
Abstract
Giant axonal neuropathy (GAN) is caused by loss of function of the gigaxonin protein. On a cellular level GAN is characterized by intermediate filament (IF) aggregation, leading to a progressive and fatal peripheral neuropathy in humans. This study sought to determine if re-introduction of the GAN gene into GAN-deficient cells and mice would restore proper cytoskeleton IF homeostasis. Treatment of primary skin fibroblast cultures from three different GAN patients with an adeno-associated virus type 2 (AAV2) vector containing a normal human GAN transgene significantly reduced the number of cells displaying vimentin IF aggregates. A proteomic analysis of these treated cells was also performed, wherein the abundance of 32 of 780 identified proteins significantly changed in response to gigaxonin gene transfer. While 29 of these responding proteins have not been directly described in association with gigaxonin, three were previously identified as being disregulated in GAN and were now shifted toward normal levels. To assess the potential application of this approach in vivo and eventually in humans, GAN mice received an intracisternal injection of an AAV9/GAN vector to globally deliver the GAN gene to the brainstem and spinal cord. The treated mice showed a nearly complete clearance of peripherin IF accumulations at 3 weeks post-injection. These studies demonstrate that gigaxonin gene transfer can reverse the cellular IF aggregate pathology associated with GAN.
Introduction
Giant axonal neuropathy was characterized over 25 years ago as an inborn error in intermediate filament organization (Pena et al. 1983), which was later found to be from mutations in the gigaxonin gene and resulting loss of functional gigaxonin protein (Bomont et al., 2000). More than 40 mutations have been identified in GAN patients, including deletions, insertions, missense and nonsense mutations, which lead to loss of function of gigaxonin (Bomont et al., 2000, 2003; Bruno et al., 2004; Demir et al., 2005; Houlden et al., 2007; Koop et al., 2007; Kuhlenbäumer et al., 2002; Leung et al., 2007). Gigaxonin is a broadly expressed Cul3 ubiquitin ligase adaptor protein, normally present at extremely low levels throughout the brain (Cleveland et al., 2009). Known direct targets of gigaxonin are MAP8, MAP1B, and TCB-C, which are involved with IF regulation (Allen et al., 2005; Ding et al., 2002, 2006; Wang et al., 2005). In the absence of gigaxonin, the regulation of IFs is disrupted, leading to increased IFs that can form inclusion bodies. The composition of these inclusions varies based on the specific IF composition in any given cell type. Vimentin, peripherin, neurofilaments (NF-H, NF-M, NF-L), alpha-internexin, GFAP, nestin, desmin, and keratin are IFs known to accumulate in mice and/or humans lacking gigaxonin (Berg et al., 1972; Dequen et al., 2008; Pena et al., 1983). Abnormal aggregates of keratin in hair are likely responsible for the typical curled hair observed in most patients (Bomont and Koenig, 2003; Pena et al., 1983; Prineas et al., 1976). The vimentin aggregates in fibroblasts are located in the perinuclear space close to the microtubule organizing center (Bomont and Koenig, 2003). Gigaxonin is also a substrate for ubiquitination and degradation by its own Cul3 ubiquitin ligase complex, providing a level of self-regulation at the protein level (Zhang et al., 2005).
Unfortunately, the underlying function(s) of gigaxonin and the mechanism by which the protein regulates IFs is poorly understood. The structural/functional domains of gigaxonin include an N-terminal BTB domain (BR-C, ttk, and bab), a centrally located conserved BACK domain, and six kelch motifs in the C-terminal part. Gigaxonin has a suspected role as a substrate-specific adaptor for the BCR (BTB/Kelch protein-cullin-3-RING box protein1) E3 ubiquitin ligase complex (Furukawa et al., 2003). As such an adaptor, it may facilitate proteasome targeting and degradation of various substrate proteins such as MAP1B, MAP8, and TBC-B via interactions with its C-terminal kelch domains (Ding et al., 2002, 2006; Wang et al., 2005). It has been postulated that these substrates accumulate in the absence of gigaxonin, and this hypothesis was partly confirmed in GAN knock-out (KO) mice (Allen et al., 2005; Ding et al., 2006; Wang et al., 2005) but not in human cells (Cleveland et al., 2009). However, no direct link has been established to explain how this leads to disregulation of IFs.
Conceptually, the unbalanced regulation of IFs could be corrected by re-introducing gigaxonin. In such a scenario, it is also conceivable that restoring normal IF regulation could allow cells to resolve IF inclusions and reverse existing pathology. Carriers of mutant gigaxonin alleles (humans and mice) express approximately 50% levels of gigaxonin and are phenotypically and pathologically normal, suggesting that restoration of lower-than-normal gigaxonin levels could still be efficacious. While gross overexpression of gigaxonin has a conceptual risk for toxicity, the protein-level autoregulation of gigaxonin abundance should correct for mild overexpression. Therefore, a broad therapeutic index of gigaxonin expression levels is expected. For these reasons, we propose that GAN is an excellent candidate for a gene replacement therapy. This study sought to determine if re-introduction of gigaxonin into GAN-deficient cells would restore proper cytoskeleton IF homeostasis, reversing the cellular disease phenotype of IF aggregation. In this study, skin fibroblast cultures from three different GAN patients were transduced with an adeno-associated virus type 2 (AAV2) vector containing a normal human GAN transgene or a green fluorescent protein (GFP) transgene as a control. Reduction in vimentin aggregation following viral delivery was scored microscopically. To further characterize the global intracellular changes associated with gigaxonin re-introduction, the proteome of these cells was analyzed after viral delivery, identifying and measuring the relative abundance of proteins using a differential proteomics method based on the iTRAQ strategy in combination with two-dimensional liquid chromatography mass spectrometry (2D-LC-MS) on a MALDI TOFTOF MS platform. Finally, to assess the potential application of this approach in vivo and eventually in humans, GAN-KO mice were injected with an AAV9/GAN vector and scored for reduction of the neuronal IF peripherin in the brainstem and spinal cord. This study sought to understand intracellular changes that occur in response to the re-introduction of gigaxonin into GAN-null cells, both in vitro and in vivo.
