
Abstract
Despite an expansive wealth of research following the discovery of the DMD gene 25 years ago, there is still no curative treatment for Duchenne muscular dystrophy. However, there are currently many promising lines of research, including cell-based therapies and pharmacological reagents to upregulate dystrophin via readthrough of nonsense mutations or by upregulation of the dystrophin homolog utrophin. Here we review genetic-based therapeutic strategies aimed at the amelioration of the DMD phenotype. These include the reintroduction of a copy of the DMD gene into an affected tissue by means of a viral vector; correction of the mutated DMD transcript by antisense oligonucleotide-induced exon skipping to restore the open reading frame; and direct modification of the DMD gene at a chromosomal level through genome editing. All these approaches are discussed in terms of the more recent advances, and the hurdles to be overcome if a comprehensive and effective treatment for DMD is to be found. These hurdles include the need to target all musculature of the body. Therefore any potential treatment would need to be administered systemically. In addition, any treatment needs to have a long-term effect, with the possibility of readministration, while avoiding any potentially detrimental immune response to the vector or transgene.
Introduction
Despite an expansive wealth of research following the discovery of the DMD gene, there is still no curative treatment for DMD. However, there are currently many promising lines of research, including cell-based therapies and pharmacological reagents to upregulate dystrophin via readthrough of nonsense mutations or the upregulation of the dystrophin homolog utrophin (Goyenvalle et al., 2011; Pichavant et al., 2011). However, this review concentrates on genetic-based therapeutic strategies aimed at the amelioration of the DMD phenotype. These include reintroduction of a copy of the dystrophin gene into an affected tissue by means of a viral vector, correction of the mutated DMD transcript (“exon skipping”), and direct modification of the DMD gene at the chromosomal level (“genome editing”). All these approaches have many hurdles to overcome if a comprehensive and effective treatment for DMD is to be found. All muscles including cardiac, skeletal, and smooth muscle need to be targeted; reports have suggested that dystrophin levels of more than 20% in mice are needed to protect muscles from exercise-induced damage (van Putten et al., 2012) and a minimum of 30% of normal dystrophin levels needs to be present uniformly in all myofibers to prevent muscular dystrophy in humans (Neri et al., 2007). Therefore any potential treatment would need to be administered systemically. In addition, any treatment needs a long-term effect, with the possibility of readministration while avoiding any potentially detrimental immune response to the vector or transgene. Here we review the progress made in these key areas of genetic therapies for DMD.
Viral Gene Therapy
The natural ability of viruses to enter cells and deliver genetic information has been manipulated by many research groups to develop disabled viruses that can deliver transgenes to muscle at high efficiency. Many viruses, including adenovirus, lentivirus, and adeno-associated viruses (AAV), have been intensively investigated for gene addition strategies. However, although adenoviral vectors can be used to transfer the entire 11-kb dystrophin cDNA, poor transduction efficiencies are achieved in adult muscle. The use of adenoviral vectors is further hampered by their being immunogenic, with about 50% of the population positive for neutralizing antibodies against adenovirus. Although lentiviral vectors also have a large insert size and are able to accommodate the mini-dystrophin gene, direct injection into muscle results in poor transduction efficiencies. As a result, lentivectors are currently being used for the stable transduction of myogenic stem cells (Sampaolesi et al., 2006) for the development of cell-based therapies for DMD. However, of these vectors, AAV is currently the most promising candidate, showing reduced immunogenicity and efficient levels of muscle transduction by both direct and systemic administration. Progress and prospects for AAV for DMD gene therapy are discussed below.
AAV gene therapy
AAVs are single-stranded DNA parvoviruses that require helper virus for replication. Recombinant AAVs (rAAVs) have all viral genes removed and replaced with the promoter/transgene of interest flanked only by the viral inverted terminal repeats (ITRs). rAAV can be produced efficiently at high titers by the provision of viral genes in trans. At present, nine primate AAV serotypes have been described, which demonstrate diverse tissue tropisms. Many serotypes including AAV1, -2, -6, -8, and −9 have now been demonstrated to efficiently transduce skeletal muscle after direct injection (Inagaki et al., 2006; Zincarelli et al., 2008). Importantly, for application for DMD gene therapies, AAV6 and in particular AAV8 and AAV9 have been shown to transduce both cardiac and skeletal muscle at high efficiencies after systemic gene transfer, allowing for the first time the potential for body-wide gene transfer (Gregorevic et al., 2004; Wang et al., 2005; Foster et al., 2008; Zincarelli et al., 2008). A number of studies have also demonstrated stable gene expression after direct injection of rAAV; for up to 7 years in canine and nonhuman primate models. These factors, along with the apparent lack of immunogenicity to both transgene and virus in small animal models, have made rAAV vectors promising for DMD gene therapy. However, there is a limitation in the packaging capacity of foreign DNA into AAV vectors. The AAV capsid is able to package a genome of up to 5 kb and therefore it is not possible to transfer the 11-kb full-length dystrophin cDNA into AAV vectors. To overcome this limitation, truncated but functional mini- and micro-dystrophin cDNAs have been engineered by several research groups and have been tested in both murine and canine models of DMD.
