N-acetylglucosamine to O-linked mannose (Yoshida et al., 2001; Zhang et al., 2002). LARGE synthesizes laminin-binding repeating disaccharide units of [–3-xylose–α1,3-glucuronic acid-β1–] (Inamori et al., 2012). The functions of fukutin and FKRP are not yet fully elucidated. Recently, mutations in collagen IV α1 (COL4A1) (Labelle-Dumais et al., 2011) and ISPD (encoding isoprenoid synthase domain containing) (Roscioli et al., 2012; Willer et al., 2012) are identified as new causes of these diseases.
α-DG is the only known protein functionally glycosylated by the aforementioned glycosyltransferases. It is a cell-surface glycoprotein receptor that interacts with high affinity to several extracellular matrix components, including laminin (Ervasti and Campbell, 1993; Gee et al., 1993; Yamada et al., 1994), agrin (Gee et al., 1994), perlecan (Peng et al., 1998), neurexin (Sugita et al., 2001), and pikachurin (Sato et al., 2008). It is heavily substituted by O-linked glycans, particularly of the O-linked mannosyl glycosylation (Chiba et al., 1997; Sasaki et al., 1998; Smalheiser et al., 1998). Hypoglycosylation of α-DG in dystroglycanopathies is evidenced by loss of IIH6C4 or VIA4-1 immunoreactivity, two antibodies that recognize the carbohydrate epitope present on α-DG. A functional consequence of hypoglycosylation is the loss of ligand-binding activity to extracellular matrix molecules such as laminin and perlecan (Grewal et al., 2001; Kano et al., 2002; Michele et al., 2002; Longman et al., 2003; Takeda et al., 2003; Kim et al., 2004; Liu et al., 2006), and pikachurin (Kanagawa et al., 2010; Hu et al., 2011).
Overexpression of LARGE leads to hyperglycosylation of α-DG in that IIH6C4- and VIA4-1-recognized protein species migrate slower than normal on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Barresi et al., 2004). Interestingly, LARGE overexpression also restores laminin-binding activity in cells isolated from Largemyd mice, and generates ligand-binding IIH6C4 immunoreactive species in cells isolated from patients with WWS, MEB, and FCMD. The ability to hyperglycosylate α-DG and “rescue” its laminin-binding activity is unique to LARGE and its homolog LARGE2 (Barresi et al., 2004; Brockington et al., 2005; Fujimura et al., 2005; Grewal et al., 2005). Local overexpression of LARGE in the POMGnT1 knockout leg muscle resulted in hyperglycosylation of α-DG (Kanagawa et al., 2009). These studies raise the hope of a one-for-all common LARGE gene therapy for dystroglycanopathies.
Adeno-associated viral (AAV) vectors of serotypes 6, 8, and 9 can give rise to widespread transduction in multiple tissues, including skeletal muscle following intravenous or intraperitoneal injections (Gregorevic et al., 2004; Wang et al., 2005; Bostick et al., 2007). Remarkably, self-complementary adeno-associated virus serotype 9 (scAAV-9) is capable of penetrating the blood-brain barrier in mice to target the brain (Foust et al., 2009). When injected in newborns, AAV9 vectors efficiently transduced skeletal muscle in dogs (Yue et al., 2008). Widespread gene delivery to the skeletal muscle, central nervous system, and the retina makes AAV9 vectors a promising choice for gene delivery in dystroglycanopathies.
In this study, we tested the feasibility of skeletal muscle gene therapy by overexpressing LARGE in POMGnT1 knockout and Largemyd mice using an AAV9 vector, AAV9-LARGE. Functional glycosylation of α-DG in skeletal muscles was restored and muscular dystrophy phenotype was significantly ameliorated. Our work provides in vivo evidence that LARGE overexpression is a viable approach for gene therapy in congenital muscular dystrophies belonging to the dystroglycanopathy type.
Results
Efficient transduction of skeletal muscle by systemic injection of AAV9-EGFP virus in the newborn