Methods
Patients fibroblast cultures
Primary skin fibroblasts were maintained in MEM-alpha (Gibco no. 12561) containing penicillin and streptomycin with 10% fetal bovine serum (FBS), at 5% CO2 and 37°C. P1 has a homozygous missense mutation (p. Ala49Glu), P2 has heterozygous mutations (p. S52G and C393X), and P3 has a homozygous deletion (c.1647-8680_*258del, encompassing exons 10 and 11). These patients were all clinically verified to be GAN patients with typical symptoms. P2 was described previously (Bomont et al., 2000), and an Ala49Thr (similar to P1) has been described (Bruno et al., 2004). The normal human fibroblast line was transformed with hTERT.
Vector preparation
rAAV vectors were produced using a triple-transfection method in HEK293 cells as previously described (Gray et al., 2011a). Highly pure recombinant virus was recovered using sequential step and continuous CsCl gradients, then the peak fractions were dialyzed in phosphate-buffered saline (PBS) containing 5% D-sorbitol. Viral titers were determined by dot blot or quantitative polymerase chain reaction (qPCR) as described (Gray et al., 2011a). The recombinant vectors in these studies were single-stranded AAV using AAV2 inverted terminal repeats, packaged in either an AAV2 or AAV9 capsid. The CMV-GAN construct used a cytomegalovirus (CMV) enhancer/promoter, myc-tagged gigaxonin coding sequence, and SV40 polyA. The GFP construct used the same CMV enhancer/promoter, enhanced GFP coding sequence, and SV40 polyA.
Vector treatments in vitro, immunocytochemistry, and vimetin aggregate scoring
For proteomic analysis, cells were grown in 10 cm plates. Cells were seeded 2.5×105 per plate, in triplicate to three plates. After 1 day, 5×1010 vector genomes (vg) of either ssAAV2/CMV-GAN or ssAAV2/CMV-GFP were added to each plate. Three days post-infection, cells were harvested with trypsin, washed with PBS, and cell pellets were frozen at −80° C until use.
For immunocytochemistry, 10,000 cells per well were seeded onto poly-L-lysine coated coverslips (BD no. 354085) in 24-well plates on day 0. On day 1, 5×109 vg of AAV2 vector, or a corresponding amount of PBS containing 5% sorbitol, was added per well. On day 2 the media was changed. On days 4–5 (3–4 days post-infection), cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. Cells were then washed with PBS and incubated twice for 10 min at room temperature with PBS containing 0.1% Triton-X100. Blocking was done for 1 hr at room temperature with PBS containing 3% goat serum and 0.1% Triton-X100. Primary antibody incubation was for 1 hr at room temperature with PBS containing 3% goat serum, 0.1% Triton-X100, 1:1000 dilution of mouse anti-vimentin (Millipore no. CBL202), and 1:500 dilution of rabbit anti-GFP (Millipore no. 3080). After three washings with PBS containing 0.1% Triton-X100, secondary antibody incubation was for 1 hr in PBS containing 3% goat serum, 0.1% Triton-X100, goat anti-mouse Alexa594 (Invitrogen no. A11032), and goat anti-rabbit Alexa488 (Invitrogen no. A11008). After washing twice with PBS, coverslips were mounted onto slides using ProLong Gold Antifade mounting media with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen no. P-36931). The presence of vimentin aggregates were scored visually, with >500 cells typically assessed per coverslip.
qPCR analysis of vector genomes and gigaxonin mRNA expression
RNA was recovered from the cells using the RNA Mini Plus kit (Qiagen); tissue lysis was performed with the TissueLyser (Qiagen), and automated purification was carried out on a Qiacube (Qiagen). RNA was treated to remove any residual genomic DNA as follows: RNA was incubated for 10 min at 37°C with 20 U Protector RNase Inhibitor (Roche), 1 × DNase I reaction buffer (NEB) and 2 U of DNase I (NEB). This was followed by an additional incubation of 10 min at 75°C with EDTA added to a final concentration of 2.5 mM for DNaseI inactivation. cDNA was synthesized from the purified RNA using the Transcriptor First Strand cDNA Synthesis kit (Roche), following the manufacturer's instructions, then used directly as template for reverse transcription (RT)-qPCR. RT-qPCR amplifications and analysis were carried out on a Roche Lightcycler 480, following the manufacturer's instructions and as described (Gray et al., 2010). All primer sets used SyBR green for quantitation except for the GAPDH primer, which used a primer and hydrolysis probe combination.
To quantify the myc-tagged gigaxonin transgene separate from the endogenous gigaxonin, primers were designed so that the upstream primer annealed to the myc tag. An internal set of gigaxonin primers were also used that would equally recognize the endogenous and transgene gigaxonin. qPCR of cDNA samples was used to determine the absolute numbers of myc-gigaxonin, total gigaxonin, and GAPDH transcripts per unit volume in each sample. Transcript quantification is reported as the absolute number of gigaxonin transcripts per microliter divided by the absolute number of actin transcripts per microliter in the sample. Primers for myc-tagged human gigaxonin were as follows: FORWARD: 5′-tgatcagcgaggaggacctgg-3′; REVERSE: 5′-gttgatccatcatcttttggagg-3′. Internal gigaxonin primers recognizing the endogenous and transgene gigaxonin were as follows: FORWARD: 5′-ggttatgagagagatcctgga-3′; REVERSE: 5′-cgtgatggaggcagtaatgta-3′. The human GAPDH reactions use the following primers and probe: FORWARD: 5′-agccacatcgctcagacac-3′; REVERSE: 5′-gcccaatacgaccaaatcc-3′; PROBE: UPL#60 (Roche cat no. 04688589001). A melting curve analysis was performed at the end of each SyBR green qPCR run, following the manufacturer's instructions, to verify the purity of the PCR product.