rAAV micro-dystrophin gene transfer
The development of mini- or micro-dystrophin genes is based on observations in patients with Becker muscular dystrophy (BMD), who despite often having large deletions, present with a mild phenotype or can even be asymptomatic (England et al., 1990; Matsumura et al., 1994; Beroud et al., 2007). A number of groups have conducted extensive studies in the mdx and mdx/utr double-knockout mouse models after AAV micro-dystrophin gene transfer (Wang et al., 2000; Fabb et al., 2002; Harper et al., 2002; Sakamoto et al., 2002; Gregorevic et al., 2006; Yue et al., 2006). Maintenance of the N-terminal actin-binding and β-dystroglycan-binding domains (part of the DGC) is essential for the functionality of any micro-dystrophin protein. A number of functional micro-dystrophins have successfully been developed that contain large deletions in the rod and C-terminal domains.
When expressed at a sufficient level via direct injection or via the production of transgenic mouse lines, many of these micro-dystrophin proteins have been demonstrated to improve, but not completely normalize, a range of markers of the dystrophic phenotype. Restoration of the dystrophin-associated protein complex, stabilization of muscle degeneration, and improvements in muscle function including normalization of specific force and improvement in resistance to eccentric contractions have been demonstrated after delivery of micro-dystrophin at different stages of disease progression (Crawford et al., 2000; Harper et al., 2002; Sakamoto et al., 2002; Foster et al., 2008; Wang et al., 2000, 2009). In addition to skeletal muscle, it is also necessary to treat cardiac muscle, as correction of skeletal muscle function in the absence of cardiac expression is likely to exacerbate cardiac dysfunction (Townsend et al., 2008). Studies using both AAV6 and AAV9 to transduce the myocardium of mdx mice with micro-dystrophin have demonstrated high levels of gene transfer associated with improvements in fibrosis, echocardiography, and protection from dobutamine stress-induced acute heart failure (Townsend et al., 2007; Bostick et al., 2008, 2009, 2011; Shin et al., 2011; Lai and Duan, 2012; Schinkel et al., 2012). As loss of dystrophin expression from smooth muscle results in changes to the vasculature (Loufrani et al., 2001, 2002) and systemic AAV has been shown to transduce smooth muscle, it should also be considered that stabilization of the vasculature may contribute to improvements observed in skeletal and cardiac muscle function (Ito et al., 2006).
Despite the current wealth of research assessing the function of current micro-dystrophins, rationale and design for improvements in gene configuration are still ongoing. A number of groups have assessed the inclusion of further functional domains in the micro-dystrophin construct. The addition of spectrin-like repeats 16/17 allowed normalized relocation of neuronal nitric oxide synthase (nNOS) at the sarcolemma, associated with improved muscle function and exercise performance (Lai et al., 2009; Zhang and Duan, 2012). In addition, a number of improvements have been made to one of the most promising micro-dystrophins, ΔR4-R23/ΔCT, originally described by Harper and colleagues in 2002. Sequence optimization and addition of helix 1 of the coiled-coil domain of the C-terminal domain to ΔR4-R23/ΔCT improved resistance to eccentric contractions in the mdx mouse (Foster et al., 2008; Koo et al., 2011a). Although ΔR4-R23/ΔCT has been shown to restore specific force and to improve resistance to eccentric contractions, the Chamberlain group has demonstrated disruption to the myotendinous junction, formation of “ringed fibers,” and aberrant formation of neuromuscular junctions in the mdx mouse (Banks et al., 2008, 2009, 2010) after gene transfer with ΔR4-R23/ΔCT. Replacement of hinge 2 with hinge 3 within the construct led to improved prevention of muscle degeneration and stabilized neuromuscular junctions (Banks et al., 2010).
Alternatively, dual AAV vectors are currently being developed to deliver larger 6- to 8-kb mini-dystrophin genes to dystrophic muscle. Typically, the gene is split into two independent AAV vectors. Either the two DNA sequences share a homologous recombinogenic region, termed overlapping vectors, or vectors contain splice donor/acceptor sites to allow trans-splicing between the two gene portions. Both methods have been well characterized and have been demonstrated to result in mini-dystrophin expression after both direct intramuscular and systemic intravenous injection, with mice showing improved muscle function in comparison with micro-dystrophin gene transfer (Lai et al., 2005; Ghosh et al., 2007, 2008, 2011; Odom et al., 2011; Zhang and Duan, 2012). Although it would appear that the optimal mini/micro-dystrophin gene is still evolving it is clear that these studies give significant promise that AAV mini/micro-dystrophin gene transfer may be a clinically applicable strategy.
rAAV gene transfer in large animal models and clinical trials
In small animal studies rAAV vectors have demonstrated limited immunogenicity to either the AAV capsid or to the transgene. Indeed, delivery of rAAV to the liver is being extensively investigated as a means to induce tolerance to immunogenic transgenes (Mingozzi et al., 2007a; Martino et al., 2009; Sharland et al., 2011; Zhang et al., 2012). Although rAAV is capable of transducing canine skeletal and heart muscle at high efficiency (Koo et al., 2011b; Bish et al., 2012), rAAV transfer into large animal models and in clinical trials has demonstrated that rAAV can elicit variable immunological outcomes (Mingozzi and High, 2007; Mingozzi et al., 2007b; Wang et al., 2007a,b; Yuasa et al., 2007; Yue et al., 2008; Ohshima et al., 2009; Mendell et al., 2010a,b). The potential for immunity to both transgene and capsid is likely to result in prevention/reduction of gene expression and prevention of readministration of the vector.
