
Abstract
Inefficient alveolar wound repair contributes to the development of pulmonary fibrosis. Hepatocyte growth factor (HGF) is a potent growth factor for alveolar type II epithelial cells (AECII) and may improve repair and reduce fibrosis. We studied whether targeted gene transfer of HGF specifically to AECII improves lung fibrosis in bleomycin-induced lung fibrosis. A plasmid encoding human HGF expressed from the human surfactant protein C promoter (pSpC-hHGF) was designed, and extracorporeal electroporation-mediated gene transfer of HGF specifically to AECII was performed 7 days after bleomycin-induced lung injury in the rat. Animals were killed 7 days after hHGF gene transfer. Electroporation-mediated HGF gene transfer resulted in HGF expression specifically in AECII at biologically relevant levels. HGF gene transfer reduced pulmonary fibrosis as assessed by histology, hydroxyproline determination, and design-based stereology compared with controls. Our results indicate that the antifibrotic effect of HGF is due in part to a reduction of transforming growth factor-β1, modulation of the epithelial–mesenchymal transition, and reduction of extravascular fibrin deposition. We conclude that targeted HGF gene transfer specifically to AECII decreases bleomycin-induced lung fibrosis and may therefore represent a novel cell-specific gene transfer technology to treat pulmonary fibrosis.
Introduction
Various cellular and molecular mechanisms have been suggested to be critical in the pathogenesis of pulmonary fibrosis. Some evidence suggests the epithelial–mesenchymal transition (EMT) as an important mechanism that may contribute to the accumulation of fibroblasts and differentiation to myofibroblasts in pulmonary fibrosis (Willis et al., 2005). More recently, persistent activation of intraalveolar procoagulant activity and increased deposition of extravascular fibrin have been shown to increase the fibrotic response in the IPF lung (Antoniou et al., 2007).
Repeated injuries to alveolar type II epithelial cells (AECII) (Sisson et al., 2010) and their failure to reepithelialize is thought to be one of most important factors triggering the development of IPF. In the alveolar epithelial repair process many different cytokines and growth factors are involved. Hepatocyte growth factor (HGF) is a potent mitogenic factor in various organs including the lung (Tsao et al., 1993; Singh-Kaw et al., 1995). In the lung HGF plays an important role both in the developmental stage and in the adult lung; during fetal lung development HGF is implicated in the regulation of mesenchyme–epithelium interactions (Itakura et al., 1997). HGF is a pulmotrophic factor, as it stimulates proliferation of respiratory epithelium during postpneumonectomy compensatory lung growth (Sakamaki et al., 2002). Furthermore, HGF has antiapoptotic effects on epithelial cells (Bardelli et al., 1996) and induces proliferation of AECII. It is therefore a promising candidate to support the repairing capacities of AECII and to improve alveolar epithelial repair in the injured lung. We showed that electroporation-mediated gene transfer of HGF directly to the bleomycin-injured lung reduces pulmonary fibrosis (Gazdhar et al., 2007).
However, to increase efficiency of electroporation-mediated gene transfer and to minimize possible side effects of this treatment a more targeted technique of gene transfer is needed. We therefore developed a noninvasive gene transfer technique that allows us to specifically target AECII by using a surfactant protein C promoter system, and present data showing a marked reduction in pulmonary fibrosis in the bleomycin-induced lung fibrosis model after cell-specific hHGF gene transfer. Moreover, we present possible mechanisms that may contribute to the beneficial effect induced by cell-specific HGF gene transfer to AECII in this model.
Materials and Methods
Plasmid
The plasmid pSpC-hHGF was constructed by inserting human HGF cDNA (2.1 kb) into the backbone of the 3.7-kb human SpC promoter, the reverse tetracycline-controlled trans-activator (rtTA) coding sequence, and 0.45-kb simian virus 40 (SV40) poly(A). Empty vector pSpC (not carrying hHGF) served as control. Ninety percent homology between rat and human HGF has been reported (Liu et al., 1993) and hence human HGF was chosen. For gene delivery, the plasmid was suspended in endotoxin-free water. More details are provided in the online supplement (supplementary data are available online at
Animals
Male Fischer F344 rats (220–240 g) were obtained from Charles River Laboratories (Sulzfeld, Germany). Experiments were performed in accordance with the standards of the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (
Instillation of bleomycin
On day 1 of the protocol, F344 rats (220–240 g) were anesthetized by inhalation of 4% isoflurane in a glass chamber, intubated with a 14-gauge intravenous catheter (Insyte, Madrid, Spain), and instilled intratracheally with bleomycin (1.28 U/rat) in a volume of 500 μl to both lungs.
In vivo HGF gene transfer
Seven days after intratracheal instillation of bleomycin, the animals were randomly divided into two groups (each, n=10). One group was treated by intratracheal administration of the empty vector, pSpC (control group), the second group with the plasmid carrying the HGF sequence, pSpC-hHGF (350 μl, 1 μg/μl) followed by extracorporeal electroporation of both lungs, using a pulse generator (Inovio, Oslo, Norway). Additional details are provided in the online supplement.
Assessment
All animals were killed on day 14 as described below; n=4 from each group were taken for stereology, and n=6 each were taken for histology and biochemical analysis. On day 14 (7 days after hHGF gene transfer) animals were anesthetized, thiopental (50 mg/kg body weight) was administered intraperitoneally, and the animals were ventilated via a tracheostomy with a rodent ventilator (Harvard Apparatus, Holliston, MA). Except for lungs assigned to design-based stereology, the pulmonary vessels were flushed with 20 ml of 0.9% saline under pressure (20 cmH2O). The heart–lung block was explanted and tissue samples were collected for further analysis.
Histology immunohistochemistry, and immunofluorescence
After routine hematoxylin and eosin staining, tissue sections were evaluated with the scoring system of Ashcroft and colleagues (1988) to evaluate the extent of fibrosis by a trained pathologist blinded to the study. Formalin-fixed tissue sections were deparaffinized in a xylene series and rehydrated through a decreasing ethanol series. Sections were then incubated with goat anti-human HGF antibody, rabbit polyclonal to E-cadherin, rabbit polyclonal to α-smooth muscle actin, or rabbit polyclonal to SpC at appropriate dilutions overnight at 4°C followed by appropriate second antibody and stained. For better visualization of the costaining, double-immunofluorescence imaging was performed and observed under a confocal microscope. More details are provided in the online supplement.
In situ hybridization
Tissue sections were subjected to proteinase K treatment, prehybridized, and later hybridized with either sense or antisense complementary RNA (cRNA) digoxigenin (DIG)-labeled probes in a humid chamber at 55°C for 18 hr. Detection was achieved with an alkaline phosphatase-conjugated anti-DIG antibody. More details are provided in the online supplement.
Hydroxyproline assay
Lungs were analyzed for collagen content as initially described by Woessner (1961). A standard curve was generated on the basis of known concentrations of reagent-grade hydroxyproline (Sigma-Aldrich, St. Louis, MO). More details are provided in the online supplement.