To compare newborn versus adult systemic injections, we constructed an AAV9–enhanced green flourescent protein (EGFP) vector using the chicken β-actin promoter and injected it at two developmental stages into newborn heart and adult tail vein. When examined for expression of GFP fluorescence (two months and one month after injection for newborn and adult respectively), bright GFP fluorescence was observed in skeletal muscle (Fig. 1A, B, D, and E) and in the heart (Fig. 1G and H) for both injections. However, GFP fluorescence was much brighter in newborn injections for both diaphragm and leg muscle than adult injections (compare Fig. 1A and D with B and E). Both injections also resulted in low but detectable GFP expression in some cardiomyocytes (Fig. 1G and H) and the brain (not shown). Overall GFP fluorescence in the diaphragm was much brighter than that in the leg muscle. There was no apparent difference in GFP fluorescence levels amongst various leg muscles including the quadriceps and gastrocnemius muscles. Interestingly, newborn injections labeled few hepatocytes while adult injections labeled most cells in the liver (compare Fig. 1J and K). Uninjected animals exhibited only background green fluorescence in the diaphragm, leg muscle, and the heart (Fig. 1C, F, and I). These results indicate that systemic injection of AAV9-vectors in newborns can mediate highly efficient gene delivery to the skeletal muscle but avoid liver tropism.
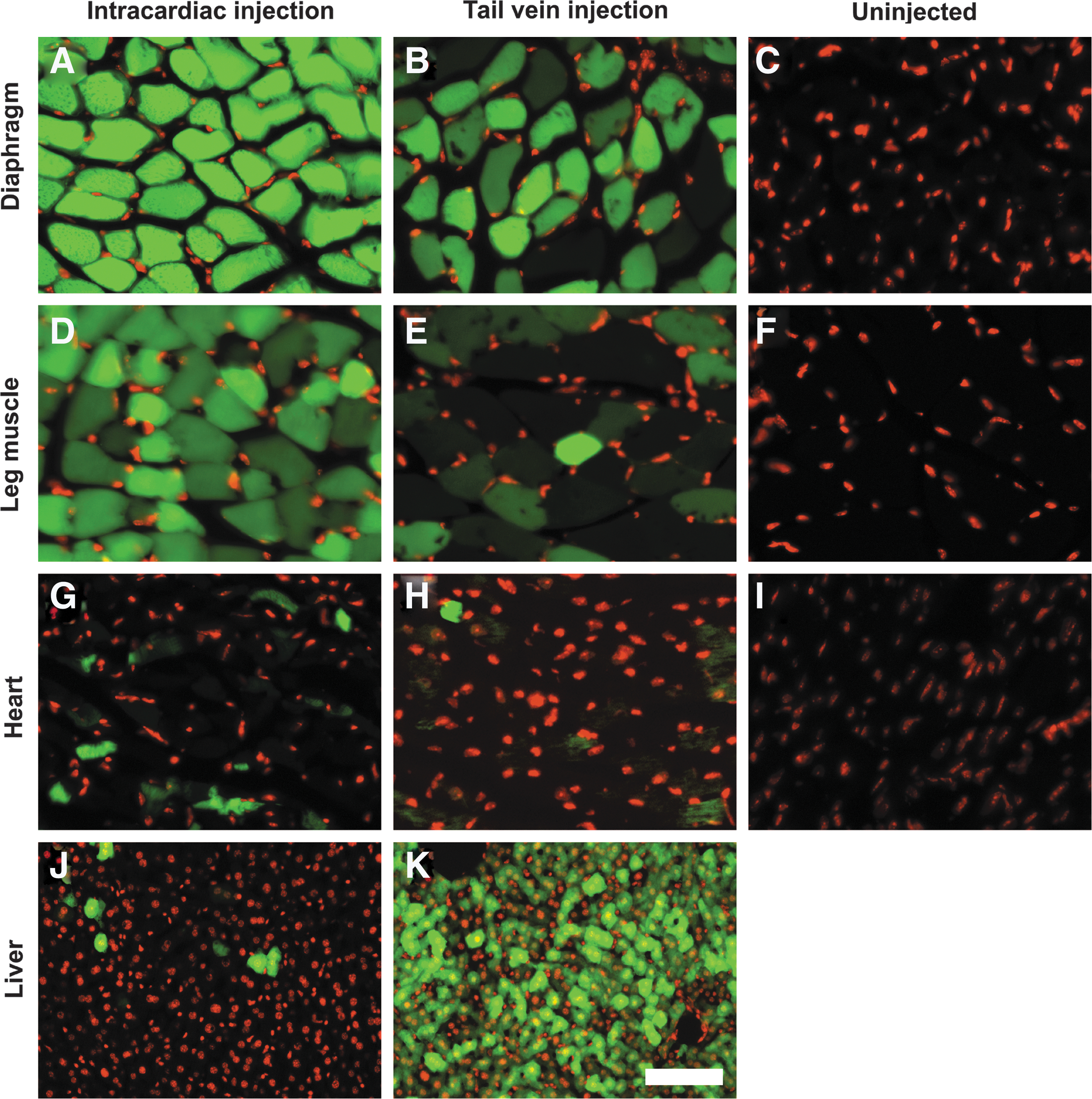
Intracardiac injection of AAV9-EGFP virus in newborns efficiently infected skeletal muscle but not the liver. AAV9-EGFP virus was injected into the heart of postnatal day 5 mice
AAV9-LARGE virus treatment confers functional glycosylation of α-DG
To evaluate the feasibility of LARGE overexpression in gene therapy, AAV9-LARGE, a vector for LARGE overexpression driven by the chicken β-actin promoter, was constructed. The virus was injected intracardially into newborn POMGnT1 knockout and Largemyd mice. Control mice (wild-type or heterozygous animals) that were infected with AAV9-LARGE virus did not show any apparent phenotypic signs. To examine whether AAV9-LARGE virus treatment restored functional glycosylation, we carried out an IIH6C4 Western blot on glycoproteins isolated from leg-muscle lysates using wheat germ agglutinin (WGA)-agarose (Fig. 2A). IIH6C4 is an antibody that recognizes the ligand-binding carbohydrate epitope of α-DG. While IIH6C4 immunoreactivity was observed at about 150 kDa for the controls (lane 7), a weaker IIH6C4 immunoreactivity was observed at about 75 kDa for POMGnT1 knockout mice (lane 3) because α-DG is hypoglycosylated in POMGnT1 knockout mice. Upon AAV9-LARGE virus treatment, IIH6C4 immunoreactivity in POMGnT1 knockout samples was located not only at about 75 kDa as in untreated but also at much greater molecular mass range extending greater than 250 kDa(lanes 1 and 6). IIH6C4 immunoreactivity was not detected in Largemyd samples (lanes 5). AAV9-LARGE treatment resulted in IIH6C4 immunoreactivity from 150 kDa to molecular masses much greater than the control (lanes 2, 4, and 8). As expected, β-DG was detected with the anti-β-DG antibody in all samples indicating that a similar amount of protein was loaded for all samples.
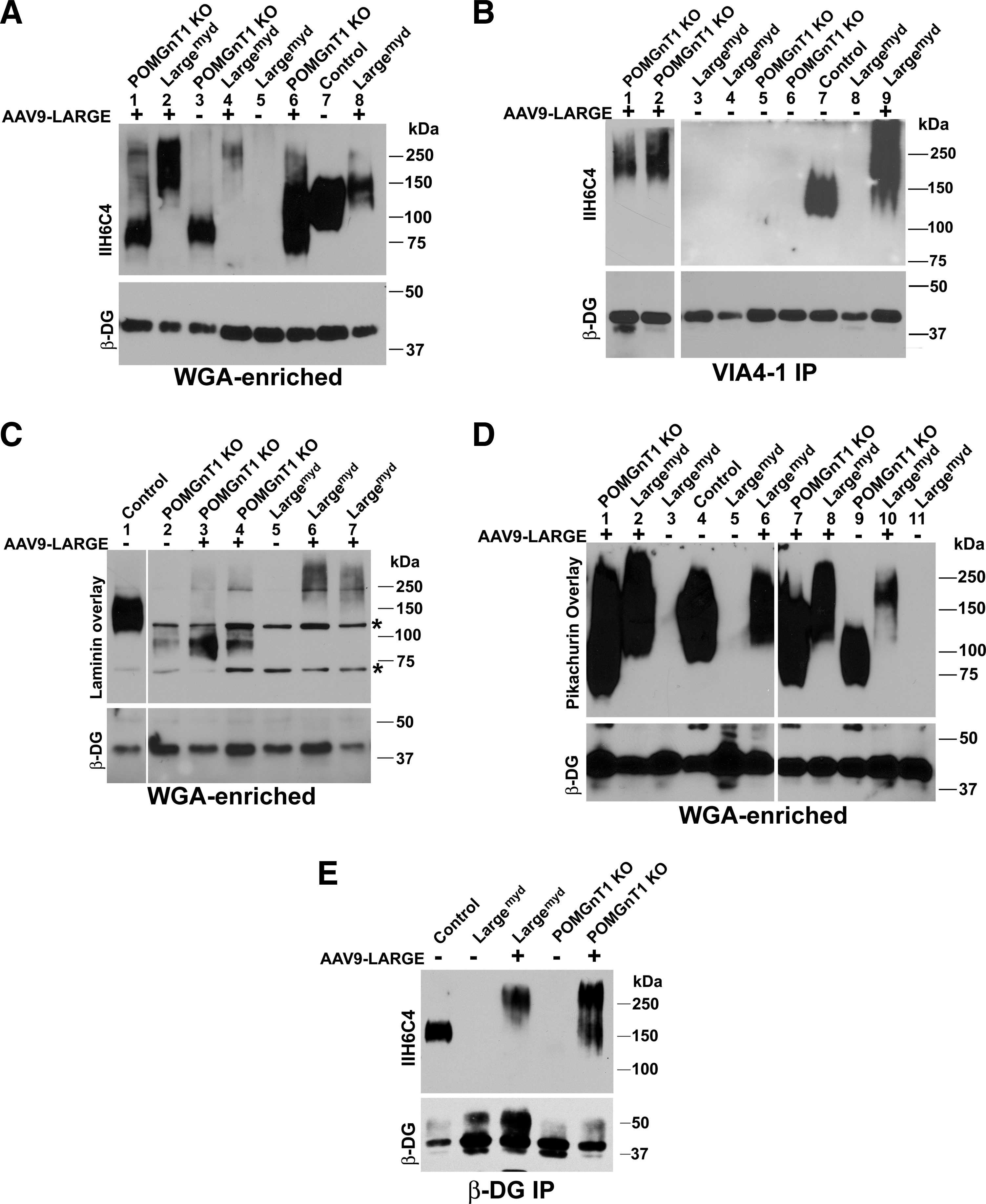
AAV9-LARGE virus treatment partially restored functional glycosylation in skeletal muscle. AAV9-LARGE virus was injected intracardially into postnatal day 3–5 in animals. Immunoblot with IIH6C4 antibody and ligand (laminin and pikachurin) overlay experiments were carried out on glycoproteins isolated from muscle lysates with wheat germ agglutinin (WGA)-agarose. IIH6C4 immunoblot was also carried out on VIA4-1 immunoprecipitates isolated from lysates.