Cytotoxic immune responses to capsid and transgene have been exhibited after intramuscular injection of rAAV2, rAAV6, and rAAV8 vectors carrying various transgenes including micro-dystrophin in canine models of DMD (Wang et al., 2007a,b; Yuasa et al., 2007; Yue et al., 2008; Ohshima et al., 2009). These immune responses have not been typically encountered in small animal models, but have been observed in juvenile and neonatal muscular dystrophy dogs and random-bred wild-type dogs (Wang et al., 2007a,b; Kornegay et al., 2010). However, transient immunosuppression has been shown to allow expression of micro-dystrophin in canine models for up to 6 months (Wang et al., 2007b; Shin et al., 2012). High levels of micro-dystrophin expression have also been demonstrated in nonhuman primates after direct injection and vascular delivery of an rAAV8 vector, although efficiency was reduced by 50% when animals had preexisting antibodies to AAV8 (Rodino-Klapac et al., 2010).
A number of clinical trials using rAAV vectors have now been conducted; including trials for hemophilia factor IX (FIX), limb girdle muscular dystrophy type 2D, and DMD. These studies have been complicated by the generation of both cytotoxic and humoral immune responses to the capsid of various rAAVs (Manno et al., 2006; Mingozzi and High, 2007; Mingozzi et al., 2007b; Mendell et al., 2010a; Bowles et al., 2012), the development of which appears to be dose dependent in humans (Mingozzi et al., 2009). Transient immunosuppression at the time of vector administration (as discussed previously in canine studies) may prevent activation of CD8+ and CD4+ T cells. However, encouragingly, a successful clinical trial for FIX delivery has been reported by Nathwani and colleagues (2011), in which AAV8 vectors were administered in the absence of profound immunosuppression and therapeutic levels of FIX protein were achieved.
The presence of neutralizing antibodies before administration can also hinder or prevent gene transfer. Several studies in animal models demonstrated that transient immunosuppression at the time of vector administration effectively allows administration in the presence of preexisting neutralizing antibodies (Halbert et al., 1998; Manning et al., 1998; Jiang et al., 2006; Riviere et al., 2006). An alternative strategy for the avoidance of preexisting antibodies was employed in an rAAV trial for DMD. Patients received AAV2.5 CMV micro-dystrophin via intramuscular injection into a biceps muscle. AAV2.5 is a variant of AAV2 that contains five amino acids (one insertion and four substitutions) in the VP1 region from AAV1. In addition to retaining the beneficial properties of both AAV2 (ease of purification) and AAV1 (increased transduction efficiency in muscle), it has been demonstrated that in mice preexisting antibodies to either AAV1 or AAV2 did not prevent successful administration of rAAV2.5 vector (Bowles et al., 2012). However, two of five patients were demonstrated to have significant levels of preexisting antibodies that were capable of neutralizing rAAV2.5. In these patients the number of vector genomes detected was much lower than was found in other patients. Although conducted in a small number of patients it is indicative that the use of chimeric AAV serotypes alone may not be sufficient to overcome the issue of preexisting immunity to AAV in human populations.
Micro-dystrophin expression was detected in only two of six patients and at low levels (one and three fibers, respectively), although vector DNA was detected in all muscle biopsies. A possible explanation is that the transduction efficiency of AAV2.5 vectors in human muscle may be low. However, unexpectedly, dystrophin-specific T cells were detected in four patients. Further analysis revealed that some patients exhibited preexisting dystrophin-specific T cells and that gene transfer may have stimulated a memory T cell response in these patients. In addition, micro-dystrophin-specific T cells were also detected within at least one patient. The detection of both CD4+ and CD8+ dystrophin-specific T cells within these patients provides one explanation for the low levels of dystrophin expression that were observed. This unexpected observation, in particular the presence of preexisting dystrophin-specific T cells, is in contrast to the proposed induction of tolerance to dystrophin by the presence of spontaneous revertant fibers (Ferrer et al., 2000). However, what is unclear is why preexisting antibodies are detected in some patients and not others. It is speculated that this may be related to disease severity, levels of inflammation within the muscle, and timing of the revertant fiber “event.” It should be noted that dystrophin immunity has not been reported as an issue in the “exon-skipping” clinical trials for DMD; however, neither has the immune status of these individuals currently been reported.