Human HGF rat HGF, plasminogen activator inhibitor-1, and transforming growth factor-β1 determination
Multiplex assay system to measure HGF, vascular endothelial growth factor, and plasminogen activator inhibitor-1 levels: For HGF levels a Bio-Plex suspension array system (Bio-Rad Laboratories, Basel, Switzerland) was used. Plasminogen activator inhibitor-1 (PAI-1) and vascular endothelial growth factor (VEGF) were measured with a Milliplex MAP (multianalyte profiling) rat cardiovascular disease (CVD) panel 1 kit and 24-plex panel (Millipore, Bedford, MA), respectively, in accordance with the manufacturer's instructions. More details are provided in the online supplement.
ELISA for rat HGF and transforming growth factor-β1: Lung homogenates were analyzed for rat HGF and transforming growth factor (TGF)-β1 measurements, and ELISA kits for TGF-β1 (R&D Systems, Abingdon, UK) and rat HGF (B-Bridge, Cupertino, CA) were used in accordance with the manufacturers' protocols. HGF and TGF-β1 levels were normalized for protein content values and are given as picograms per milligram of total lung protein.
Stereological analysis light and electron microscopy assessments
All methods applied in this study regarding the quantification of pulmonary structure were based on the American Thoracic Society/European Respiratory Society (ATS/ERS) standards for quantitative morphological analysis of the lung (Hsia et al., 2010). Whole lungs (four lungs per group) were used for unbiased stereology. Using a cascade sampling design, lung structure was analyzed at various levels with increasing magnification to quantify the severity of pulmonary fibrosis (Ochs, 2006; Knudsen and Ochs, 2011). A detailed description, including a definition of each stereological parameter, is included in the online supplement.
MSB staining technique for fibrin
To stain fibrin on the paraffin sections, the standard MSB (Martius yellow, scarlet, and blue) technique was applied as described by Lendrum and colleagues (1962). The sections were dewaxed and a celestin blue–hematoxylin sequence was used to stain the nuclei. More details are provided in the online supplement.
Real-time RT-PCR analysis
For RT-PCR, probes labeled with fluorophores 6-carboxyfluorescein (FAM) and tetramethylrhodamine (TAMRA) (Microsynth, Balgach, Switzerland) were used for detection of urokinase plasminogen activator (uPA). Fold change was subsequently calculated according to the formula 2–ΔΔCp. More details are provided in the online supplement.
Statistical analysis
If not otherwise stated, data are presented as means±SD. Statistical analysis was done by the nonparametric Mann–Whitney rank sum test, using GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA). The results were considered significant at p<0.05. A nonparametric Spearman test for correlation followed by a linear regression in case of statistical significance was performed for correlation of human hHGF and TGF-β1 levels.
Results
hHGF is specifically expressed in alveolar type II epithelial cells after electroporation-mediated hHGF gene transfer in vivo
Extracorporeal electroporation-mediated hHGF gene transfer was performed 7 days after intratracheal bleomycin instillation, using pSpC-hHGF. In situ hybridization and immunohistochemistry performed 7 days after electroporation-mediated hHGF gene transfer showed expression of hHGF at the RNA and protein levels. Both mRNA (Fig. 1a, inset: sense probe) and hHGF protein expression (Fig. 1b) were detected and specifically expressed in AECII as shown by costaining experiments performing surfactant C staining (Fig. 1c–e). In accordance with the stereological data (see below) and as shown in Fig. 1e, not all AECII were transfected with hHGF.
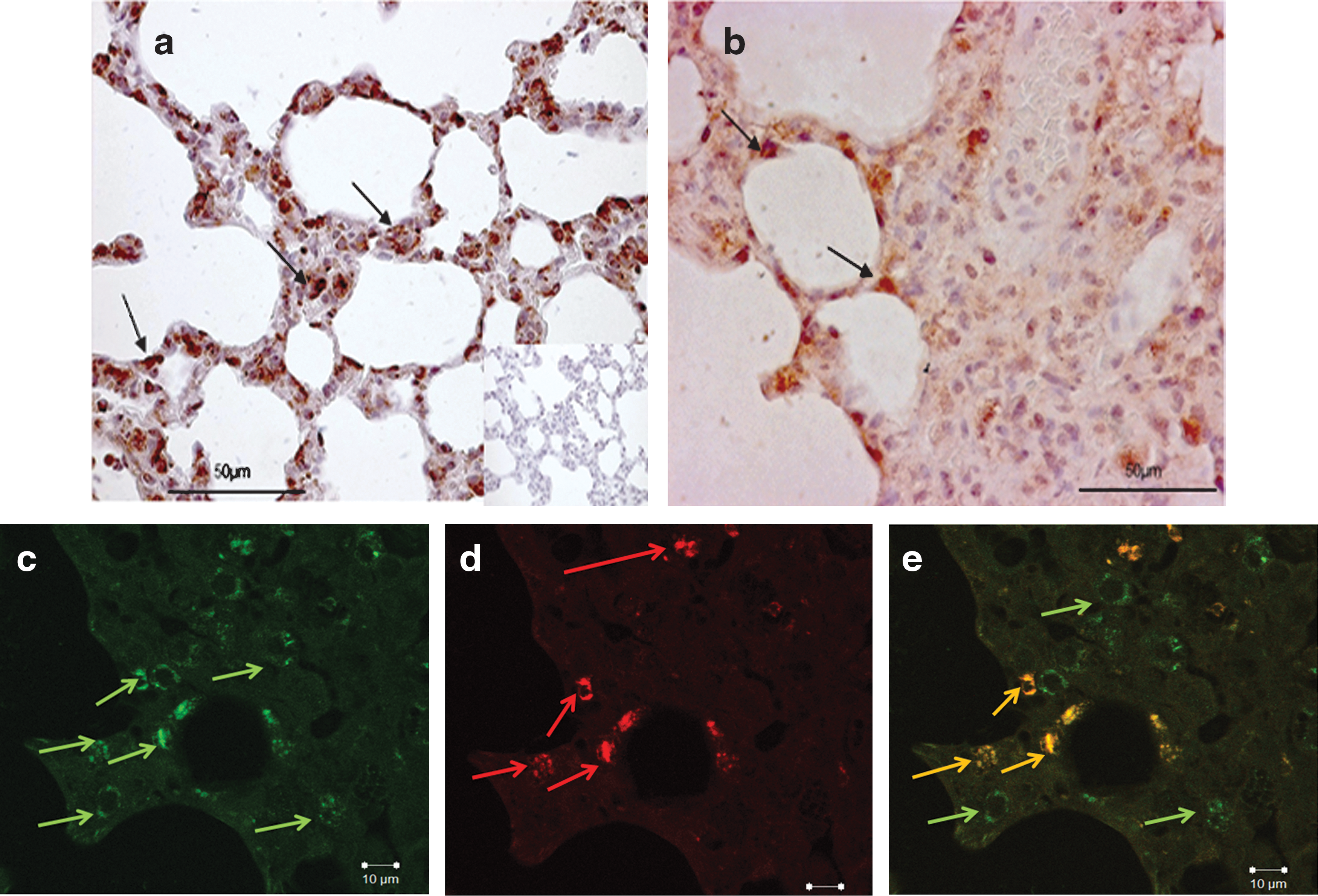
hHGF mRNA and protein expression in bleomycin-injured rat lung after electroporation-mediated cell-specific gene transfer of hHGF (pSpC-hHGF).
hHGF levels in lung homogenates from pSpC-hHGF- and pSpC-treated rats were measured to quantify hHGF expression in bleomycin-injured lung. In pSpC-hHGF-treated rat lungs, the mean hHGF level was 0.33±0.24 pg/mg of total lung protein, whereas no hHGF could be detected in rats treated with the empty vector (pSpC) 7 days after electroporation-mediated hHGF gene transfer (p<0.01). Rat HGF levels were also increased in the treatment group (26.43±24.50 vs. 9.90±3.23 pg/mg of total lung protein in the control group) as measured by ELISA (mean±SD).