To further determine whether AAV9-LARGE virus treatment restored functional glycosylation of α-DG, we carried out immunoprecipitation of leg-muscle lysates with VIA4-1, another antibody commonly used to characterize functional glycosylation of α-DG, and detected LARGE-glycosylated proteins by immunobloting with IIH6C4 (Fig. 2B). IIH6C4 immunoreactivity was detected at 150 kDa in the VIA4-1 immunoprecipitate from the control lysate (lane 7). In immunoprecipitates of POMGnT1 knockout (lanes 5 and 6) and Largemyd (lanes 3, 4, and 8) muscles, no IIH6C4 immunoreactivity was detected. Upon AAV9-LARGE virus treatment, IIH6C4 immunoreactivity was observed as a smear extending from 150 kDa to the beginning of the separating gel for both POMGnT1 knockout (lane 1 and 2) and Largemyd (lane 9) muscles. As a control, β-DG was readily detected in the lysates. Together, these results indicate that treatment with AAV9-LARGE virus partially restored IIH6C4 and VIA4-1 immunoreactivity in both mutant mice.
We also evaluated glycosylation of α-DG by VIA4-1 immunofluorescence staining (Fig. 3). While the diaphragm and quadriceps muscle of control mice showed bright VIA4-1 immunofluorescence (Fig. 3A and A’), POMGnT1 knockout (Fig. 3J and J’) and Largemyd (Fig. 3D and D’) mice showed only background fluorescence. AAV9-LARGE treatment partially restored VIA4-1 immunofluorescence (Fig. 3G, G’, M, and M’). Some myofibers in AAV9-LARGE virus-treated mice showed brighter VIA4-1 immunofluorescence than the control. As expected, laminin immunofluorescence intensity was similar in the diaphragm of control, POMGnT1 knockout and Largemyd mice with and without AAV9-LARGE treatment (Fig. 3C, F, I, L, and O). Largemyd mice showed some regions with apparent collapsed laminin immunofluorescence patterns (data not shown). The morphology of AAV9-LARGE-treated Largemyd mouse diaphragms was mostly normal. Similar results were obtained for laminin immunofluorescence staining of quadriceps muscle (Fig. 3C’, F’, I’, L’, and O’). Further, β-DG immunofluorescence was comparable in all diaphragm and quadriceps samples (Fig. 3B, E, H, K, N, B’, E’, H’, K’, and N’). These results indicate that AAV9-LARGE virus treatment partially restored functional glycosylation of α-DG in POMGnT1 knockout and Largemyd mice.

AAV9-LARGE virus treatment increased VIA4-1 immunoreactivity in skeletal muscle of POMGnT1 knockout and Largemyd mice. Diaphragm
Binding of extracellular matrix molecules such as laminin by α-DG is dependent on its glycosylation and is diminished in POMGnT1 knockout and Largemyd mice. To determine whether the IIH6C4 immunoreactive species from AAV9-LARGE virus-treated mutant animals were capable of ligand-binding, laminin (Fig. 2C) and pikachurin (Fig. 2D) overlay experiments were carried out. In the laminin overlay experiment, laminin binding was observed for the control (Fig. 2C, lane 1). However, remnant laminin binding was observed in the POMGnT1 knockout samples between 75–100 kDa (lane 2). The AAV9-LARGE virus treatment resulted in observable laminin binding to proteins at a much greater molecular mass range (150 kDa and above) in addition to the binding pattern observed between 75–100 kDa (lanes 3 and 4). For Largemyd mice, no laminin binding was observed (lane 5). AAV9-LARGE virus treatment resulted in laminin binding at high molecular mass ranges from 150 to 250 kDa and higher (lanes 6 and 7).
Pikachurin overlay assay exhibited a much more robust binding signal than the laminin overlay assay (Fig. 2D). Pikachurin binding was observed at molecular mass of about 100–250 kDa in the control (lane 4), a greater molecular mass range than observed for IIH6C4 immunoreactivity and laminin binding perhaps due to robust binding between pikachurin and α-DG. In POMGnT1 knockout mice, pikachurin binding was detected between 75 kDa to 130 kDa range at a reduced level and reduced molecular mass range (lane 9) from the control. Upon AAV9-LARGE virus treatment, pikachurin binding was found at molecular mass ranges from 75 kDa to 250 kDa and higher (lanes 1 and 7), similar to the location of IIH6C4 immunoreactivity. In Largemyd mice, no ligand binding was detected (lanes 3, 5, and 11). However, AAV9-LARGE virus treatment generated pikachurin binding at molecular mass ranges from 100 kDa to 250 kDa and higher (lanes 2, 6, 8, and 10), similar to the location of IIH6C4 immunoreactivity. As expected, β-DG was readily detected in all samples.
To further evaluate whether α-DG is glycosylated by AAV9-LARGE treatment, we carried out immunoprecipitation with an anti-β-DG antibody followed by immunoblotting with the IIH6C4 antibody (Fig. 2E). IIH6C4 immunoreactivity was detected at 150 kDa in control but not in POMGnT1 knockout and Largemyd samples. By contrast, IIH6C4 immunoreactivity was detected at molecular mass range greater than 150 kDa for AAV9-LARGE-treated POMGnT1 knockout and Largemyd samples. Together, these results indicate that AAV9-LARGE virus treatment led to α-DG glycosylation and generated ligand binding IIH6C4 immunoreactive carbohydrate species in POMGnT1 knockout and Largemyd mouse skeletal muscles.
Severity of muscular dystrophy is ameliorated by AAV9-LARGE treatment
The diaphragm of the control mouse exhibited typical morphology of muscle fibers with nuclei located in the periphery (n=7, Fig. 4A and C). AAV9-LARGE virus treatment did not cause any noticeable changes (n=3, Fig. 4B and D). Largemyd mice exhibited severe necrosis of muscle fibers (see asterisk) with many centrally located nuclei (see arrows) in all animals examined (n=7, Fig. 4E and G). Inflammation was evident in necrotic regions. AAV9-LARGE virus treatment significantly improved the histological appearance of the diaphragm in Largemyd mice (Fig. 4F and H). AAV9-LARGE-treated Largemyd animals that exhibited IIH6C4 immunoreactivity were completely devoid of large areas of necrosis. We counted centrally and peripherally located nuclei from five randomly selected images taken from four AAV9-LARGE-treated Largemyd and four untreated animals. AAV9-LARGE-treated Largemyd animals had much fewer centrally located nuclei than untreated (2.67%±1.31% vs. 27.33%±1.36% [mean±SEM], p=0.0000062, Student's t test).