Strategies to reduce immunotoxicity for rAAV gene therapy
Although this AAV clinical trial resulted in few dystrophin-positive fibers it has brought to the fore some important considerations for AAV micro-dystrophin therapies. In addition to the use of transient immunosuppression, there are currently many strategies being developed to reduce the immunogenicity of rAAV vectors for use in clinical trials. The choice of AAV serotype is of the utmost importance, with the optimal vector showing high efficiency after systemic gene transfer and low immunogenicity. At present, AAV8 appears to be a promising candidate; efficient gene transfer has been demonstrated in canine, nonhuman primate (NHP), and clinical trials, with limited immune complications (Rodino-Klapac et al., 2010; Koo et al., 2011b; Nathwani et al., 2011), and the prevalence of neutralizing antibodies is comparatively low in the human population (Boutin et al., 2010). As immunogenicity appears to be dose dependent (Mingozzi et al., 2009), strategies to reduce viral load should also be employed. This may be partly achieved through choice of serotype; however, studies have also demonstrated that sequence optimization of the transgene is also of benefit in reducing viral load (Foster et al., 2008), and the use of muscle-restrictive promoters and the inclusion of microRNA target sequences within the expression cassette can minimize ectopic expression of the transgene and prevent expression in hematopoietic cell lineages (Brown et al., 2006; Annoni et al., 2009; Koo et al., 2011b). Koo and colleagues demonstrated long-term expression in a canine model of DMD after rAAV8 gene transfer of a sequence-optimized micro-dystrophin under the control of a muscle-restrictive promoter in the absence of any immune suppression (Koo et al., 2011b). Route of administration also appears to play an important role in the development of an immune response to vector and transgene, as along with the benefit of reaching multiple muscle groups, systemic delivery also is less immunogenic than direct intramuscular injection (Toromanoff et al., 2010). Isolated limb perfusions for the delivery of vectors to an entire limb have successfully been developed in canine models and NHPs (Toromanoff et al., 2008, 2010; Qiao et al., 2009; Rodino-Klapac et al., 2010), with the ultimate goal being systemic delivery of rAAV to all muscles. Fan and colleagues have carried out a safety and feasibility study of high-pressure limb perfusion of saline in patients with muscular dystrophy and successfully set out a safe protocol for this procedure that will serve as a basis for the translation of rAAV gene transfer into clinical trials (Fan et al., 2012).
Correction of the Mutated DMD Transcript
Transcript-editing therapy using antisense oligonucleotide-induced exon skipping
Genomic deletions of one or more exons are the cause of about 70% of DMD cases; genomic deletions can also result in the less severe disease, Becker muscular dystrophy (BMD) (Aartsma-Rus et al., 2006). Deletions that cause disruption of the open reading frame (ORF) of the DMD gene cause complete loss of expression of dystrophin and a DMD phenotype (Fig. 1a), whereas deletions that maintain the ORF lead to the milder BMD phenotype. Inhibition of the splicing of specific exons, by so-called exon skipping using antisense oligonucleotides (AOs), can induce exclusion and skipping of targeted frame-shift exons, leading to restoration of disrupted reading frames and synthesis of shortened BMD-like dystrophin protein (see Fig. 1b). This protein will be partially functional because it contains both the N- and C-terminal domains, which have important signaling functions between the extracellular matrix and cytoskeleton. Transcript editing using AO-induced exon skipping to produce functional truncated dystrophin protein, first described by Dunckley and colleagues in 1996, has been demonstrated in vitro (Graham et al., 2004), and further shown to ameliorate the severe muscle damage in animal models of DMD (Lu et al., 2005; Alter et al., 2006; McClorey et al., 2006a; Malerba et al., 2009, 2011a,b; Yokota et al., 2009).