Cell-specific hHGF gene expression in alveolar type II epithelial cells reduces lung fibrosis
Targeted cell-specific hHGF gene transfer markedly reduced lung fibrosis compared with the control group as assessed 14 days after bleomycin instillation and 7 days after hHGF gene transfer. Whereas severe inflammation and fibrotic changes were detected in the lungs of control animals (Fig. 2a), considerable improvement could be observed in hHGF-treated animals (Fig. 2b). The fibrotic changes in the lung were further quantified using the Ashcroft score. Cell-specific gene transfer of hHGF reduced the score from 3.8±0.7 in the control group to 2.2±1.1 in the hHGF-treated group (p=0.02) (mean±SD) (Fig. 2c). In accordance, the collagen content, as assessed by hydroxyproline assay, was significantly reduced in the animals treated by cell-specific hHGF gene transfer compared with the control group (1663±1746 vs. 3829±1694 μg/gm of wet lung tissue; p=0.035) (mean±SD) (Fig. 2d).
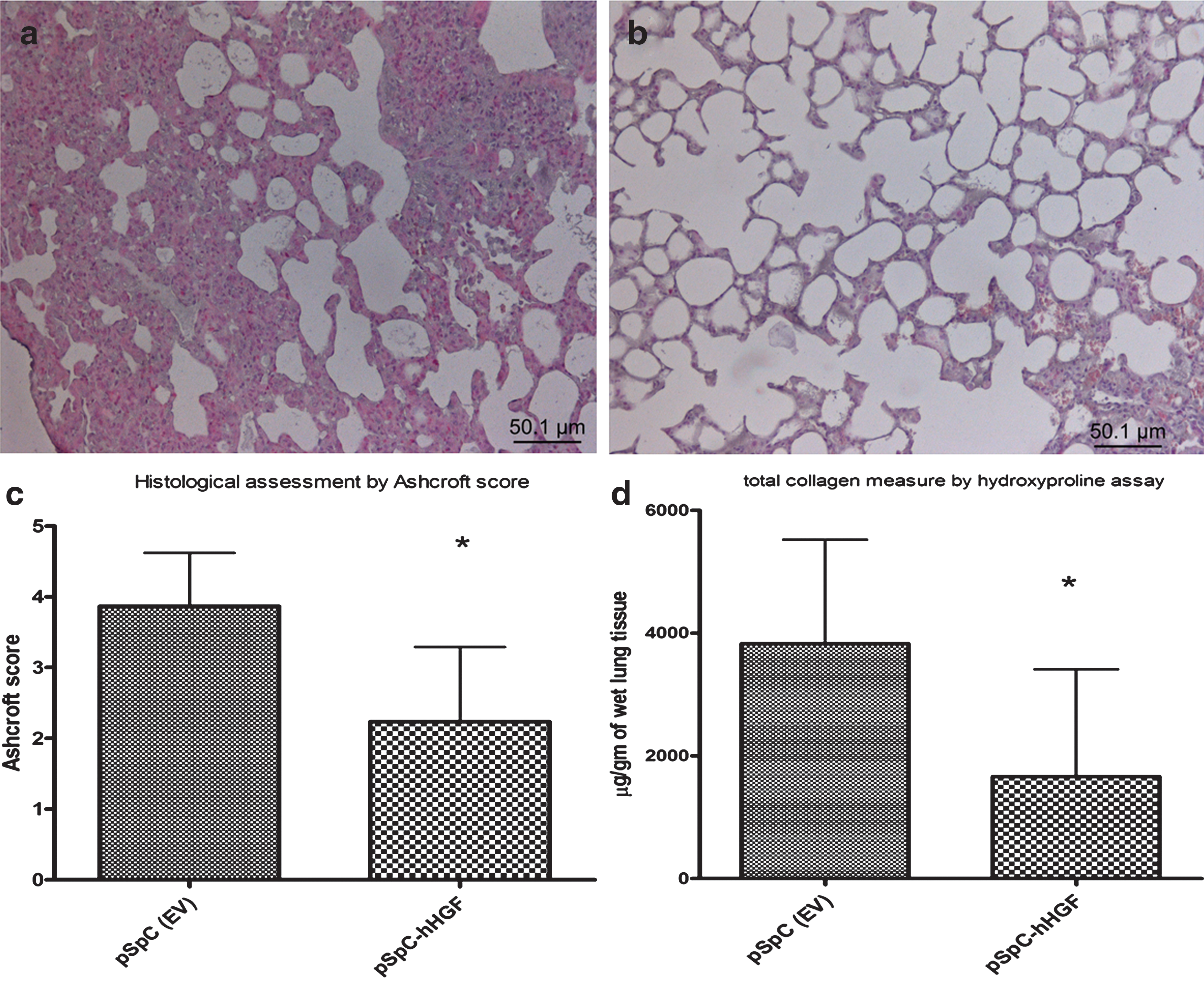
Electroporation-mediated cell-specific gene transfer of hHGF reduces bleomycin-induced pulmonary fibrosis.