iTRAQ reagent labeling and peptide separation
Proteins were extracted by resuspending each cell pellet in 200 μL of lysis buffer, containing 0.05 M triethyl ammonium bicarbonate, 7 M urea, 2 M thiourea, and protease inhibitor cocktail (Roche). The cells were sonicated, and the supernatant was collected after centrifugation at 13,000×g for 20 min at 4°C. An acetone precipitation was performed prior to the protein concentrations measurement by the Bradford protein assay (Thermo Scientific). A summary of the experimental workflow is shown in Supplementary Figure S2 (Supplementary Data are available online at
The labeled peptide mixture was separated on an Ettan liquid chromatography system (GE Healthcare), using a Luna C18 column (2.0 mm i.d.×150 mm length×5 μm, 100 Å column; Phenomenex), as described by Mussche et al. (2012). The buffer compositions were 20 mM HCOONH4, 2% ACN at pH 10 for buffer A and 20 mM HCOONH4, 80% ACN at pH 10 for buffer B. The flow rate was 0.2 mL/min, and the elution gradient was as follows: 7 min 100% A, 60 min 0%–50% B, 15 min 50%–100% B, 10 min 100% B, and 3 min 100% A. Fractions were collected every minute. After drying, the fractions were redissolved in 0.1% formic acid for nano-LC/MS analysis.
Nano-LC/MS analysis
Off-line LC-MS was done as described by Mussche et al. (2012). Briefly, analytical reversed phase separation of the labeled peptide mixture was performed on an Ultimate Plus Dual-gradient Capillary/Nano-LC system (Dionex-LC Packings) using a C18 column (75 μm i.d.×150 mm length, 3 μm, 100 Å; Dionex) at a flow rate of 250 nL/min. The mobile phases were 5% ACN/0.05% trifluoroacetic acid (TFA), pH 3 for buffer A and 80% ACN/0.05% TFA, pH 3 for buffer B. Peptides were resolved by gradient elution using a gradient of 0%–50% buffer B over 25 min, 50%–100% buffer B over 10 min, 5 min of 100% buffer B, and 10 min of 100% buffer A. Fractions were collected every 30 sec directly on a MALDI target plate. The MALDI plates were analyzed by a 4800+ MALDI-TOFTOF Proteomics Analyzer (AB SCIEX). A plate calibration was performed using the 4700 calibration mixture with a mass tolerance of 0.5 m/z. The MSMS mode was calibrated with a mass tolerance of 0.1 m/z. Additionally, MS spectra were internally calibrated with Glu-fibrinopeptide (1552.62 m/z). MS data were acquired in the reflector positive mode over a mass range of 900–3500 m/z, by accumulation of 1000 laser shots per spectrum. The eight highest precursor masses in each MS spectrum with an S/N threshold of minimum 50, were selected for fragmentation, and the fraction-to-fraction mass tolerance was set to 200 ppm. MS/MS data were acquired with 2000 laser shots per spectrum, using 1 keV collision energy and air as the collision gas. Each plate was analyzed twice by reinterpretation of the MS spectra with a method containing an exclusion list from initial MS/MS data to avoid analyzing peptides more than once.
Database searching and data analysis
Data analysis was performed with the software package Mascot Daemon (Perkins et al., 1999; version 2.3, Matrix science). All MS/MS spectra were processed against the UniProtKB/SwissProt Homo sapiens database. Search parameters included iTRAQ labeling at the N-terminus and lysine residues, cysteine modification by methyl methanethiosulfonate, digestion by trypsin (including two missed cleavages allowed), and methionine oxidation as a variable modification. Normalization of the ratios was performed by the median ratio. A significance threshold of p<0.05 was used for peptide identification and an ion score cutoff of 30. Only unique peptides were selected for quantification, and protein ratios were calculated by the weighted average of the peptide ratios. Using the build in decoy function, searching a concatenated database containing both forward and shuffled decoy sequences, allowed estimation of the false discovery rate (Reidegeld et al., 2008). The ratio of GAN/GFP was considered in the three patients and accepted as significant regulated when the ratios of all three patients samples showed a significant value at the 95% confidence level (p<0.05).
The function, interactions and localization of the differential regulated proteins were studied based on information in databases such as Uniprot, neXtProt, and STRING (Snel et al., 2000), only considering experimentally confirmed interactions.
Western blotting of fibroblast samples
For validation of the proteins found by iTRAQ, fibroblast protein extracts of each sample (proteins sonicated in lysis buffer) were separated by tricine-SDS-PAGE on a 16.5% polyacrylamide gel. Proteins were transferred by electroblotting (3 hr at 60 V) onto a nitrocellulose membrane (Invitrogen). Immunoblotting was carried out with primary antibodies against the galectin-1 (ab25138, Abcam), PARK7/DJ1 (Abcam ab18257), and β-actin (Abcam). The intensity of the protein bands was quantified using the Quantity One imaging software (Bio-Rad).
Mouse studies
All investigations were approved by the University of North Carolina–Chapel Hill Institutional Animal Care and Use Committee. Ten microliters of ssAAV9/CMV-GFP (8×1010 vg) or ssAAV9/CMV-GAN (7.2×1010 vg) vector were injected into the cisterna magna. The AAV9/GFP injections were done in 19–20 month-old heterozygous GAN mice, and the ssAAV9/GAN injections were done in KO GANΔex1 (Dequen et al., 2008) or GAN Δex3–5 (Ding et al., 2006) mice. Three weeks following vector injection, each mouse was sacrificed and transcardially perfused with PBS or PBS containing 1 U/mL heparin (Abraxis).