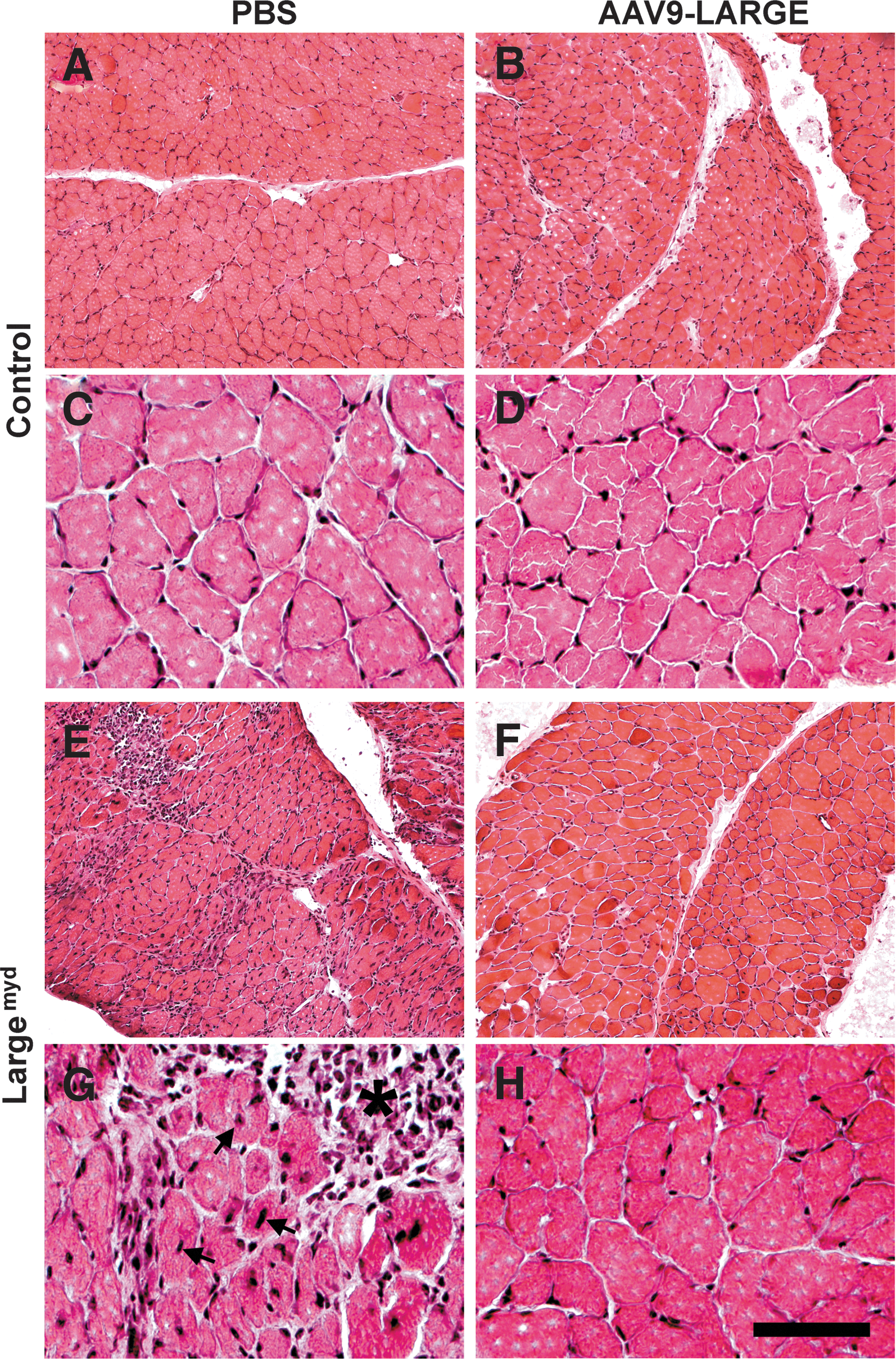
AAV9-LARGE virus treatment ameliorated muscular dystrophy in the diaphragm of Largemyd mice. Diaphragm sections of 3–4-month-old mice were stained with hematoxylin and eosin (H&E).
Similarly, the quadriceps muscle of Largemyd mice showed many regions with necrotic fibers (see asterisk) and centrally located nuclei (see arrows) (n=7, Fig. 5C and D). By contrast, AAV9-LARGE-treated mice did not exhibit any regions with a large number of necrotic fibers (Fig. 5E and F). Similar results were obtained for the gastrocnemius muscle (data not shown). Centrally and peripherally located nuclei were counted for the quadriceps muscle from five randomly selected images taken from four AAV9-LARGE treated Largemyd and four untreated animals were counted. The number of centrally located nuclei were reduced in AAV9-LARGE-treated animals (12.93%±5.16% vs. 35.13%±3.38% [mean±SEM], p=0.0057, Student's t test). Variability in recovery of IIH6C4 immunoreactivity appeared to correlate with phenotypic rescue. While the quadriceps muscle of one animal (Fig. 2A, lane 2) had 5.76% of the nuclei located centrally, the quadriceps muscle of another animal (Fig.2A, lane 4) had 28.21% of the nuclei located centrally. In general, the beneficial effect of the AAV9-LARGE treatment appeared to be less robust in the quadriceps muscle than the diaphragm as evidenced by a less robust reduction in centrally located nuclei.
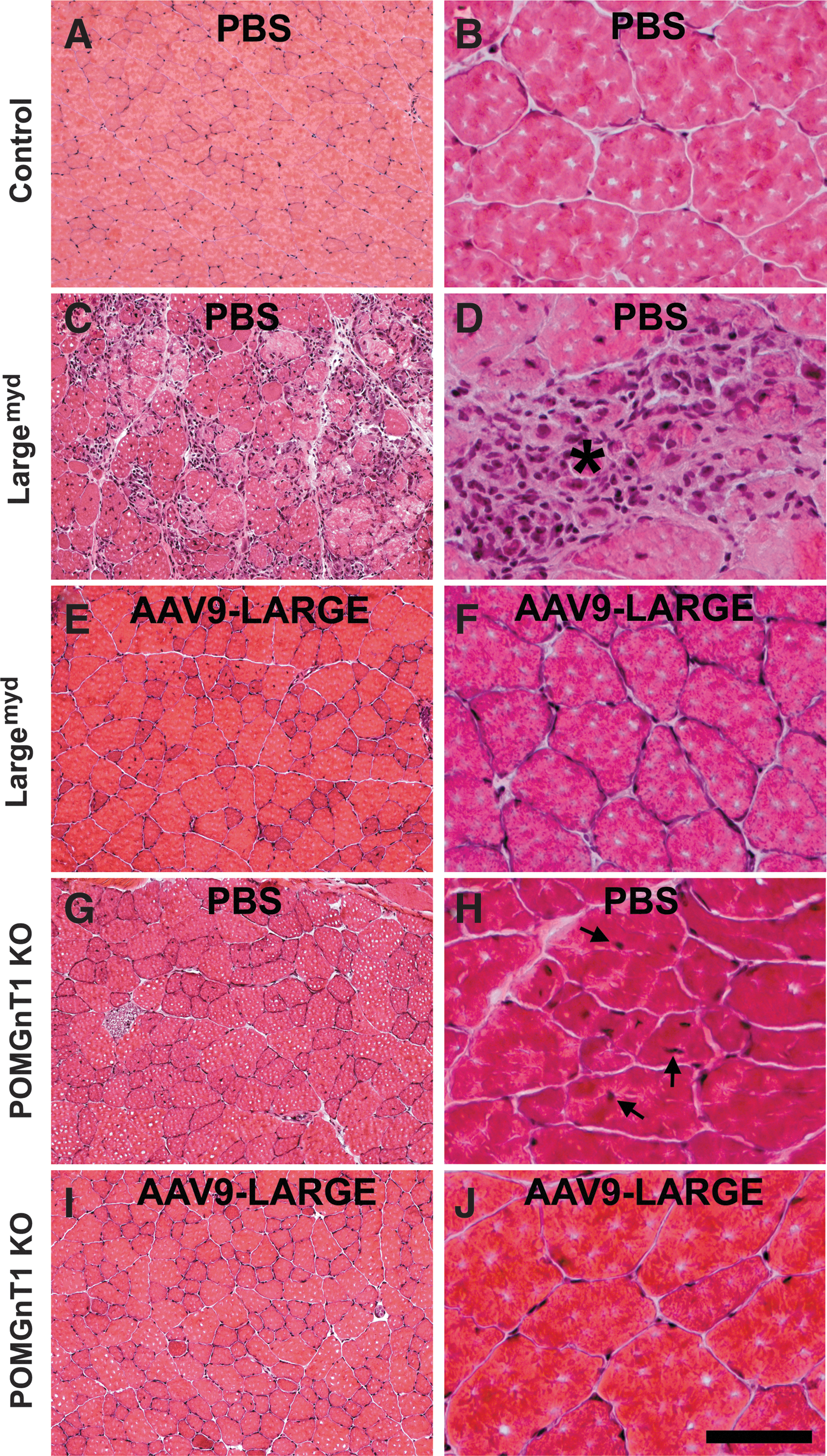
AAV9-LARGE virus treatment ameliorated muscular dystrophy in the quadriceps muscle of Largemyd and POMGnT1 knockout mice. Quadriceps muscle sections of 3–4-month-old mice were stained with H&E.