Stereological analysis
General lung architecture
Stereological data are given in Supplementary Table S1. The volume fraction of septal wall tissue [V
V(sep/par)] as well as the mean thickness of alveolar septal walls [
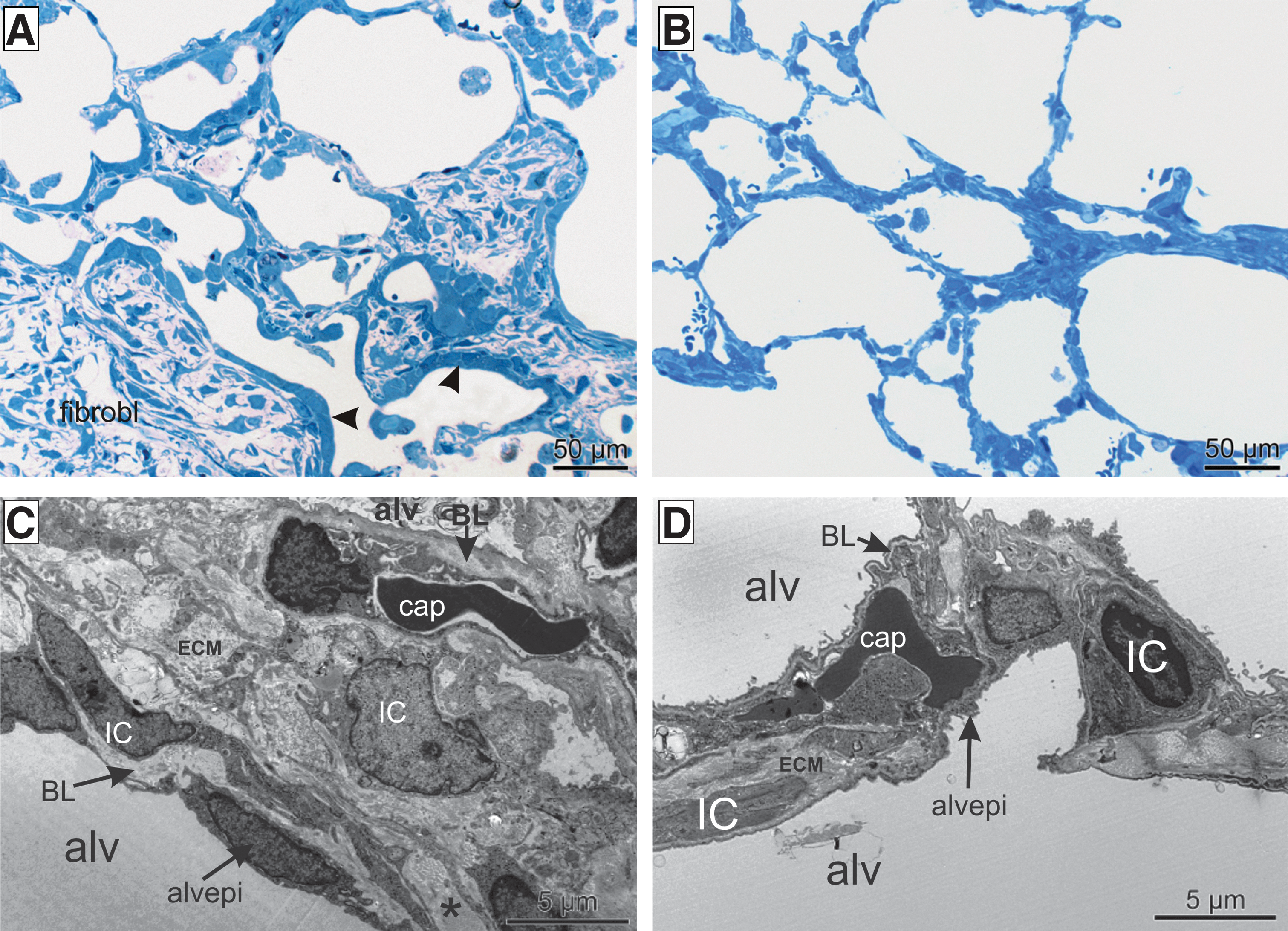
Light
Detailed stereological data: Treatment by targeted cell-specific hHGF gene transfer led to a significant reduction in alveolar septal wall
Parameters related to alveolar type II epithelial cells
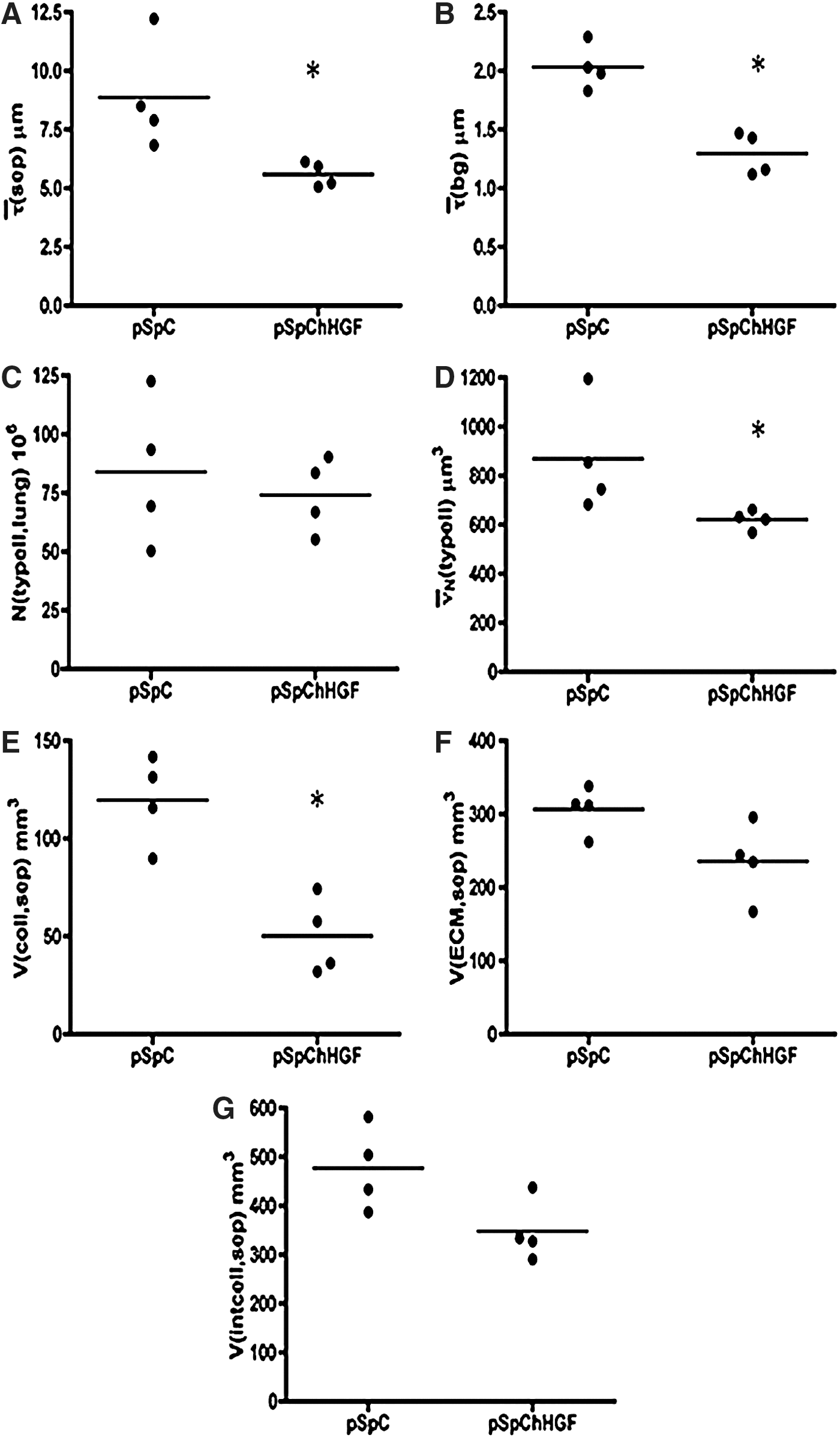
Stereological data are summarized in Supplementary Table S2. Neither the total number of AECII per lung [N(typeII,lung)] (Fig. 4C) nor the total surface area of alveolar epithelium covered with AECII [S(typeII,lung)] differed between the two groups. However, the number-weighted mean volume of AECII [
Parameters related to alveolar septal walls
Stereological data are summarized in Supplementary Table S3. The composition of the alveolar septa was analyzed to identify the septal wall components that are responsible for the reduction in thickness of the alveolar septa after cell-specific hHGF gene transfer (Fig. 4E–G). The total amount of collagen fibrils [V(coll,sep)] was significantly reduced within the alveolar septal walls after hHGF gene transfer (Fig. 4E). Regarding the amount of ECM [V(ECM,sep)] and interstitial cells [V(intcell,sep)], there was a trend toward reduction (Fig. 4F and G). Neither the total volume of the alveolar epithelium [V(alvepi,sep] nor the total volume of capillaries [V(cap,sep)] differed between the two groups. Taken together, the reduction in the thickness of the alveolar septal walls in the hHGF-expressing group was due to a reduction in the interstitial components, in particular the ECM and collagen fibrils. Furthermore, a statistically significant decrease in the blood–gas barrier thickness [
hHGF reduces TGF-β1 levels in the lungs
Because TGF-β1 is a major profibrotic factor, we determined TGF-β1 levels in the lung tissue homogenate after HGF gene transfer. TGF-β1 levels were reduced in animals treated with pSpC-hHGF compared with the control animals (10.7±6.52 vs. 27.3±12.80 pg/mg of total lung protein; p<0.05) (mean±SD). Moreover, we found a clear correlation between hHGF and TGF-β1 levels after hHGF gene transfer (p<0.01, correlation coefficient r=–0.98; linear regression R 2=0.81) (Fig. 5), indicating that reduced TGF-β1 levels may contribute to the antifibrotic effect of HGF after cell-specific electroporation-mediated gene transfer.