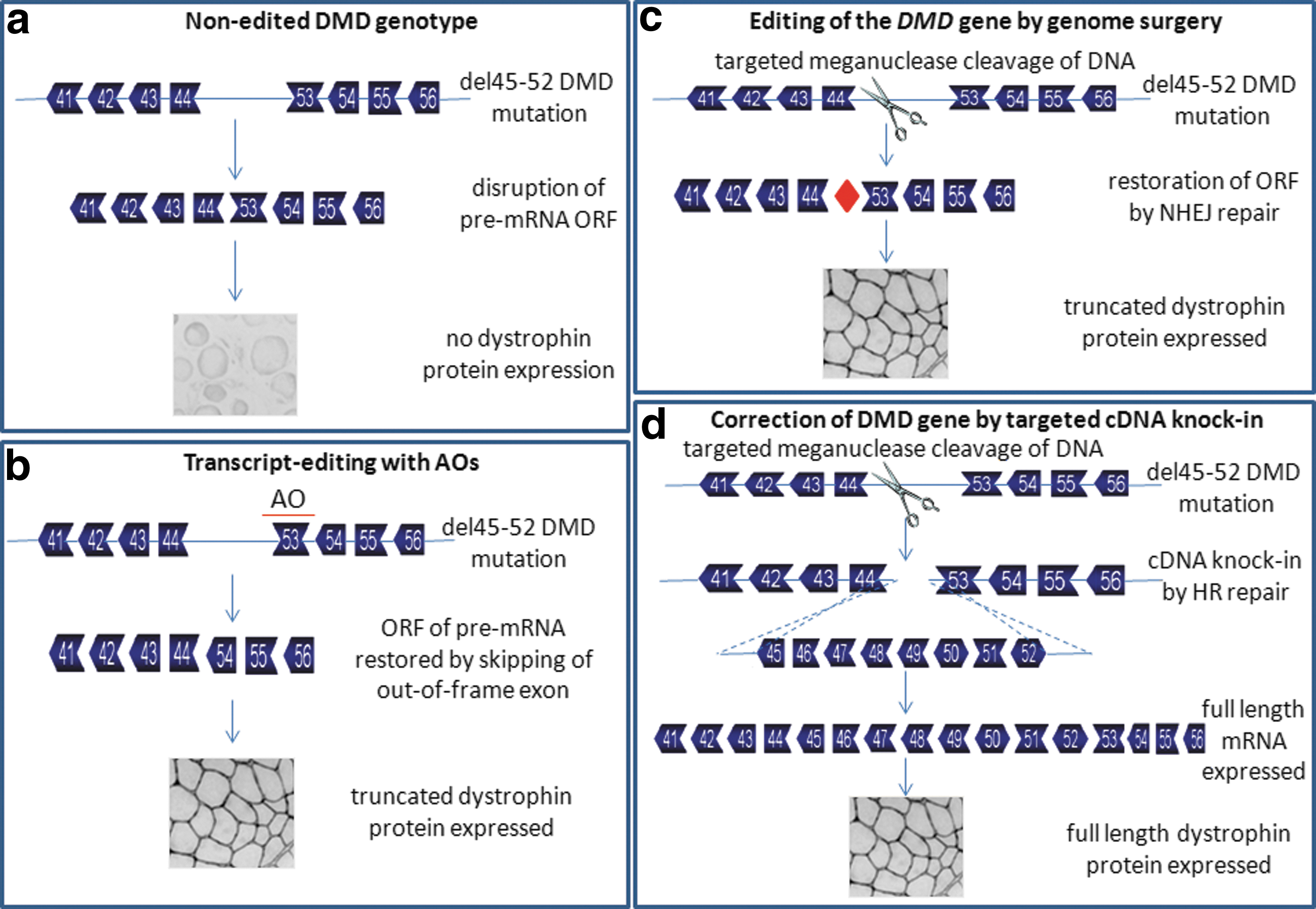
Correction of DMD mutations by transcript editing and genome surgery. Depicted are the theories behind exon skipping editing of mutated DMD transcripts and modification of the DMD gene itself by genome surgery.
Exon skipping has been shown to be translatable from animals to humans, both in vitro in DMD patient cells (van Deutekom et al., 2001; Wilton et al., 2007; Aartsma-Rus et al., 2009; Popplewell et al., 2010) and muscle explants (McClorey et al., 2006b; Arechavala-Gomeza et al., 2007), and latterly in clinical trials. After successful phase 1 trials (van Deutekom et al., 2007; Kinali et al., 2009), phase 2 trials of two different chemistries of AOs for the targeted skipping of exon 51 of the DMD gene have been completed. PRO051, a 2′-O-methyl phosphorothioate (2′OMePS) AO, gave dose-dependent restoration of dystrophin production that resulted in a modest, but not statistically significant, improvement in 6-min walk test results after 12 weeks of extended treatment (Goemans et al., 2011). The 12-week systemic delivery of the second AO, AVI-4658, which has a phosphorodiamidate morpholino (PMO) backbone, resulted in dose-dependent restoration of dystrophin production; functionality was established by the detection of other dystrophin-associated proteins at the sarcolemma (Cirak et al., 2011). Both chemistries showed no adverse effects and phase 3 trials are now commencing (
A potential problem with AO-induced transcript editing is that of targeted delivery to (1) the body-wide skeletal musculature, because the majority of administered AO is taken up and cleared by the liver and kidney, meaning much of the dose is wasted; and (2) specifically the heart, because no dystrophin expression has been observed in cardiac muscle after systemic delivery of AO (Malerba et al., 2011a,b). This is largely because naked AOs are unable to enter the relatively undamaged cardiomyocytes. Many delivery systems have been developed to improve uptake of AOs by skeletal muscle, including polymers (Williams et al., 2008), nanoparticles (Rimessi et al., 2009; Sirsi et al., 2009), microbubbles (Alter et al., 2009), and viral vectors (Goyenvalle et al., 2004; Denti et al., 2008; see the next section [U1 or U7 snRNA-Mediated Exon Skipping] for more detail). However, polymers, nanoparticles, and microbubbles achieve only poor efficiency in the delivery of AOs, and viral vectors, despite showing promise in mice, were less efficient in dogs, probably as a result of the more complex immune system in higher animals (Gregorevic et al., 2009). PMOs have an advantage over other chemistries in that they can be conjugated to cell-penetrating peptides (CPPs); these peptide-conjugated PMOs (PPMOs) dramatically enhance deliverability and produce efficient and persistent skipping action in the heart (Wu et al., 2009; Yin et al., 2009). However, preclinical studies of AVI-5038, a PPMO targeted to skip exon 50 of the DMD gene, showed that weekly administration over 12 weeks produced significant toxicological effects, particularly in relation to the kidney. There are a number of alternative peptide conjugates that appear to be less toxic, and are undergoing rapid preclinical development (Yin et al., 2010).