For the POMGnT1 knockout mice, no concentrated focal areas of necrosis were observed for the diaphragm or quadriceps muscle, but centrally located nuclei were frequently observed in the quadriceps muscle (n=4, Fig. 5G and H). The AAV9-LARGE virus treatment reduced the number of centrally located nuclei in the quadriceps muscle (Fig. 5I and J) (n=4, 2.09%±0.29% vs. 6.42%±1.54% [mean±SEM], p=0.017, Student's t-test).
AAV9-LARGE-treated mice exhibit fewer signs of muscle regeneration
Muscular dystrophy is associated with the presence of regenerating muscle fibers. We therefore carried out immunofluorescence staining with antibodies against monoclonal rat antineural cell adhesion molecule (NCAM) and developmental myosin heavy chain (MHCd), two markers highly expressed in regenerating but not mature myofibers (Bentzinger et al., 2005). In addition, 5-bromo-2’-deoxyuridine (BrdU) was injected 3 hr before the mice were sacrificed to label proliferating cells. In the diaphragm (Fig. 6A and B) and quadriceps muscle (Fig. 6J and K) of controls, no NCAM- and MHCd-positive myofibers were observed. BrdU-labeled nuclei were not found (Fig. 6C and L). By contrast, NCAM- and MHCd-positive fibers were frequently observed in the diaphragm (Fig. 6D and E) and quadriceps muscle (Fig. 6M and N) of Largemyd mice. Clusters of BrdU-labeled nuclei were also frequently found (Fig. 6F and O). By contrast, very few NCAM- and MHCd-positive fibers and BrdU-labeled nuclei were observed in the diaphragm (Fig. 6G, H, and I) and quadriceps muscle (Fig. 6P, Q, and R) of AAV9-LARGE treated Largemyd mice, indicating that AAV9-LARGE treatment reduced signs of muscle regeneration in Largemyd mice.

AAV9-LARGE virus treatment reduced the number of regenerating fibers in skeletal muscle of Largemyd and POMGnT1 knockout mice. Diaphragm
For POMGnT1 knockout mice, NCAM and MHCd-positive fibers were frequently found in the quadriceps muscle (Fig. 6S and T), although at much less frequency than Largemyd mice. AAV9-LARGE virus treatment resulted in a reduction of NCAM- and MHCd-positive fibers (Fig. 6V and W). Interestingly, BrdU-labeled nuclei were rarely observed in POMGnT1 knockout and AAV9-LARGE-treated POMGnT1 knockout mice, indicating lack of significant cell proliferation in adult POMGnT1 knockout muscles (Fig. 6U and X). These data indicate that AAV9-LARGE virus treatment significantly reduced the numbers of regenerating fibers in POMGnT1 and Largemyd mice.
AAV9-LARGE treatment prevented fibrosis in skeletal muscle of Largemyd mice
One consequence of severe muscular dystrophy is fibrosis. To evaluate whether AAV9-LARGE treatment prevented fibrotic changes, sections were also immunofluorescence-stained with antibodies against tenascin-C and collagen I, two markers specific for increased fibrosis in muscle (Ringelmann et al., 1999) (Fig. 7). The diaphragm and the quadriceps muscle of Largemyd mice showed dramatically increased immunoreactivity to tenascin-C (Fig. 7E and G) and collagen I (Fig. 7F and H) over the controls (Fig. 7A–D), indicating presence of significant fibrosis. AAV9-LARGE virus-treated Largemyd mice were similar to controls and exhibited much less immunoreactivity to tenascin-C and collagen I than untreated mice (Fig. 7I–L). These results indicated that AAV9-LARGE virus treatment reduced fibrosis in the mutant mice. POMGnT1 knockout mice did not show any apparent increase in immunoreactivity to antibodies against tenascin-C and collagen I indicating the absence of severe fibrosis. AAV9-LARGE treatment did not show any noticeable effect in these animals. These results indicate that there were fibrotic changes in the diaphragm and quadriceps muscle of Largemyd mice and that AAV9-LARGE virus treatment mostly prevented these changes.
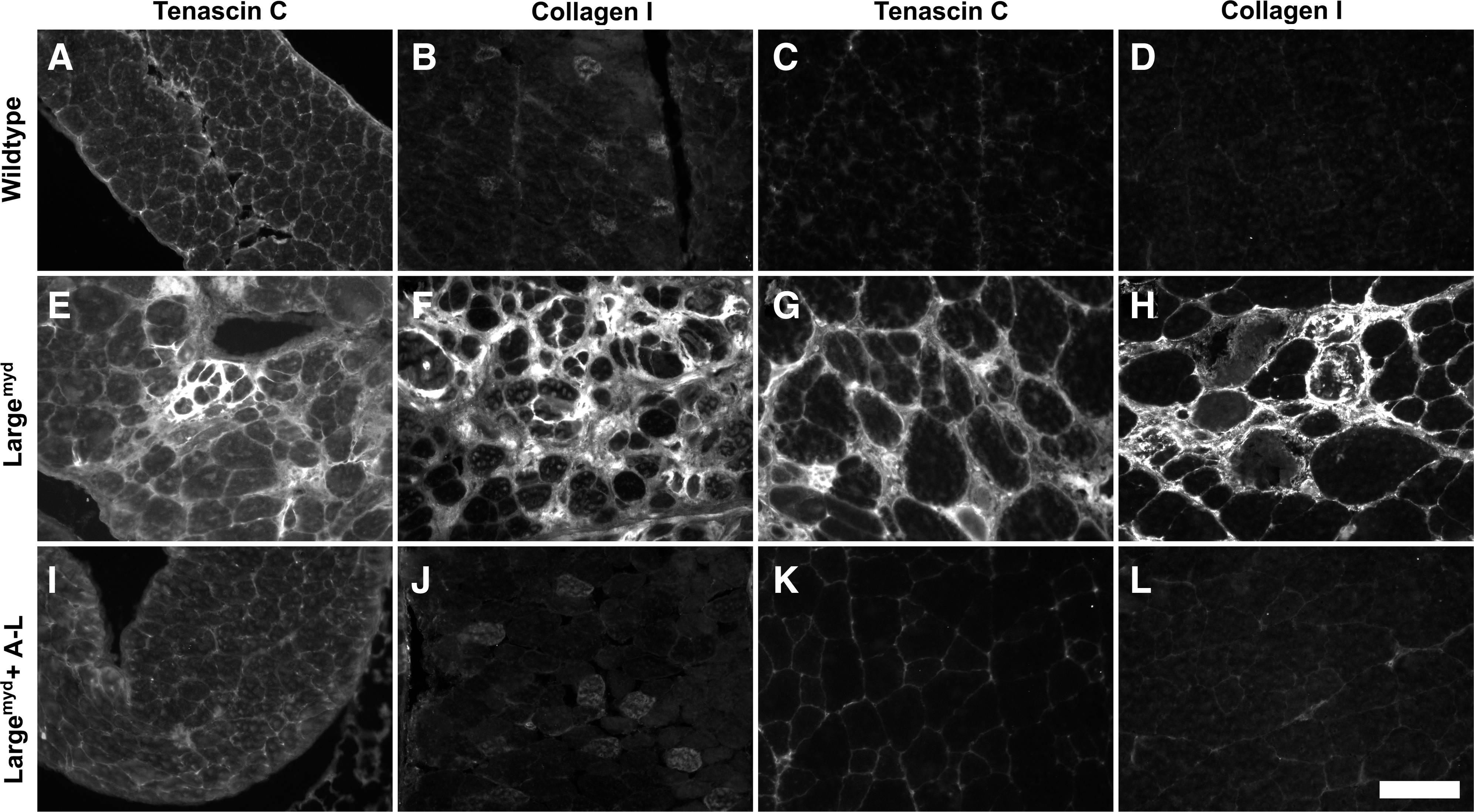
AAV9-LARGE virus treatment reduced fibrosis in skeletal muscle of Largemyd mice. Diaphragm and quadriceps muscle sections from 3–4-month-old mice were immunofluorescence stained with antibodies against tenascin-C
AAV9-LARGE virus-treated Largemyd and POMGnT1 knockout mice exhibit improved muscular function
Because of widespread transduction of the AAV9 viral vector, a vertical pole test and a treadmill exercise assay that evaluate overall motor function were carried out to evaluate whether AAV9-LARGE gene therapy improves muscular function of the mice. In the vertical pole test (Fig. 8A), the mice were placed at one end of a horizontally placed pole, which was gradually lifted to a vertical position so that the mice were on top of the pole. The ability of the mice to climb down and off the pole was scored. Largemyd mice scored poorly (median score=1.3) when compared to the control animals (wild-type and heterozygous animals, median score=13, p<0.0001, Mann-Whitney U Test, U=140). AAV9-LARGE virus treatment moderately improved the scores on the vertical pole test of Largemyd mice (median score=2.95, p=0.0097, Mann-Whitney U Test, two-tailed, U=60.5).