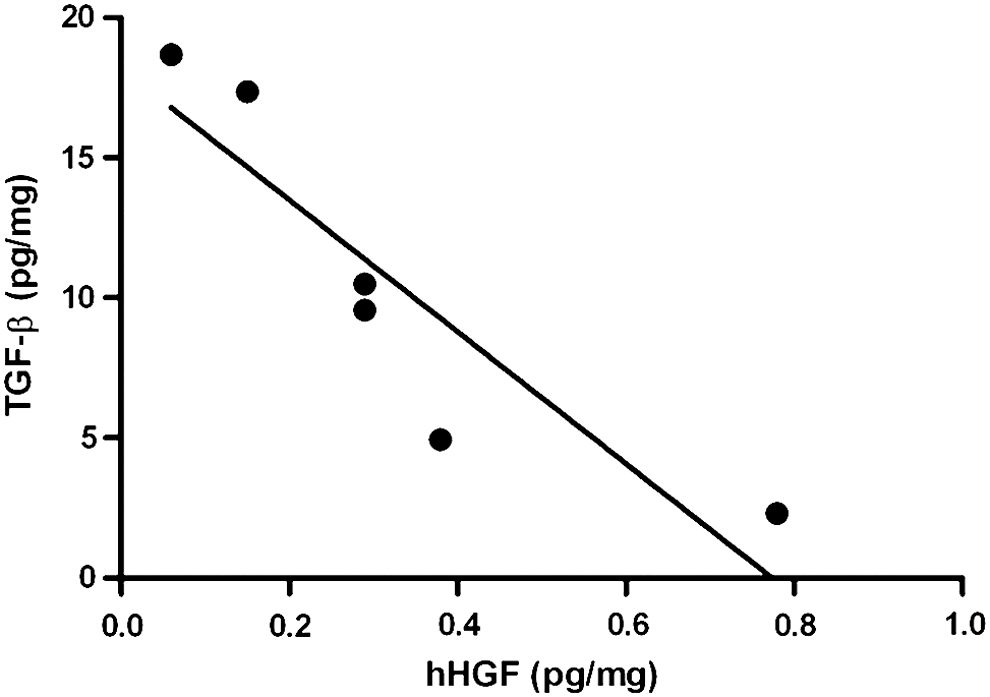
TGF-β1 levels were elevated in the group treated with empty vector; however, in the hHGF-treated group the TGF-β1 levels were reduced. A correlation analysis of TGF-β1 and hHGF levels in the treated group clearly indicate that the level of TGF-β1 was reduced when the HGF level was higher, showing a significant negative correlation (p<0.01, correlation coefficient r=–0.98; linear regression: R 2=0.81).
hHGF regulates EMT and increases alveolar epithelial cell proliferation
To study the effect of hHGF on the epithelial–mesenchymal transition in the bleomycin-injured lung, we stained for two typical markers indicating modulation of the EMT: α-smooth muscle actin (α-SMA) for (myo)fibroblasts and the adhesion protein E-cadherin for epithelial cells.
α-SMA was markedly decreased in pSpC-hHGF-treated animals (Fig. 6h), whereas highly positive signals were observed in the control group (Fig. 6g). E-cadherin immunoreactivity was localized mainly at the cell membrane in the control lungs (pSpC) (Fig. 6a; and Fig. 6c at higher magnification), whereas in the pSpC-hHGF-treated lungs E-cadherin-positive signals shifted from the cell membrane to the cytoplasm (Fig. 6b; and Fig. 6d at higher magnification). These results indicate that cell-specific hHGF gene transfer may modulate the EMT, resulting in a decrease in (myo)fibroblasts in the fibrotic lung tissue.