As there is such an array of mutations that cause DMD, personalized molecular medicine for each skippable DMD deletion is necessary. This would require the optimization and clinical trial work-up of many specific AOs. By targeting deletion hotspots (e.g., exons 45–55) with cocktails of AOs or chemically linked AOs, multi-exon skipping may result. Development of a multi-exon skipping formulation would have the potential to treat approximately 65% of patients with DMD (Beroud et al., 2007). Such a strategy has been shown to work in vivo in mdx mice (Adams et al., 2007) and golden retriever muscular dystrophy (GRMD) dogs (Yokota et al., 2011), but has not yet been achieved in DMD patient cells (van Vliet et al., 2008). The likelihood of multi-exon skipping being achieved with AOs alone is generally regarded to be extremely slim.
As AOs act on the transcript rather than the gene, their modification of gene expression is not permanent; the continual delivery of AO to the nucleus is therefore required for a meaningful therapeutic effect (Vacek et al., 2003). Because of biological turnover of AOs, regular readministration would be needed clinically. The financial cost of AO-induced exon skipping as a therapy may yet prove to have a major impact on the translation of this technology to the clinic. In addition, it is not yet known what adverse effects regular readministration of AOs may have in patients. The use of viral vectors to carry AOs into the nucleus would address these issues; viral vector transfer of AO sequence into the host genome would allow the continual expression of functional amounts of AO within the nucleus.
U1 or U7 small nuclear RNA-mediated exon skipping
AO activity would be enhanced by its correct subcellular localization within the nucleus and its inclusion into the spliceosome; this can be facilitated by linking it to small nuclear RNAs (snRNAs). Stable cellular expression of modified U1 or U7 nonspliceosomal snRNAs can result in sustained, sequence-specific modification of the targeted mRNA structure. U1 or U7 snRNAs, which are normally involved in the processing of the 3′ end of histone mRNA, were used to express chimeric RNAs containing antisense sequences to the acceptor and donor splice sites of exon 51. Transfection of these chimeric snRNAs and their corresponding genes into DMD myoblasts carrying del48–50 resulted in high levels of exon 51 skipping and rescue of dystrophin expression (de Angelis et al., 2002). To allow high-efficiency gene transfer into skeletal muscle in vivo, U1 or U7 snRNAs expressing antisense sequences to the donor and acceptor splice sites of exon 23 have been packaged into AAV vectors. The single administration in mdx mice of this AAV vector resulted in persistent, specific exon 23 skipping, sustained production of functional dystrophin protein and amelioration of disease phenotype. This strategy produced body-wide dystrophin expression including in the heart for the lifetime of the mice after a single administration (Denti et al., 2006). This work has been extended to the utrophin/dystrophin double-knockout (dKO) mouse, a more severe and progressive mouse model of DMD. A single intravenous injection of scAAV9-U7ex23 into these mice virtually normalized dystrophin expression in the musculature body-wide, including the heart. This resulted in considerable improvements in muscle function, dystrophic pathology, and dKO mouse life span (Goyenvalle et al., 2012a). These findings suggest great potential for AAV-U7 in systemic treatment of the DMD phenotype.
U7 snRNA-mediated exon skipping has made advances in the development of a multi-exon skipping strategy of exons 45 to 55. A combination of antisense sequences in a single AAV vector was able to produce multi-skipping of at least three exons in vitro in DMD patient myoblasts and in vivo in human DMD (hDMD) mice (Goyenvalle et al., 2012b). With development, it is possible that exon 45 to exon 55 skipping will nr achievable with AAV vectors encoding multiple U7snRNAs, but it is anticipated that the efficiency of multi-exon skipping will be extremely low.
Correction of DMD Mutations by Genome Editing
Modification of the DMD gene itself, so that mutations are corrected or the reading frame is restored, is encompassed by the general term genome editing. This has been attempted by a number of strategies, with various degrees of success.
Oligonucleotide-mediated genome repair
Oligonucleotide-mediated genome repair has been used to correct a number of diseases caused by point mutations (Igoucheva et al., 2001; Tagalakis et al., 2001). This is achieved by engineering mismatches between a targeting oligonucleotide vector and the genome sequence so that endogenous DNA repair mechanisms change single bases in genomic DNA (Liu et al., 2002a). Oligonucleotide-mediated genome repair could be applicable to the 15% of DMD cases with point mutations. The in vitro and in vivo correction of point mutations within mdx mice and GRMD dogs, using chimeric RNA/DNA oligonucleotides (RDOs) and also oligodeoxynucleotides (ODNs), has been demonstrated at the genomic, transcript, and protein levels (Bertoni, 2005; Bertoni et al., 2005). Ex vivo repair of the splice site point mutation genomically and transcriptomically in mdx5cv mouse myoblasts, using single-stranded DNA oligonucleotides, resulted in full-length dystrophin protein expression (Maguire et al., 2009). This would allow the production of corrected progenitor cells capable of restoring functional dystrophin in muscle fibers on transplantation into mdx5cv mice. Research is now concentrating on improving the low efficiency and poor persistence of repair seen, by using RNA interference (RNAi) to suppress expression of the recombinase inhibitor proteins (Maguire et al., 2009), and single-stranded ODNs with modified peptide nucleic acids (Kayali et al., 2010). As for AO-induced exon skipping, oligonucleotide-mediated genome repair would require individual oligonucleotide development for each DMD point mutation.