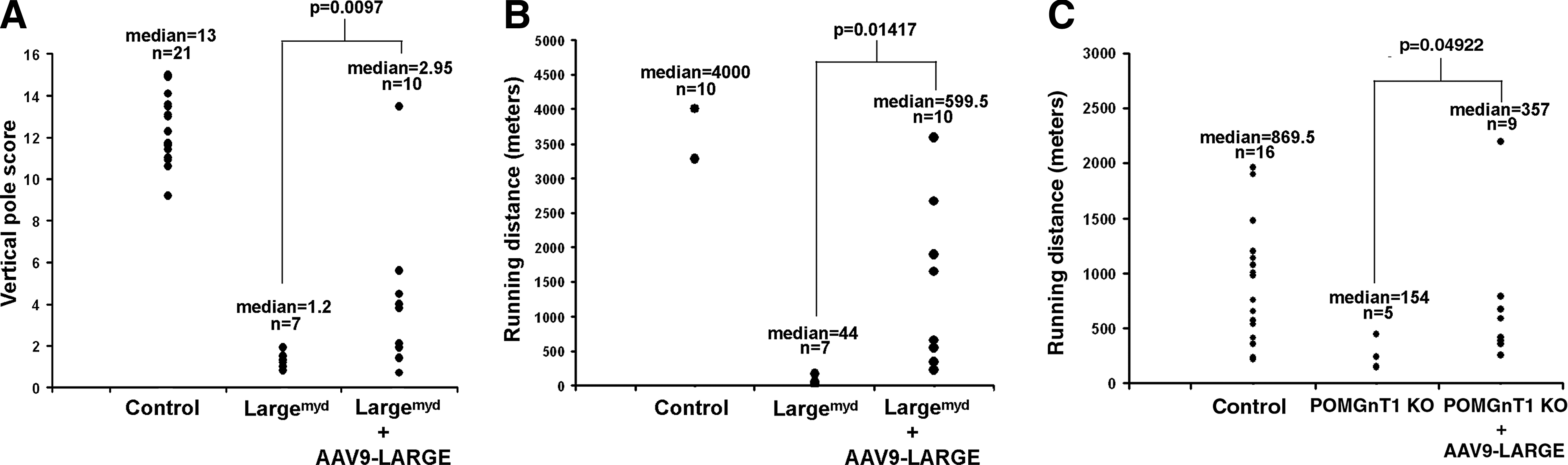
Improved motor function in Largemyd and POMGnT1 knockout mice after AAV9-LARGE virus treatment. Three- to five-month-old mice were evaluated for their performance on vertical pole test and treadmill running.
On the treadmill, Largemyd mice exhibited poor running ability when compared to control mice (Fig. 8B). While most control mice (wild-type and heterozygous) can run much longer than 4000 meters (the test was stopped at 4000 m), the median running distance for Largemyd mice was only 44 meters with a mean of 60.5 meters. AAV9-LARGE treatment improved the running capability of Largemyd mice with a median running distance of 599.5 meters with a mean distance of 1205 meters (p=0.01417, Student's t Test).
POMGnT1 knockout mice did not exhibit apparent deficits in the vertical pole test (p=0.28393). They also performed much better than Largemyd mice on the treadmill; thus the treadmill speed was increased to 20 meters/min. However, their running distance was significantly reduced from littermate control (wild-type and heterozygous) mice (Fig. 8C). The median running distance for POMGnT1 knockout mice was 154 meters with a mean of 224 meters. AAV9-LARGE treatment improved running ability to a median of 357 meters with a mean of 618 meters (p=0.04922, Student's t test). These results indicate that the overall motor performance of Largemyd and POMGnT1 knockout mice improved upon AAV9-LARGE treatment.
Discussion
The discovery that overexpressing LARGE promotes recovery of IIH6C4 immunoreactive and laminin-binding protein species in cells deficient in LARGE, as well as those with mutations in other CMD-causing genes, provided the rationale to use LARGE in gene therapy for all congenital muscular dystrophies caused by defective α-DG glycosylation. This study tested the feasibility of AAV9-mediated delivery of LARGE gene therapy in two models of congenital muscular dystrophy: POMGnT1 knockout and Largemyd mice. Systemic delivery of AAV9-LARGE virus in the newborn led to partial recovery of IIH6C4 immunoreactivity in the skeletal muscle and ligand-binding activity in both mouse models. The severity of the muscular dystrophic phenotype was significantly ameliorated.
This study provides proof-of-concept that LARGE gene therapy mediated by AAV9 vectors can improve the muscle histology. Additionally, AAV9-LARGE-treated Largemyd mice showed significantly improved muscular function as revealed by improved performance on a vertical pole test and treadmill running. Interestingly, AAV9-EGFP vector transduced the diaphragm more efficiently than the quadriceps muscle. Consistent with this, the muscular dystrophic phenotype was ameliorated more significantly in the diaphragm than in the quadriceps muscle. This has significant implications because respiratory failure is a major cause of death for CMD patients and rescue of dystrophy in the diaphragm would be helpful to prevent respiratory failure. Thus, if successfully translated into clinics, AAV9-LARGE gene therapy has the potential to improve muscular functions of CMD patients of dystroglycanopathies and significantly lengthen their lifespan.
The phenotypes previously reported for POMGnT1 knockout (Liu et al., 2006) and Largemyd mice (Holzfeind et al., 2002) were not readily comparable because of differences in genetic background. In this study, POMGnT1 knockout allele and Largemyd allele were crossed into the same genetic background. The muscular dystrophic phenotype in Largemyd mice was more severe than in POMGnT1 knockout mice, a finding not surprising given that there was residual ligand binding activity by α-DG in skeletal muscles of POMGnT1 knockout but not in those of Largemyd mice (Kanagawa et al., 2009). However, POMGnT1 knockout mice die more readily during the early postnatal period. The differential phenotypic expression in severity of muscular dystrophy and postnatal survival may reflect genetic redundancy of Large with Large2. While Large2 is expressed in organs other than brain, eye, and muscle (Fujimura et al., 2005), there is no known protein O-mannose β1,2 N-acetylglucosaminyltransferase activities in addition to POMGnT1.
It has been assumed that the only IIH6C4 immunoreactive and ligand-binding hyperglycosylated protein species produced by LARGE overexpression is α-DG (Barresi et al., 2004; Brockington et al., 2005; Fujimura et al., 2005; Patnaik and Stanley, 2005; Bao et al., 2009). However, LARGE expression causes glycosylation of proteins other than α-DG since overexpressing LARGE in clonally expanded neural stem cells deficient in DG led to glycosylation of proteins, which were recognized by the IIH6C4 antibody (Zhang et al., 2011). It will be interesting to determine whether LARGE overexpression in skeletal muscle fibers caused glycosylation of proteins other than α-DG and whether their glycosylation by LARGE overexpression played any role in the apparent amelioration of the skeletal muscular dystrophy phenotype.