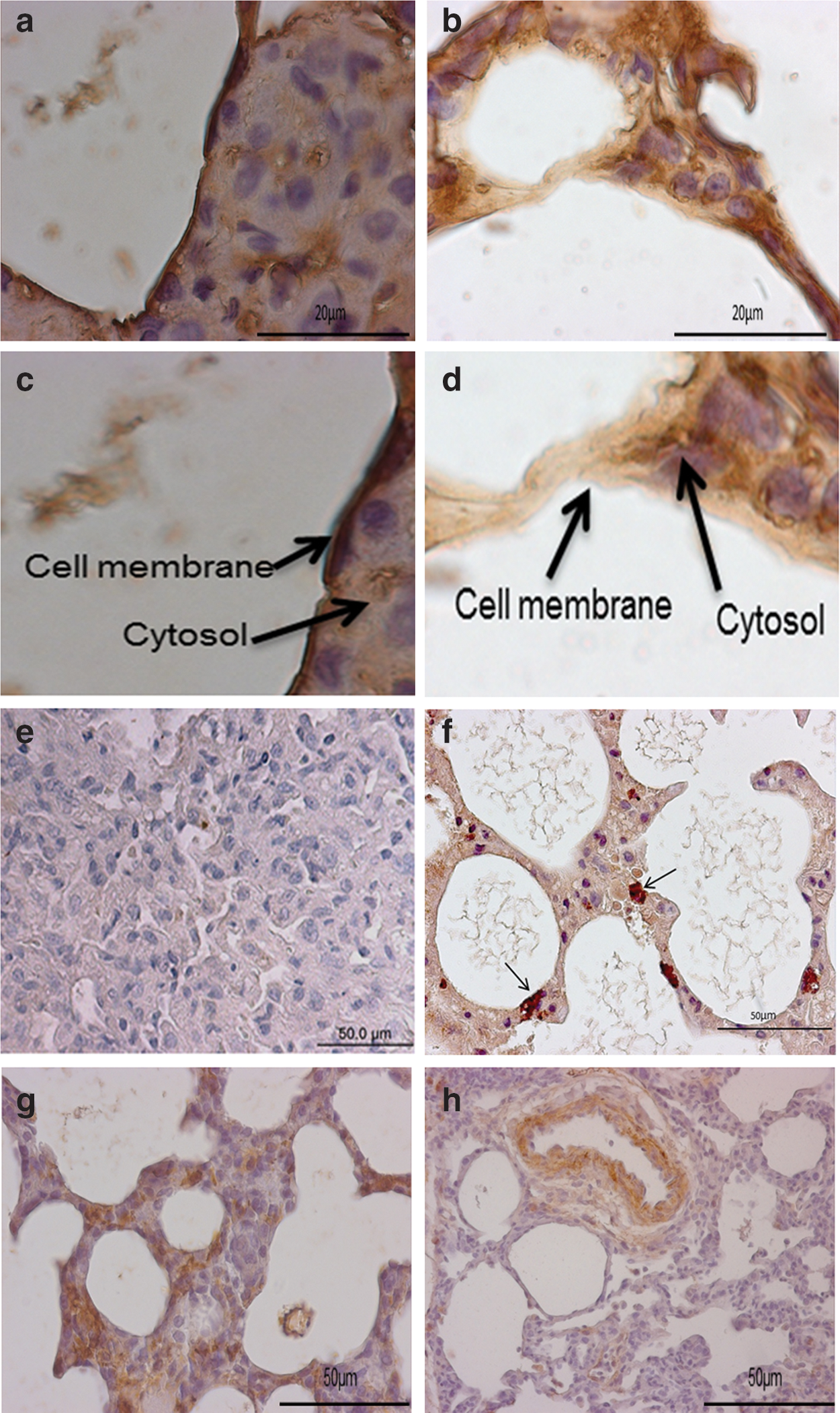
E-cadherin was observed by immunostaining on the cell surface in control animals (pSpC)
Furthermore, hHGF-treated lungs showed increased Ki67 positivity in AECII (Fig. 6f) compared with the control group (Fig. 6e), indicating that HGF induces alveolar epithelial cell proliferation in the bleomycin-injured lung.
hHGF induces fibrinolytic activity and reduces alveolar fibrin deposition
Because dysbalance in alveolar coagulation and fibrinolysis was shown to contribute to the development of pulmonary fibrosis, we studied the presence of fibrin in bleomycin-injured lungs after HGF gene transfer. Alveolar fibrin depositions were markedly reduced after hHGF gene transfer (Fig. 7b) compared with controls (Fig. 7a), based on MSB staining (arrows indicate extravascular fibrin). We further studied the expression of uPA and PAI-1 after HGF gene transfer as selected markers of alveolar coagulation and fibrinolysis. Analysis of mRNA expression of uPA by real-time RT-PCR revealed that uPA was increased after cell-specific hHGF gene transfer (p=0.02) (Fig. 8a). Moreover, the protein level of PAI-1 was markedly reduced after hHGF gene transfer compared with control (681.2±392.3 vs. 11,608±7502 pg/ml; p=0.002) (mean±SD) (Fig. 8b). Upregulation of uPA and downregulation of PAI-1 indicate a profibrinolytic environment in the injured lung after hHGF gene transfer that may contribute to the reduced accumulation of fibrin that was observed after HGF gene transfer.
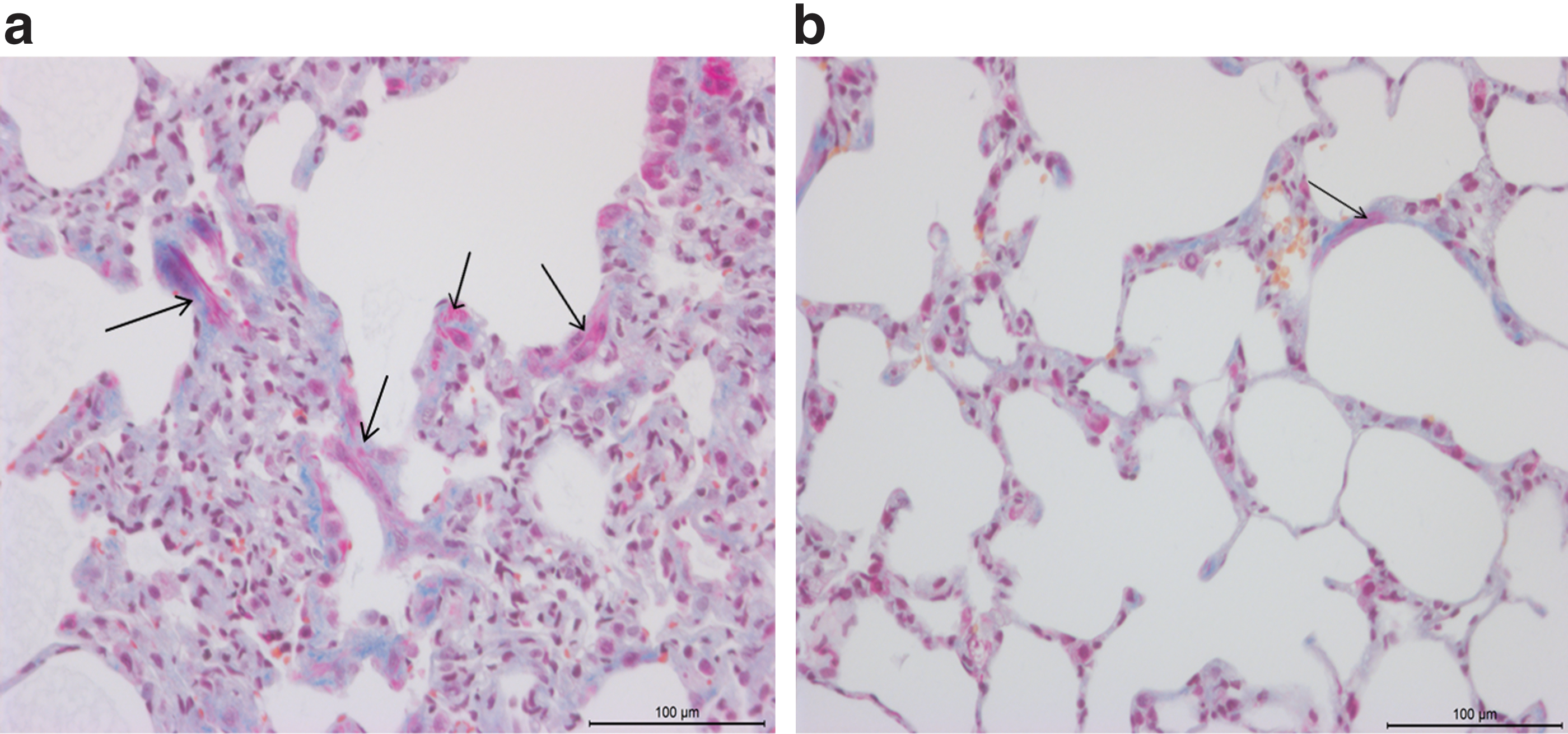
Extravascular and intercellular fibrin deposition was observed in the control group
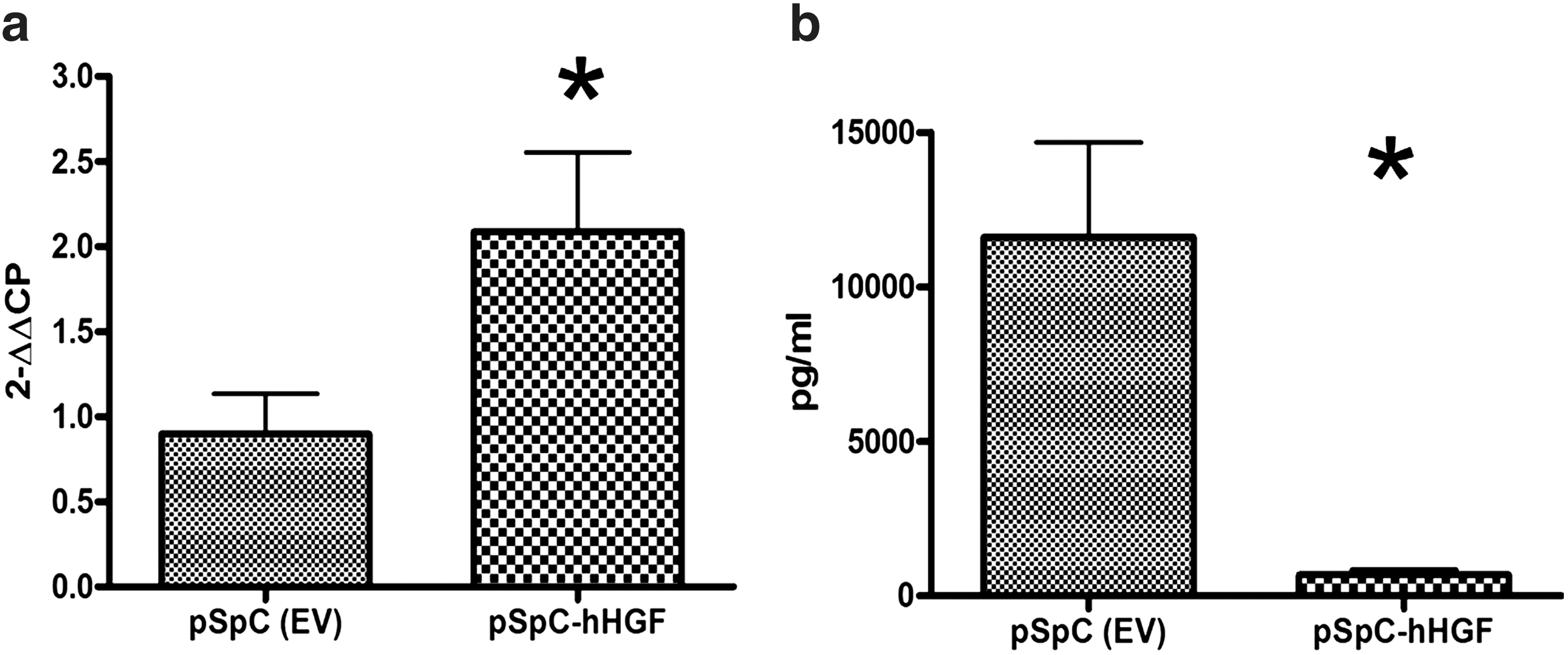
Urokinase plasminogen activator (uPA) mRNA levels were significantly increased after hHGF gene transfer as compared with the control group (pSpC) (*p=0.02)
hHGF does not influence VEGF levels in lung homogenate
No significant differences in VEGF levels between the hHGF-treated group and the control group were detected in lung homogenates as measured by Bio-Plex (5909±6277 pg/ml in the control group vs. 5405±2574 pg/ml in the treated group; NS) (mean±SD).
Discussion