Immunohistochemistry to detect peripherin aggregates
After 48 hr of fixation in freshly made PBS with 4% paraformaldehyde, the entire brain and portions of the cervical and lumbar spinal cords were sectioned at 40 μm using a Leica vibrating microtome. Every fifth section was processed for immunohistochemistry (IHC). Samples were incubated for 1 hr at room temperature in blocking solution (10% goat serum, 0.1% triton X-100, 1 × PBS), then incubated 48–72 hr at 4°C in primary antibody solution (3% goat serum, 0.1% triton X-100, 1 × PBS, and primary antibody). Primary antibodies were as follows: rabbit anti-GFP (Millipore no. AB3080, 1:500), rabbit anti-peripherin (Millipore no. AB1530, 1:500), mouse anti-neurofilament heavy chain (NF-H, Covance no. SMI-32P, 1:1000). After washing three times in PBS, secondary amplification was performed using a VectaStain ABC Elite Kit (Vector Labs, no. PK-6101 for rabbit and PK-6102 for mouse, as appropriate) with 3,3′-diaminobenzidine tetrachloride (DAB; Polysciences, Inc. no. 04008) substrate and nickel-cobalt intensification of the reaction product.
DAB-processed brain sections were digitized using a Scan-Scope slide scanner (Aperio Technologies). Virtual slides were viewed using ImageScope software package (v. 10.0; Aperio Technologies), which allowed sections to be viewed at equivalent magnifications.
Western blot of mouse tissue
To validate expression of the gigaxonin transgene following intracisternal vector delivery, a small portion of the caudal brainstem was homogenized and subjected to SDS-PAGE, then blotted. The myc-tagged gigaxonin band was detected with a goat anti-gigaxonin antibody (N-18, Santa Cruz no. 49688, 1:300) or a mouse anti-myc antibody (Millipore no. 05–724, 1:1000).
Results
Reduction of vimentin aggregates following gigaxonin gene transfer
The first part of this study sought to investigate the effects of AAV/gigaxonin gene transfer into GAN patient fibroblasts, in terms of the IF organization and potentially broader effects on the proteome (Fig. 1). Dermal fibroblasts taken from human GAN patients show cytoskeletal disorganization and aggregation of the IF vimentin when grown in culture, and these accumulations may be enhanced by culturing them in low serum media (0.1% FBS) or growing cells to confluence for 3 days (Bomont and Koenig, 2003). We have observed that when these cells are grown on poly-L-lysine coated coverslips the accumulations are enhanced even in 10% FBS-containing media. Untransformed fibroblasts from three GAN patients and a normal human fibroblast line were seeded, then infected the next day with AAV2 vectors (CMV-GFP, CMV-GAN, or PBS “mock”) at an multiplicity of infection (MOI) of 5×104 vg/cell. The media was changed 24 hr after infection and then the cells were grown in complete media (10% FBS) for an additional 2–3 days (3–4 days post-infection). Cells were then fixed and immunolabeled for vimentin, and the percentage of vimentin aggregate-containing cells were scored.
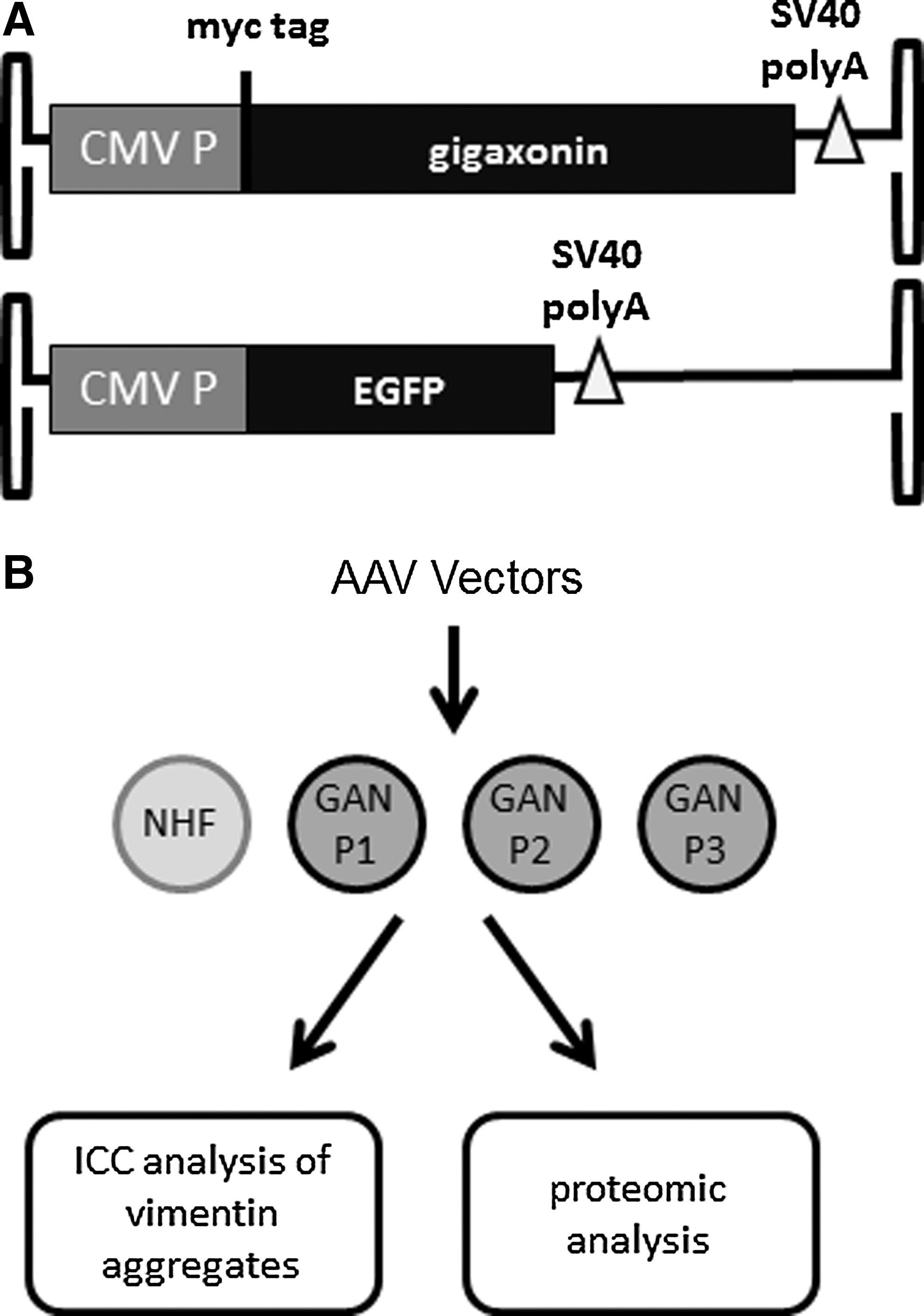
Study design.