LARGE synthesizes laminin-binding repeating disaccharide units of [–3-xylose–α1,3-glucuronic acid-β1–] (Inamori et al., 2012). Some studies indicate that LARGE overexpression leads to modifications of multiple glycans including O-linked mannosyl glycans, complex N-, and mucin O-glycans of α-DG (Patnaik and Stanley, 2005; Aguilan et al., 2009). Others indicate that LARGE modifies a phosphoryl glycosylation branch on O-linked mannose (Yoshida-Moriguchi et al., 2010). We have found that LARGE overexpression causes differential glycosylation of α-DG and other proteins. Importantly, glycosylation of α-DG by LARGE requires the presence of O-mannose (Zhang and Hu, 2012). However, proteins other than α-DG can be glycosylated on O-mannose as well as N-glycans (Zhang and Hu, 2012). In this study, AAV9-LARGE treatment resulted in glycosylation of α-DG in POMGnT1 knockout and Largemyd mice as expected. Phenotypic rescue thus involves hyperglycosylation of α-DG. Since hyperglycosylation of non-α-DG proteins by LARGE overexpression is capable of mediating laminin binding, we speculate that phenotypic rescue may also involve hyperglycosylation of other proteins as well. Identifying these proteins and their contribution to phenotypic rescue will be addressed in the future. Future studies will also need to address whether LARGE overexpression ameliorates phenotypes in O-mannosyl glycosylation deficiency.
For FCMD mutations, pathogenic exon-trapping by retrotransposon insertion into the 3’-untranslated region of the FKTN gene can be corrected in vitro by antisense oligonucleotides against splicing sites (Taniguchi-Ikeda et al., 2011). Although only transient, local adenoviral transfer of LARGE in Largemyd mice recovers functional glycosylation and rescues dystrophic phenotype (Barresi et al., 2004). Together, these results reinforce the idea of the feasibility of gene therapy for congenital muscular dystrophies. Systemically injected AAV9 vectors can enter the central nervous system (CNS) to infect neurons in the brain and the retina in addition to skeletal muscle (Foust et al., 2009). They are ideal vehicles for gene therapy in congenital muscular dystrophy with CNS involvement. In addition, it has been reported that LARGE overexpressing transgenic lines do not have any phenotypic abnormalities (Brockington et al., 2010). Recently, a very interesting report demonstrates that transgenic overexpression of LARGE in the differentiated myofibers rescues muscular dystrophy phenotype and restores function at the neuromuscular junction (Gumerson et al., 2012). In this study, AAV9-LARGE virus infection in the newborn POMGnT1 knockout and Largemyd mice ameliorated their muscular dystrophic phenotype. Future studies are needed to determine whether LARGE gene therapy would ameliorate phenotypes in the brain and the retina and in other mouse models of dystroglycanopathy and whether it can reverse muscular dystrophy in adults.
Materials and Methods
Animals
Protocols for using laboratory mice were approved by the Institutional Committee for Use and Care of Laboratory Animals at State University of New York Upstate Medical University and adhered to the guidelines of the National Institutes of Health. All efforts were made to minimize pain and suffering of animals.
Largemyd mice were obtained from Jackson Laboratories (Bar Harbor, ME). POMGnT1 knockout mice were generated at Lexicon Genetics (The Woodlands, TX) (Liu et al., 2006). Previously reported POMGnT1 knockout (Liu et al., 2006) and Largemyd (Holzfeind et al., 2002) mice were on different genetic backgrounds, and thus the severity of muscular dystrophic phenotypes was not readily comparable. To obtain animals with similar genetic background, heterozygous POMGnT1+/− and Large+/− mice were crossed to obtain double heterozygous (POMGnT1+/−Large+/−) mice. Animals used for this study were generated by crosses of double heterozygotes. Out of 463 newborn mice genotyped from P0–10, 8 were POMGnT1−/−Large−/− (double mutant) mice, a frequency far below the expected 1/16 with none surviving to 1 month (only litters in which every newborn was genotyped were included in this tally). Twenty-three were POMGnT1−/−Large+/+or+/− (POMGnT1 knockout) mice, a frequency also well below the expected one-fourth, and 11 survived to adulthood. Sixty-six were POMGnT1+/+or+/−Large−/− (Largemyd) mice and most survived to adulthood. Three hundred and sixty-six were POMGnT1+/+or+/−Large+/+or+/− mice at the expected frequency with no observable abnormality in histology and behavior and thus were used as controls. All experiments were performed in POMGnT1 knockout (POMGnT1−/−Large+/− or+/+), Largemyd (POMGnT1+/− or+/+Large−/−), and control (POMGnT1+/− or+/+Large+/− or+/+) animals.
To measure running capability, mice were placed into individual lanes of a treadmill (Model 1055SRM Exer-3/6; Columbus Instruments, Columbus, OH). The animals were acclimated to the treadmill with the belt and shock grids off for 10 min. The shock grids were then turned on and the belt started at 5 meters/min for 5 min for the mice to warm up. The speed was then ramped up at the acceleration of 1 meter/min2 to 10 meters/min for Largemyd mice and to 20 meters/min for POMGnT1 knockout mice. When a mouse spent greater than 3 sec on the shock grid without attempting to run, the shock grid was turned off and running distance recorded.
The vertical pole test was carried out similarly to Paylor et al. (1998) with minor changes. A pole 1.75 cm in diameter and 50 cm in length was covered by cloth tape. The mice were placed at one end of the horizontally placed pole, which was then shifted to vertical position. The performance of the mice was scored as follows: fell before the pole reached halfway to vertical position=0; fell before the pole reached vertical position=1; after reaching vertical position, fell in 0–10 sec=2, 11–20 sec=3, 21–30 sec=4, 31–40 sec=5, 41–50 sec=6, 51–60 sec=7; remained on for 60 sec but climbed down on top half of the pole=8; climbed down to the lower half of the pole=9; climbed all the way down the pole and got off the pole in 51–60 sec=10, 41–50 sec=11, 31–40 sec=12, 21–30 sec=13, 11–20 sec=14, 1–20 sec=15.
Viral vectors
Adeno-associated viral vectors (serotype 9) for expression of enhanced green fluorescent protein (EGFP) (AAV9-EGFP, 3.2×1013 GC/ml) and LARGE (AAV9-LARGE, 2.6×1013 GC/ml) driven by the chicken β-actin promoter were constructed at Vector Biolabs (Philadelphia, PA) on a fee-for-service basis.
Animal surgery and vector injection
For intracardial injection at newborn, postnatal day 2–5 animals were anesthetized by hypothermia on ice. A Monoject insulin syringe fitted with a 28G needle (The Kendall Company, Mansfield, MA) was used to inject 50 μl of the viral vector directly into the heart. The injected mice were returned to the dam after recovery under a heat lamp.
For tail vein injection into adult animals, mice were placed in a restraint after being warmed by a heat lamp for about 5 min. The tail was cleaned by swabbing with 70% alcohol and then injected intravenously with 100 μl of the viral solution. The animals were returned to their cages after injection.