We demonstrate that targeted cell-specific, electroporation-mediated gene transfer of hHGF specifically to AECII is an efficient nonviral gene transfer method in the bleomycin-induced lung injury and fibrosis model in the rat. Cell-specific hHGF gene transfer performed 7 days after bleomycin-induced lung injury resulted in considerable reduction of lung fibrosis. Stereological assessment showed a marked reduction in the thickness of the alveolar septal walls, mainly by reduction of collagen fibrils and ECM in the interstitial space. Cell-specific hHGF gene transfer induces downregulation of profibrotic TGF-β1 modulation of the EMT and an increase in profibrinolytic activity by upregulation of uPA and marked downregulation of PAI-1 in the bleomycin-injured lung. These effects, among others, may significantly contribute to the antifibrotic effect observed after cell-specific hHGF gene transfer in the bleomycin-injured rat lung.
IPF is a fatal lung disease characterized by accumulation of excessive collagen, (myo)fibroblasts, and remodeling of extracellular matrix, leading to destruction of lung architecture and a decline in lung function (du Bois and Wells, 2001; Gross and Hunninghake, 2001). AECII are considered to be precursor cells of the alveolar epithelium and are characterized by their ability to rapidly proliferate and differentiate into AECI. In the IPF lung, however, AECII become hyperplastic and show evidence of abnormal epithelial repair, in particular in areas of fibroblast foci (Adamson et al., 1988, 1990). One report suggests that targeted injury of AECII induces fibrosis in the mouse model (Sisson et al., 2010). Hence inefficient restoration of the alveolar epithelium is crucial for the development of pulmonary fibrosis, and thus enhancement of alveolar epithelial repair after injury is a promising novel strategy to treat pulmonary fibrosis. Here, we aimed at overexpression of an epithelial growth factor, hHGF, targeting specifically the AECII. hHGF was expressed in AECII by means of a surfactant protein C promoter, in order to support alveolar epithelial repair after bleomycin-induced lung injury.
HGF is a potent mitogen and induces epithelial cell proliferation, motility, and morphogenesis and stimulates DNA synthesis in AECII (Shiratori et al., 1995; Day et al., 2002). Its therapeutic potential in the bleomycin lung injury model has also been reported (Mason et al., 1994; Watanabe et al., 2005; Gazdhar et al., 2007). However, the short half-life of the HGF protein diminishes its potential as a future treatment. Gene therapy is a promising novel approach for lung disease, but the lack of safe and nontoxic, reproducible techniques has been a major problem. We therefore developed a novel, nonviral gene transfer technique using electroporation-mediated gene transfer in vivo.