Approximately 53% of the fibroblasts were transduced (average of nine trials, range 30% to 71%), as gauged by the number of cells GFP-positive in the AAV2/GFP control group. No suitable anti-gigaxonin antibodies were available to immuno-label for cells expressing the gigaxonin transgene, and immunocytochemistry against the myc tag also failed to detect the gigaxonin protein. Treatment with the AAV2/GAN vector resulted in a significant reduction in the percentage of cells containing vimentin aggregates, as compared to the mock-infected and AAV2/GFP-infected cells (Fig. 2). Analysis of the mRNA from the cells by RT-qPCR indicated a 3.8-, 1.1-, and 12.4-fold (P1, P2, and P3 respectively) average increase in gigaxonin mRNA in GAN-treated patient cells versus mock-treated patient cells. The amount of gigaxonin mRNA seen in mock-treated patient cells was equivalent when comparing P1 and P2 to normal human fibroblasts, but the P3 mRNA levels were lower. This is understandable since P1 and P2 had missense mutations that were unlikely to affect the mRNA expression and stability, whereas P3 had a homozygous deletion at the 3′ end of the gene. The overall average amount of gigaxonin mRNA in GAN-treated cells was 6.2-, 2.6-, and 5.7-fold (P1, P2, and P3 respectively) above that detected in normal human fibroblasts. These results indicated that gigaxonin was being mildly overexpressed at 3 days post-infection in this experiment.

Reduction of vimentin aggregates after AAV/gigaxonin gene transfer. Cultured GAN patient fibroblasts were treated with in parallel with vehicle buffer (mock), AAV2/GAN vector (GAN), or AAV/GFP vector (GFP), then fixed and immunostained for vimentin after 3–4 days.
There was a small decrease in the overall number of fibroblasts following the AAV2/GAN treatment, suggesting the possibility of overexpression-related toxicity, but it isn't clear if this was due to the gigaxonin gene or high doses of AAV used (p=0.03 compared to mock treatment and p=0.69 compared to GFP treatment, Supplementary Fig. S1). No vimentin aggregates were observed in any of the normal human fibroblasts, and no obvious change in vimentin was observed in the normal human fibroblasts following AAV2/GAN treatment. These results indicate that gigaxonin can function to reduce vimentin IF aggregations and restore normal vimentin organization within 3–4 days of infection.
Proteome analysis
Currently, there is an incomplete understanding of the cellular function of gigaxonin, and gaps of knowledge exist regarding both its direct binding partners and its downstream targets. To assess the more global consequence of gigaxonin re-introduction into GAN patient fibroblasts, we undertook a proteomic analysis of the cells following AAV2/GAN or AAV2/GFP gene transfer. Protein extracts from the total cell lysate from six cultured fibroblast cell line samples (three GAN-infected and three GFP-infected patient cultures) were trypsinized and iTRAQ-labeled, followed by reversed phase (RP)-RP LC and MALDI-TOFTOF MS/MS analysis for identification and quantitation. All RP-LC MALDI-TOFTOF runs were searched against the UniProtKB Homo sapiens database, resulting in identification of 14,460 peptides leading to 780 proteins. Proteins with all six iTRAQ labels were considered in the final list. The iTRAQ protein ratios reported here are weighted averages calculated from the ratios of the individual peptides determined for each protein with the accompanying SD and p value. From the total of 780 proteins identified with >95% confidence, 31 proteins were found to be significantly regulated (p<0.05) between GAN- and GFP-transfected cells in all three patients (Table 1). Changes seen between different samples will not be due to any AAV-specific effects since each AAV2/GAN-treated culture was compared with an AAV2/GFP-treated culture from the same patient.
Uniprot accession number.
Percentage of protein sequence coverage.
Number of peptides.
Patient 1, ratio of GAN-transfected to GFP-transfected cells (p<0.05).
Patient 2, ratio of GAN-transfected to GFP-transfected cells (p<0.05).
Patient 3, ratio of GAN-transfected to GFP-transfected cells (p<0.05).
Proteins identified in this study but not altered between GAN patients and controls (Mussche et al., 2012).
The differentially regulated proteins were further analyzed according to their function, interaction and localization, based on information in databases such as UniProt, neXtProt, and STRING (Snel et al., 2000). Some possible interesting interactions with components of the ubiquitin-proteosome system are listed in Supplementary Table S1. A set of proteins previously described to be differentially regulated in GAN patients versus controls was found not altered in response to the AAV2/GAN treatment (Supplementary Table S2). A list of all proteins detected is provided as Supplementary Table S3. Interestingly, we found no change in the overall levels of vimentin in response to the AAV2/GAN treatment.