Spliceosome-mediated RNA trans-splicing
Trans-splicing is a rare, but natural, process by which two separately transcribed mRNAs are spliced together to produce a composite transcript. Reprogramming the mRNA expressed from a mutated gene, using trans-splicing, could correct certain genetic diseases. Spliceosome-mediated RNA trans-splicing (SmaRT) uses an engineered pre-mRNA trans-splicing molecule (PTM) that binds specifically to the target pre-mRNA in the nucleus, which triggers trans-splicing repair by the spliceosome. The defective portion of the target pre-mRNA is removed and replaced with a wild-type cDNA exon contained within the PTM, and a functional gene product is transcribed. The use of SmaRT for the functional correction of a number of disease mutation models has been reported (Mansfield et al., 2000; Liu et al., 2002b; Chao et al., 2003; Tahara et al., 2004; Chen et al., 2009). Using a double trans-splicing approach, it has been shown that replacement of the mutated mdx exon 23 was achievable in the mRNA target (Lorain et al., 2010). SmaRT has the advantages that the endogenously controlled repair occurs only where the target transcript is expressed, and the small size of the PTM constructs allows their easy accommodation in current vector systems. In the context of DMD, this gene-editing strategy could be applicable to virtually all types of DMD mutation. However, even with the use of a downstream intronic splice enhancer (DICE) (Lorain et al., 2010), the efficiencies of trans-splicing are poor, and there is the potential for nonspecific trans-splicing. For this approach to be potentially viable as a therapy, further optimization would be required, as would the development of efficient targeted cellular delivery.
Gene targeting
An exciting new field of development in genome editing is gene targeting; this has provided a powerful tool for creating genetic modifications as a gene repair strategy for a variety of genetic diseases (Martin et al., 2010; Carroll, 2011; Jensen et al., 2011). Genome surgery can be achieved by using endonucleases (meganucleases [MNs], zinc finger nucleases [ZFNs], and transcription activator-like effector nucleases [TALENs]) to cut at specific sites in the DNA. The resulting double strand break (DSB) can be repaired in two major ways: by homologous recombination (HR) or nonhomologous end joining (NHEJ) (Longhese et al., 2010). HR requires the presence of an identical or nearly identical sequence to be used as a template for repair of the break (Jensen et al., 2011). MNs can be used to stimulate HR up to 10,000-fold in cultured cells (Rouet et al., 1994; Choulika et al., 1995) in comparison with HR at a noncleaved site. MNs have been used to induce HR in a variety of cell types and organisms (Paques and Duchateau, 2007), including mammalian cells, mice, plants, Drosophila, Escherichia coli, and trypanosomes (Boothroyd et al., 2009). The first report of the use of MNs to modify a chromosomal locus described high levels of gene targeting of the human XPC gene, which is involved in the disease xeroderma pigmentosum (XP) (Arnoud et al., 2007). Genome editing with ZFNs have been used to restore hemostasis, in terms of increased clotting times, in a mouse model of the disease hemophilia. When delivered directly to mouse liver, ZFNs were able to efficiently induce DSBs, which were repaired through homology-directed targeted gene insertion driven by a specific gene-targeting vector at the ZFN-specified locus (Li et al., 2011). HR gene repair has also been shown to successfully correct the severe combined immunodeficiency (SCID) mutation of the IL2Rγ gene (Urnov et al., 2005), and has led to the development of clonogenic stem cells carrying the corrected IL2Rγ gene (Lombardo et al., 2007). In the context of DMD, genome surgery has been limited to a proof-of-principle study in which an engineered MN restored the normal reading frame in dog micro-dystrophin sequences carrying a frameshift mutation through NHEJ repair. The MN induced micro-deletion or micro-insertion in the micro-dystrophin so that dystrophin expression was restored, both in myoblasts in vitro and in muscle fibers in vivo (Chapdelaine et al., 2010) (Fig. 1c). This work has been extended to show DMD gene correction repair, using a specific MN (Daboussi et al., 2012) and a repair matrix to drive targeted cDNA insertion (Popplewell et al., 2012) (Fig. 1d). Whether the genome surgery results in reading frame restoration (when a DSB is repaired by NHEJ) or in cDNA knock-in (when a DSB is repaired by HR), the change in the DMD gene will be permanent, removing the need for repeated long-term administration. It would overcome the potential immune-related problems posed by AAV vector-based strategies, and the high costs and potential toxicological risks of long-term AO use. Genome correction has the major advantage that with the right MN and targeting repair matrix, any DMD mutation would be treatable, unlike exon skipping, which is applicable only for patients with frame-shifting deletions. In addition, genome correction therapy for DMD would produce full-length dystrophin protein, whereas expression only of truncated protein is possible by exon skipping and by AAV vector-based strategies.
Future Considerations and Outlook
There have been major advances toward the development of a therapeutic treatment/cure for DMD. It is encouraging that steady progress is being achieved in the three different branches of research reviewed here, namely (1) introducing a shortened but functional replacement version of the DMD gene virally, (2) correcting the mutated DMD gene at the transcript level by exon skipping, and (3) correcting the mutated DMD gene at the genome level by genome surgery.