Histology
For analysis of viral transduction, the adult animals were deeply anesthetized by an overdose of pentobarbital (100 mg/kg body weight) and sacrificed by intracardial perfusion of 4% paraformaldehyde. Some tissues were cut into 200 μm thick sections using a vibratome. For frozen sections, fixed tissues were equilibrated with 30% sucrose for cryoprotection. Frozen tissues were cut into 10-μm-thick sections with a Cryostat and mounted onto Superfrost plus slides (Fisher Scientific, Pittsburgh, PA). For histological evaluation, the sections were fixed with 4% paraformaldehyde and stained with hematoxylin and eosin (H&E). For observation of GFP fluorescence, some sections were counterstained with 4',6-diamidino-2-phenylindole (DAPI). Fluorescence images were acquired with a Zeiss Axioskop upright fluorescence microscope equipped with a digital camera (Carl Zeiss Microimaging, Inc., Thornwood, NY). For nuclei examination, images were captured with a 40X objective.
For histological analysis of muscle of animals treated by AAV9-LARGE virus, mice were deeply anesthetized by pentobarbital followed by rapid decapitation. Quadriceps and diaphragm muscles were frozen in optical cutting temperature media and cut into 10-μm-thick sections. The sections were mounted onto Fisherbrand slides and stained by H&E.
Antibodies
Antibodies were obtained as follows: monoclonal anti-tenascin-C, collagen I β-DG (43DAG1/8D5) from Abcam (Cambridge, MA); polyclonal anti-laminin-1 from Sigma-Aldrich (St. Louis, MO); monoclonal IIH6C4, monoclonal VIA4-1, monoclonal anti-developmental myosin heavy chain (MHCd), monoclonal rat antineural cell adhesion molecule (NCAM), and monoclonal anti-BrdU from Millipore Corporation (Billerica, MA); monoclonal anti-β-DG (clone 7D11) from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA); and polyclonal anti-pikachurin from Wako Chemicals Inc. (Richmond, VA).
Immunofluorescence staining
The skeletal muscle sections were fixed with 4% paraformaldehyde for 30 min. Primary and secondary antibody incubation was performed as described previously (Hu, 2000; Hu et al., 2007). Briefly, the sections were incubated with 0.5% H2O2 for 1 hr, blocked with 3% bovine serum albumin (BSA) in phosphate buffer (pH 7.4) for 1 hr, and then incubated with primary antibodies including anti-β-DG (1 to 200 dilution), anti-laminin-111 (1 to 1,000 dilution), VIA4-1 (1 to 2000 dilution), anti-NCAM (1 to 300 dilution), anti-MHCd (1 to 300 dilution), or anti-tenascin-C (1 to 300 dilution) at 4°C overnight. After washing with phosphate buffer containing 0.1% Triton X-100, the sections were then incubated with fluorescein isothiocyanate-conjugated goat anti-mouse IgG or IgM or goat anti-rabbit IgG at 1 to 400 dilutions. After washing with phosphate buffer containing 0.1% Triton X-100, the sections were counterstained with DAPI (Sigma-Aldrich) to visualize nuclei. Some slides were processed without incubation with primary antibodies as controls.
Cell proliferation was identified by using BrdU immunolabeling. Briefly, mice were injected with BrdU (100 μg/g body weight) intraperitoneally. To identify cells in the S phase of proliferation, mice were sacrificed 3 hr after BrdU injection. For immunodetection of BrdU-labeled nuclei, sections were treated with 4 N HCl for 10 min at room temperature (RT) to denature DNA, neutralized twice for 10 min with 0.5 mM sodium borate buffer (pH 8.5). After blocking for 1 hr with 1.0% BSA in Tris-buffered saline (TBS), the sections were incubated with a mouse monoclonal IgG anti-BrdU antibody overnight at 4°C. Sections were then washed in phosphate buffered saline (PBS) and incubated for 2 hr with 1:200 RITC-conjugated rabbit anti-mouse IgG antibody and counterstained for 10 min with 0.10% DAPI (Sigma-Aldrich).
Western blot analysis and ligand overlay experiments
Leg-muscle (thigh) samples were homogenized in cold lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% TritonX-100, pH 7.4) supplemented with protein inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) and centrifuged at 16,100 g for 20 min at 4°C. The supernatants were collected. Glycoproteins were isolated by wheat germ agglutinin (WGA)-affinity gel purification from quadriceps muscle lysate. For 2 mg of total lysate proteins, 50 μl of WGA-agarose (EY Laboratories, San Mateo, CA) was added and incubated for 4 hr at 4°C. The WGA-gel was then washed three times with the lysis buffer. Bound glycoproteins were eluted by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, separated on SDS-PAGE, and electrotransferred onto polyvinylidene fluoride (PVDF) membranes.
For detection of α-DG with IIH6C4 and β-DG with monoclonal antibody MANDAG2-7D11, standard immunoblotting procedures were carried out on PVDF membranes blotted with WGA-gel isolated proteins. Briefly, the PVDF membranes were blocked with 3% BSA in Tris-buffered saline with Tween (TBST) (50 mM Tris, pH 7.4, 150 mM NaCl, 0.05% Tween-20) for 30 min and incubated with primary antibodies in TBST containing 3% BSA for 2 hr and washed with TBST. The membranes were then incubated with Goat anti-mouse IgM (or IgG) conjugated with horseradish peroxidase (1:3000) for 45 min. After extensive washing with TBST, the signal was visualized with SuperSignal west pico chemiluminescent substrate (Thermo Scientific, Rockford, IL).
For the laminin-overlay assay, laminin-1 (Invitrogen, Carlsbad, CA) was biotinylated (Vector Labs). PVDF membranes were blocked with Tris-buffered saline (TBS, 50 mM Tris, pH7.4, 150 mM NaCl) containing 3% BSA, 1 mM CaCl2, and 1 mM MgCl2 for 1 hr. The membranes were then incubated with 1.25 μg/ml biotinylated laminin-1 in TBST containing 1 mM CaCl2 and 1 mM MgCl2 overnight at 4°C. After extensive washing, the membrane was incubated with peroxidase-conjugated streptavidin for 1 hr and washed. The signal was visualized as above.
For the pikachurin overlay assay, the PVDF membrane was incubated with 3% BSA in Tris-buffered saline (50 mM Tris, pH 7.4, 150 mM NaCl, 1mM CaCl2, 1 mM MgCl2) followed by incubation with medium conditioned by HEK 293 cells expressing pikachurin (Hu et al., 2011) overnight at 4°C. After washing with TBS, membrane was incubated with a rabbit antibody against pikachurin (1:2000) for 2 hr and washed. The membrane was then incubated with goat anti-rabbit IgG conjugated with horseradish peroxidase (1:3000) for 45 min and washed. After extensive washing with TBST, the signal was visualized with SuperSignal west pico chemiluminescent substrate.
For immunoprecipitation with VIA4-1 and anti-β-DG antibodies, 3 mg of total protein lysates was mixed with 2 μg of VIA4-1 or anti-β-DG antibody and incubated for 2 hr at 4°C before adding 10 μl of protein G beads (Thermo Scientific, Rockford, IL). After gentle mixing overnight, beads were washed three times with washing buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% Triton X-100) at 4°C. Bound proteins were eluted from the beads with 5X SDS-PAGE loading dye, boiled for 5 min, separated by 7.5% SDS-PAGE, electrotransferred onto PVDF membranes, and detected with IIH6C4 antibody as described above.
Statistical analyses
Percentage of central nuclei was analyzed by Student's t-test. Chi-square analysis of original counts of nuclear location yielded similar results. Statistical analysis of vertical pole test scores was carried out by Mann-Whitney U test. Treadmill running results were analyzed by Student's t-test.
Footnotes
Acknowledgments
The authors thank Ms. Jing Li and Mr. Matthew Bauer for technical assistance in this project, Drs. Eric Olson and Mary Lou Vallano for critical reading of the manuscript, and Ms. Bonnie Lee Howell for editing the manuscript. Monoclonal anti-β-DG (clone 7D11) was obtained from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). This research was supported by National Institutes of Health grants (HD060458 and NS066582 to H.H.).
Author Disclosure Statement
The authors have no conflict of interest to disclose.