Western blot analysis was performed to validate results obtained from the proteome analysis. Galectin-1 and DJ-1 were selected, as according to the iTRAQ results these were significantly down-regulated following gigaxonin gene transfer, and galectin-1 was previously shown to be overexpressed in GAN patients compared to controls (Mussche et al., 2012). This means that this is probably an important player in GAN. Even though the degree of reduction detected in the proteomic analysis was modest (0.62–0.88 fold compared to the GFP treatment), the Western blot results confirmed the down regulation of galectin-1 and DJ-1 in the transduced fibroblasts (Fig. 3).
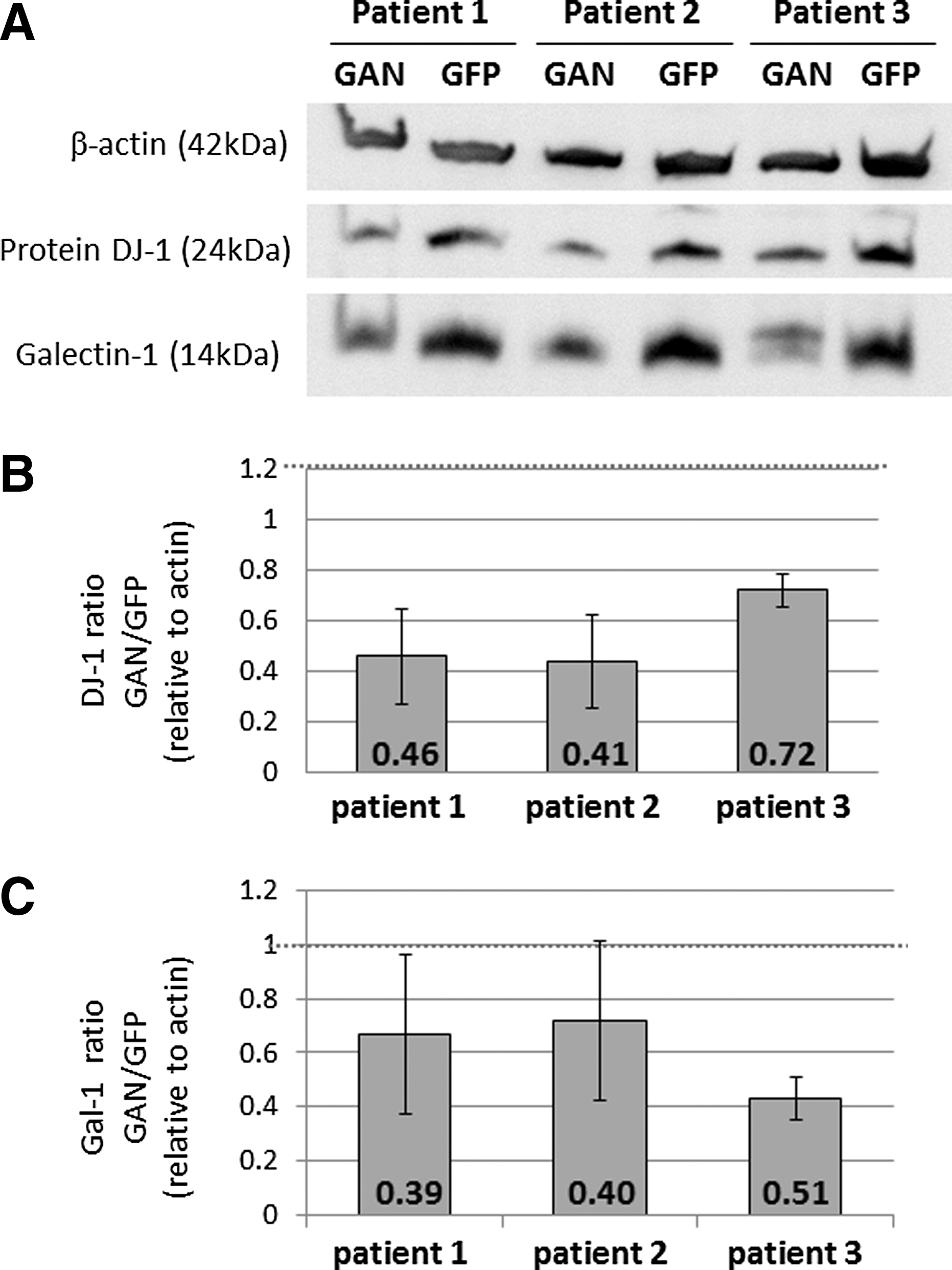
Western blot validation of changes in protein levels. Western blot experiments were performed on protein extracts of fibroblasts after treatment with AAV vectors.
In vivo rescue of peripherin IF aggregates following central nervous system AAV9/GAN gene transfer
To assess the possible translational potential of an AAV/GAN vector, a similar experiment to the in vitro vimentin IF rescue was undertaken in vivo. There are three KO mouse models described for GAN (Dequen et al., 2008; Ding et al., 2006; Ganay et al., 2011). The first was described as recapitulating many of the human disease symptoms, but subsequent rigorous analysis of all three mouse models have shown that the mice do not develop any behavioral, motor, or sensory deficits until >12 months of age, and these effects are fairly modest (Ganay et al., 2011). However, these mouse models do display IF aggregates that are a hallmark of the underlying disease pathology, including aggregations of neurofilaments, alpha-internexin, and peripherin in the central nervous system (CNS) that are visible as early as 3 months of age (Dequen et al., 2008).