The development of an rAAV micro-dystrophin gene therapy for DMD remains a real challenge. However, progress in the identification of suitable AAV serotypes for intravascular gene transfer, the continuing development of highly functional micro-dystrophin gene configurations, improvements in vector cassette design to reduce immunogenicity of the vector, along with the development of a safe clinical protocol for transvenous limb perfusion in patients with muscular dystrophy make the translation of rAAV micro-dystrophin gene transfer to the clinic a realistic possibility. However, management of toxicity and immunogenicity after rAAV gene transfer is of the utmost importance in such studies. The micro-dystrophin trial highlights the potential that a subset of patients may already be positive for dystrophin-specific T cells in addition to the possibility of preexisting neutralizing antibodies to the virus. In addition, it should be taken into consideration that dystrophic muscle may be under heightened immune surveillance due to the nature of the inflammatory DMD pathophysiology and as such may add a level of complexity compared with other monogenic diseases. One further strategy to avoid potential dystrophin immunity is to use rAAV micro-utrophin vectors. Utrophin is widely expressed in patients with DMD and should therefore not pose as a new immunogen. A study done in the dystrophin/utrophin double knockout mouse demonstrated that micro-utrophin is able to improve the physiological performance of these mice (Odom et al., 2008). In addition, a study on the biophysical interactions between micro-dystrophin/micro-utrophin and actin revealed that micro-utrophin may outperform micro-dystrophin in functional restoration of muscle properties (Lin et al., 2012). These developments suggest that the likelihood of successful rAAV-mediated gene therapy for DMD is high.
Exon skipping-mediated correction of the open reading frame of the DMD gene is likely to be the first gene therapy for DMD to reach the clinic. Indeed, trials have already progressed past phase 2 with encouraging results. Strong evidence has been provided to show targeted exon skipping, and restoration of functional dystrophin protein expression with higher doses of AOs, but no real therapeutic effect in terms of amelioration of the disease has been reported. Research is now concentrated on establishing the appropriate dosing regimen required for therapeutic effect. For exon-skipping therapy to be an available form of therapy to as many patients carrying appropriate deletions as possible, optimization of AOs for the targeted skipping of many different exons is required. This will involve a large input in terms of finance, time, and effort. To address this, international consortia of researchers are coordinating their efforts toward optimizing the remaining exons that require optimization. There is much published work on the use of PPMOs to enhance exon skipping seen in cardiac muscle, but with a cost toxicologically. The latest U.K. MDEX Consortium is concentrating on developing a novel PPMO for the skipping of exon 53 that is without adverse effect in vivo, but still targets the heart effectively. A trial using this PPMO is planned for 2013 (
Exon skipping is applicable only for frame-shifting deletions; even so, as a therapy it could have the potential to restore the reading frame for 63% of patients with DMD by single exon skipping and for 82% of patients if skipping of more than one exon is possible. The worst-case scenario is that this leaves nearly 40% of patients with DMD requiring another form of therapy if single exon skipping is achievable clinically (Table 1). This reinforces the requirement for the development of other gene-targeting strategies. Genome editing would induce permanent changes in the DMD gene, removing the need for repeated long-term administration. The benefit of genome editing as a clinical therapy would be extensive. It would overcome the potential immune-related problems posed by AAV vector-based strategies, and the high costs and potential toxicological risks of AO use. In addition, genome editing would produce full-length dystrophin protein, whereas expression of only truncated protein is possible with exon skipping and with AAV vector-based strategies. This may have a major impact on the functionality of the expressed protein, and as a consequence the success of gene editing at ameliorating the disease phenotype. However, it should be noted that progress in genome editing development has been disappointingly slow; work is now concentrating on increasing the efficiency, specificity, and delivery of gene editing as a potential therapy. The finding that DMD gene repair is possible by using a specific meganuclease together with a targeting repair matrix to drive cDNA knock-in by homologous recombination provides the basis for an exciting new area of research that should now be exploited.
DMD, Duchenne muscular dystrophy; Freq, frequency; ORF, open reading frame.
Antisense-mediated exon skipping aimed at reading frame restoration is mutation specific, and its actual applicability to deletions, duplications, and point mutations, reported in the Leiden DMD mutation database, is presented here. Single exon skipping would restore the reading frame for 63% of patients with DMD; if skipping of more than one exon is feasible, exon skipping would be a viable therapy for 82% of patients with DMD, leaving >18% of patients requiring another form of therapy. However, if skipping of only one exon is possible, more than 37% of patients with DMD would be untreatable by exon skipping. It should be noted that no consideration is made here of the varying functional bioactivities that may be associated with the different quasi-dystrophin proteins produced as a result of exon skipping.
Adapted from Aartsma-Rus et al. (2009).
Not all duplications may be viable for antisense therapy.
Excludes mutations (∼5%) not viable for antisense therapy, that is, mutations in the C-terminal domain, the N-terminal domain, and some cysteine-rich domains, and very large deletions (>36 exons).
Footnotes
Acknowledgments
The authors gratefully acknowledge the support of the Association Français contre les Myopathies, Wellcome Trust, U.K. Department of Health, Muscular Dystrophy Campaign, Duchenne Ireland, Action Duchenne, and Clinigene Network of the European Commission.
Author Disclosure Statement
G.D. and L.P. declare that they are inventors on patents for a number of DMD gene exon-skipping antisense oligonucleotide sequences. The authors declare that they have no other competing interests.