Reports have demonstrated that the physical method of in vivo electroporation-mediated gene transfer to normal and injured lung is a promising and safe technique (Gazdhar et al., 2006, 2007). However, generalized expression of hHGF in several lung cell types (including, e.g., bronchial epithelial cells) is a matter of concern because malignant transformation may occur after long-term expression. We therefore developed a cell-specific gene transfer technique specifically targeted to AECII by putting hHGF under the control of the SpC promoter in order to locally support alveolar epithelial repair induced by AECII and to avoid unwanted transgene expression in other cell types in the lung. In our previous study we have shown that electroporation-mediated gene transfer of hHGF attenuated bleomycin-induced lung fibrosis (Gazdhar et al., 2007). The procedure performed in that study, however, had some technical drawbacks due to the invasive surgical procedures involved (thoracotomy), which would make a future clinical translation difficult. Hence in this present study a modified noninvasive protocol involving placement of electrodes over the chest wall with increased number of pulses was applied, as reported (Machado-Aranda et al., 2005). By using this modified protocol we were able to achieve relevant hHGF transgene expression specifically in AECII. With this approach we were able to achieve the desired cell-specific gene transfer because HGF was detected only in the SpC-positive AECII and not in any other cell types in the lung. Moreover, it has been shown that targeted cell-specific gene transfer of hHGF in the healthy lungs of rats was not associated with any side effects, although alteration of the lung structure at the ultrastructural level were found; in particular, neither pulmonary emphysema nor signs of malignant transformation were found (Leuenberger et al., 2012).
Stereological analysis revealed that 30% of the alveolar surface area covered with AECII stained positive for HGF, asserting the efficiency of extracorporeal electroporation-mediated cell-specific gene transfer. After HGF gene transfer to AECII we observed significant and biologically relevant levels of hHGF in the rat lung tissue homogenate. Although the levels of hHGF were low, we observed reduced collagen content improving histological and stereological parameters of the bleomycin-injured lung, assuming that relatively low levels of hHGF expressed in the local microenvironement of an injured alveolus may be sufficient to induce repair processes resulting in the antifibrotic effects observed.
Under the influence of HGF the volume of destructed lung parenchyma and septal wall tissue as well as its mean thickness were reduced, demonstrating that HGF leads to reduction of fibrosis. Furthermore, TGF-β1 levels were markedly reduced after HGF treatment, with a significant negative correlation between hHGF and TGF-β1 levels. TGF-β1 is a multipotent profibrotic cytokine, regulating tissue morphogenesis and extracellular matrix production. It is involved in induction of fibrosis in various organs including the lung (Sime and O'Reilly, 2001). Elevated levels of TGF-β1 have been reported in the lungs of patients with IPF (Bergeron et al., 2003). TGF-β1 acts as a major switch in tissue repair and remodeling and also has been implicated to play an important role in the epithelial–mesenchymal transition (EMT) (Zavadil and Bottinger, 2005). Our data indicate that the antifibrotic effect of electroporation-mediated HGF gene transfer may be at least in part related to a TGF-β1-dependent mechanism.
However, the beneficial effect of cell-specific HGF gene transfer on pulmonary fibrosis may be the result of several cellular and molecular mechanisms. The reduction of profibrotic TGF-β1 after HGF gene transfer may result in the modification of antifibrotic processes such as EMT or upregulation of fibrinolytic activities as shown in our model. EMT is a complex process in which fully differentiated and functional epithelial cells transform into a mesenchymal phenotype (Kasai et al., 2005). A new emphasis has been put on the alveolar epithelium as a major site of EMT in the progress of pulmonary fibrosis; also, there is evidence indicating the epithelial origin of fibroblasts in lung fibrosis models (Adamson et al., 1988; Willis et al., 2006). TGF-β1 induces EMT by modulating E-cadherin expression leading to destabilization of structural integrity of the pulmonary epithelium. E-cadherin is an epithelial marker that plays an important role in maintaining cell–cell integrity of pulmonary epithelium and its polarization (Takeichi, 1991); loss of or downregulation of E-cadherin leads to destabilization of structural integrity of the pulmonary epithelium. TGF-β1 decreases E-cadherin expression, hence inducing EMT.
In this study we demonstrate that E-cadherin expression was observed on the epithelial membrane of AECII in untreated lungs. However, after HGF gene transfer we observed a cytosolic shift of E-cadherin from the cell membrane to the cytoplasm, as reported previously (Kim et al., 2007), resulting in reduced cell–cell adhesion and increased cell proliferation as evident by increased Ki67-positive AECII. Similar internalization of E-cadherin in response to HGF has also been demonstrated (Howard et al., 2011). Furthermore, α-SMA expression was markedly reduced after HGF gene transfer. We therefore assume that HGF is inducing epithelial cell proliferation, but preventing EMT as evidenced by reduced α-SMA-positive cells in the interstitial space (Shibamoto et al., 1994; Grotegut et al., 2006). These in vivo data are in accordance with an in vitro study using primary murine AECII (Shukla et al., 2009), in which HGF was shown to inhibit TGF-β1-induced generation of the (myo)fibroblast phenotype, indicating that HGF modulates TGF-β1-induced EMT in the lung.
HGF also contributes to angiogenesis (Hoot et al., 2010) and is known to induce VEGF expression in some cell types (Gille et al., 1998). VEGF is a potent angiogenic factor and is involved in lung development and physiology (Voelkel et al., 2006). The role of VEGF in pulmonary fibrosis, however, is controversial (Medford, 2005); studies have revealed that inhibition of VEGF or VEGF receptors attenuates bleomycin-induced lung fibrosis (Hamada et al., 2005; Chaudhary et al., 2007). In our study we did not see any significant increase in VEGF levels as measured in total lung homogenate. These data are in accordance with a clinical study in which VEGF levels were measured in the bronchoalveolar lavage of patients with IPF and did not differ between patients with IPF and control subjects (Simler et al., 2004). In addition, stereological analysis did not reveal a relevant increase in the total surface area of capillary endothelium within septal wall tissue after electroporation-mediated HGF gene transfer, indicating that relevant angiogenesis did not occur after HGF overexpression in our model.
In the course of fibrotic lung disease a dysbalance of coagulation and fibrinolysis was observed (Chambers, 2003, 2008). Upregulation of procoagulatory mediators and simultaneous downregulation of fibrinolytic factors result in a procoagulatory state leading to accumulation of fibrinous matrix in the injured alveoli. PAI-1, for instance, has been identified as a key regulator of pulmonary fibrosis as overexpression of PAI-1 leads to augmentation of fibrosis in the bleomycin model, whereas absence of PAI-1 was protective in the same model (Eitzman et al., 1996). We therefore studied the expression of fibrinolytic mediator uPA and its inhibitor, PAI-1. We show that cell-specific HGF gene transfer resulted in upregulation of uPA and downregulation of PAI-1, supporting a fibrinolytic microenvironement in the alveolar space after HGF gene transfer (Gong et al., 2003). Therefore, our data suggest that the antifibrotic effect of HGF in the bleomycin-induced lung injury model relates to modification of the balance between coagulation and fibrinolysis in the injured lung.
In conclusion, we provide a promising new cell-targeted gene transfer technique resulting in efficient hHGF gene transfer specifically to AECII, resulting in a reduction of bleomycin-induced lung injury and fibrosis in the rat model. Although gene therapy to the human lung is still far from clinical application, electroporation-mediated, targeted gene transfer to the lung or other organs may represent an efficient and safe, nonviral gene transfer technique that needs to be further developed by technical modifications and improvements for future clinical application.
Footnotes
Acknowledgments
This study was supported by the Swiss National Science foundation (SNF; T.G.). The authors thank Anna-Barbara Tschirren for technical assistance.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.