In this experiment, we introduced the GFP or GAN gene into two of the GAN-KO mouse models (GAN Δex1 and GAN Δex3–5 ) when these mice were 17–21 months old (see Supplementary Table S4 for a list of subjects and results summary). Under anesthesia, mice received a single injection into the cerebrospinal fluid of the cisterna magna. Ten microliters (7.2×1010 total vg) of AAV9/CMV-GAN was injected into KO mice or 10 μL (8×1010 total vg) of AAV9/CMV-GFP was injected into age-matched heterozygous mice, then the mice were allowed to recover. When injected into the cisterna magna in mice, AAV9 efficiently transduces large areas of the brainstem, dorsal spinal cord, motor neurons in the ventral horn, and to a much lesser extent other areas of the brain as visualized with GFP (Fig. 4A–C). In the absence of a good behavioral readout in these mice for a phenotypic rescue of function, a histological evaluation of IF aggregate resolution was pursued. Three weeks post-injection with the AAV9/CMV-GAN vector, mice were sacrificed for IHC evaluation of the IF aggregates in the brain. No IHC was done against gigaxonin because no suitable antibodies are available and IHC against the myc epitope failed. We observed nearly complete clearance of peripherin aggregates in the brainstem and dorsal lumbar spinal cord, in both the GAN Δex1 and GAN Δex3–5 KO mice (Fig. 4D–F, Supplementary Figs. S3 and S4). As expected, heterozygous mice did not show any abnormal peripherin staining and homozygous KO mice showed consistent and extensive aggregates of peripherin, at ages ranging from 8 to 24 months (Supplementary Table S4). Expression of exogenous myc-tagged human gigaxonin in the brainstem was verified by Western blot analysis of brainstem tissue from treated mice, compared to heterozygous or untreated KO mice of similar age (Fig. 4G). The amount of gigaxonin in treated mice compared to heterozygous controls indicated a higher degree of overexpression in the mice at 3 weeks post-infection compared to the mild overexpression seen in fibroblasts at 3 days post-infection (compare WB in Fig. 4G to Fig. 2). Although the study investigated peripherin aggregates in age-matched KO mice, and in some cases littermates, we wanted to verify to the best extent possible that the treated mice had IF aggregates pre-injection and that these were resolved as a result of the AAV9/GAN treatment. Because areas of the midbrain and forebrain were not efficiently targeted by AAV9 via this route of administration, they were stained for peripherin (midbrain, dorsal raphe nucleus) and neurofilament-heavy chain (NF-H, cortex and striatum) to investigate the presence of IF aggregates in these areas. Despite the absence of peripherin aggregates in the brainstem and dorsal spinal cord, the treated mice still had peripherin aggregates in the dorsal raphe nucleus and NF-H aggregates in the cortex and striatum (Supplementary Fig. S5). Although a comprehensive histopathological evaluation was not done on these mice, they did not display any adverse behavior phenotypes or gross brain pathology as a result of the treatment. These data provide strong evidence that the AAV9/GAN vector was capable of resolving long-standing peripherin IF aggregates in the CNS.

Resolution of peripherin aggregates in GAN mice. Wild-type (WT) C57Bl/6J mice were injected with 8×1010 vector genomes (vg) ssAAV9/CMV-GFP into the cerebral spinal fluid (CSF) of the cisterna magna, then GFP expression was assessed in the forebrain
Discussion
As a whole, these experiments provide critical proof-of-concept data that re-introduction of gigaxonin into affected GAN-null cells resolves IF aggregates, which are a major hallmark of the underlying disease pathology in mice and humans. This was demonstrated in vitro in cultured GAN-null fibroblasts with the IF vimentin and in vivo in GAN-null mice with the IF peripherin. Moreover, the proteomic analysis of treated GAN primary fibroblasts provides important information with regards to more global changes (besides IFs) that occur in treated cells, which provides further insights into both the mechanism of the rescue and the basic biology of gigaxonin's function. While several reasonable conclusions can be drawn from these studies, the results should be considered within the appropriate context; namely that gigaxonin was overexpressed using the CMV promoter. Also, for the proteomic analysis, it is important to consider that this analysis was restricted to a single time point of 3 days post-infection. The timing was restricted to allow enough time for gigaxonin expression without prolonged maintenance of the dividing fibroblast culture (which would dilute transgene-expressing cells). Our results indicated that gigaxonin may play a role in multiple pathways affecting cytoskeleton regulation, and thus it may take longer than practical for an in vitro experiment to observe all proteome-level changes, especially for proteins with long half-lives. Therefore we can only interpret the presence of proteome-level changes, but not the absence of change.
Proteome analysis
Mussche et al. (2012) had previously done a proteomic analysis of GAN patient fibroblasts compared to normal control fibroblasts. One interesting finding is that three proteins that were previously found to be disregulated in GAN patients compared to healthy controls again showed altered expression levels in the AAV2/GAN-treated cells, only the regulation was seen in the other direction towards normal levels. This is the case for galectin-1, aminopeptidase N (APN), and FK506-binding protein 9. Interestingly, a set of proteins previously found to be differentially regulated in GAN patients versus controls was not found to be altered in response to the AAV2/GAN treatment (Supplementary Table S2). Those results would suggest that while some of the disregulated proteins in GAN can rapidly return to homeostatic levels in response to gigaxonin reintroduction, other changes may take longer to correct or may represent irreversible changes. However, since the proteomic changes in our experiment can be correlated to resolution of IF aggregates within the 3- to 4-day experimental timeframe, we can speculate that the proteins that modulated in response to gigaxonin reintroduction are important in the GAN disease mechanism. In particular a role for galectin-1 was previously proposed for GAN, as a regulator of cytoskeleton reorganization (Mussche et al., 2012). The FK506BP9 belongs to the FK506BP family, which comprises peptidyl prolyl isomerases that function as protein-folding chaperones and are also involved in the aggregation process of alpha-synuclein clusters in Parkinson disease (Gerard et al., 2012). APN is a transmembrane zinc-binding metallo-ectopeptidase that removes amino acids from unblocked N-termini of bioactive peptides or proteins. It plays a role in cell migration and invasion of extracellular matrix, angiogenesis, and regulation of extrinsic regulatory factors of tumor growth, all conditions in which remodeling of the cytoskeleton is required. The introduction of the correct GAN-gene will activate several processes in reaction to this new gene, which is reflected in up- or down-regulation of a set of proteins (Table 1). The majority of the differentially regulated proteins in the present study were previously identified in the proteomic analysis done by Mussche et al., but were not found to be disregulated in GAN fibroblasts (indicated in Table 1; Mussche et al., 2012). This may indicate that they are associated with GAN overexpression, rather than a return to a normal state. Some of these proteins were found to be associated with several ubiquitin specific peptidases (Sowa et al., 2009), and the ubiquitin-proteasome system, as listed in Supplementary Table S1. Ubiquitin-specific peptidases are a class of deubiquitinating enzymes that help to regulate many cellular processes by controlling the amount of the protein ubiquitin, through removing ubiquitin from a large variety of substrates (D'Andrea and Pellman, 1998; Wilkinson, 2000). But targets, interactions, and specific functions of deubiquitinating enzymes are poorly understood. However, it is possible that in response to GAN overexpression they play a compensatory role by deubiquitination of targeted proteins.
Since GAN fibroblasts don't display a rise in the overall abundance of vimentin (Mussche et al., 2012) and there was no change in overall vimentin levels following AAV2/GAN treatment (Supplementary Table S3), we can speculate that the absence (and re-introduction) of gigaxonin is affecting the cytoskeletal organization of vimentin, rather than its total intracellular levels. The reduction of vimentin aggregates seen in transduced GAN fibroblasts indicates that there is a fairly rapid reorganization of the cytoskeleton. Several disregulated proteins were found associated with cytoskeleton reorganization (Table 1). Actin-related protein 2/3 complex subunit 3 functions as a component of the Arp2/3 complex, which is involved in the regulation of actin polymerization and, together with an activating nucleation-promoting factor, mediates the formation of new branched actin networks. This is important during centrosome re-orientation before cytokinesis and in cytoskeletal organization. Destrin is an actin-depolymerizing protein, which severs actin filaments (F-actin) and binds to actin monomers (G-actin) and profilin4. Transgelin is an actin-crosslinking protein found in fibroblasts and muscle. The abundance of cytoskeleton proteins that were found changed in response to gigaxonin re-expression in this study reinforces gigaxonin's role in cytoskeleton organization, but a full model for all the interactions and pathways still remains elusive.
Another pertinent observation is the up-regulation of several endoplasmic reticulum resident proteins in GAN-treated fibroblasts, in particular enzymes involved in protein synthesis, maturation, and post-translational modification, in addition to enzymes involved in endocytosis and recycling. This reflects a general return to a functional state of the fibroblast.
Potential for GAN gene therapy
GAN is a progressively fatal sensory and motor neuropathy. It begins with the loss of distal motor and sensory function, and as the disease progresses into the teenage years individuals will gradually lose the ability to walk and use their arms. The disease is normally fatal in the third decade of life, often due to respiratory dysfunction or other complications normally associated with a motor neuron disease. While GAN patients will display IF cytoskeletal disorganization in many cell types in their body, successful treatment of the spinal cord and brainstem would be hypothesized to treat the most serious disease phenotypes.
AAV9 vector administration into the vein, lumbar intrathecal space, and cisterna magna have all been validated as ways to achieve broad CNS transgene delivery in large animal models (Duque et al., 2009; Federici et al., 2012; Gray et al., 2013). In the present study, cisterna magna injections were modeled in mice to investigate a potential therapeutic approach for GAN that might be translatable to humans. Although the behavioral phenotype of these mice is very mild and therefore not amenable to assess a phenotypic rescue, they do display extensive IF aggregates. Encouragingly, there was widespread resolution of peripherin aggregates throughout the brainstem and spinal cord at 3 weeks following vector delivery. Considering that IF accumulations (including peripherin) are present as early as 3 months of age in GAN mice (Dequen et al., 2008), this result demonstrates the ability of the gigaxonin gene to resolve long-standing IF aggregates that are a hallmark of the disease pathology in vivo.
While the present studies implicate gigaxonin for a role in cytoskeleton organization, future studies are still needed to understand the exact cellular pathways affected by gigaxonin. In terms of a future gene therapy for GAN, the present study indicates the potential of gigaxonin gene replacement as a therapeutic approach for GAN. However, the use of a strong promoter and a single-stranded AAV genome is not ideal. Since gigaxonin is normally expressed at very low levels (Cleveland et al., 2009), overexpression-related toxicity is a concern and our studies suggest this possibility (see indicated changes in Table 1 and Supplementary Fig. S1), so a weak promoter may be sufficient and preferred. Self-complementary (sc)AAV vectors would greatly increase the transduction efficiency with AAV (Gray et al., 2011b; McCarty et al., 2003), which could be critical for human translation, although the 1.9-kb size of the gigaxonin gene may be challenging to fit in a ∼2.2-kb scAAV genome with the appropriate transcriptional control elements. We speculate that these improvements would play an important role in the translation of these findings to future human application.
Footnotes
Acknowledgments
We wish to acknowledge funding from Hannah's Hope Fund. We are grateful for Yanmin Yang and Jean-Pierre Julien for providing the GAN mouse models. We thank Huijing Sun for performing some of the intracisternal injections, and Haiyan Fu for showing us how to do them.
Author Disclosure Statement
The authors do not declare any conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.