
Abstract
Collagen VI gene mutations cause Ullrich and Bethlem muscular dystrophies. Pathogenic mutations frequently have a dominant negative effect, with defects in collagen VI chain secretion and assembly. It is agreed that, conversely, collagen VI haploinsufficiency has no pathological consequences. Thus, RNA-targeting approaches aimed at preferentially inactivating the mutated COL6 messenger may represent a promising therapeutic strategy. By in vitro studies we obtained the preferential depletion of the mutated COL6A2 messenger, by targeting a common single-nucleotide polymorphism (SNP), cistronic with a dominant COL6A2 mutation. We used a 2′-O-methyl phosphorothioate (2′OMePS) antisense oligonucleotide covering the SNP within exon 3, which is out of frame. Exon 3 skipping has the effect of depleting the mutated transcript via RNA nonsense-mediated decay, recovering the correct collagen VI secretion and restoring the ability to form an interconnected microfilament network into the extracellular matrix. This novel RNA modulation approach to correcting dominant mutations may represent a therapeutic strategy potentially applicable to a great variety of mutations and diseases.
Introduction
We set out to test this hypothesis, using a novel molecular approach based on exon skipping, designing a 2′-O-methyl phosphorothioate (2′OMePS) antisense oligoribonucleotide (AON) to target a common COL6A2 single-nucleotide polymorphism (SNP) occurring in cis with the de novo dominant intron 9 c.954+17_954+22del28 UCMD mutation. Transfecting patient fibroblasts with this AON induced preferential skipping of the mutated exon via SNP recognition, and caused COL6A2 messenger out-of-framing, thereby depleting the mutated transcript via NMD. Skipping quantification confirmed AON efficacy, an RNA transcript count evidenced the preferential mutated transcript depletion, and protein and morphological studies showed reduced expression of the mutated COL6A2 allele. Both histology and electron microscopy studies revealed promotion of collagen VI integration into the extracellular matrix (ECM) and focal recovery of the microfibrillar network, thus confirming the feasibility of mutated messenger depletion, and thereby dominant mutation correction, via out-of-frame exon skipping.
Materials and Methods
UCMD mutation and P221(T)AON design
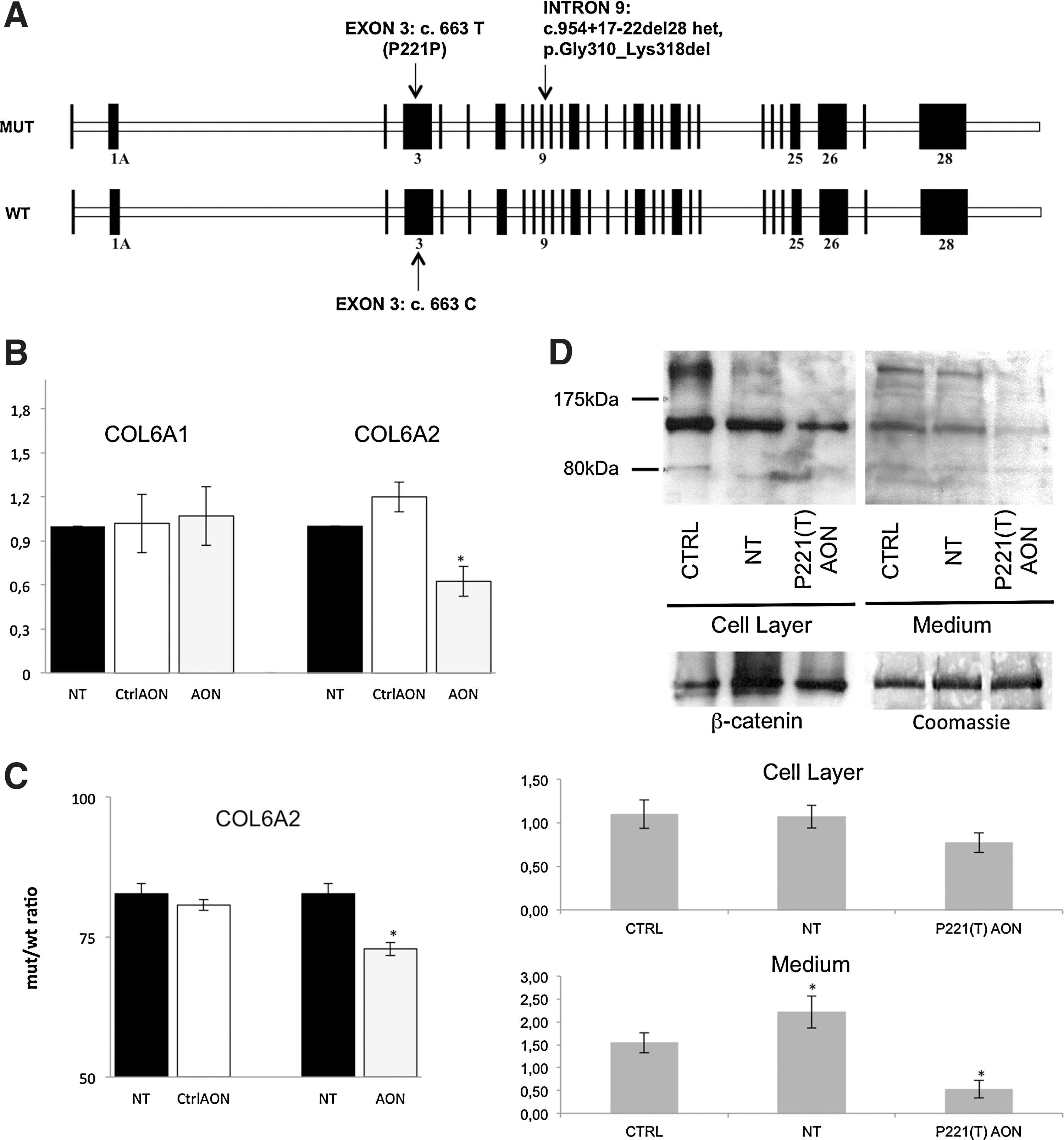
The UCMD mutation is a de novo, heterozygous 28-bp deletion within intron 9 of the COL6A2 gene (c.954+17_954+22del28) causing exon 9 skipping and predicting a triple-helix domain internal deletion (Gly310_LYS318del). In patients with UCMD, the mutation is cistronic, with the T allele of the common COL6A2 c.663C>T SNP (P221P) occurring in the out-of-frame exon 3 (Fig. 1A). A 2′OMePS-modified RNA AON [referred to as P221(T)AON] was designed, according to published guidelines (Aartsma-Rus, 2012), to target this T allele (Supplementary Fig. S1A; supplementary data are available online at
Cell culture and transfection
Primary human skin fibroblasts (obtained for diagnostic procedures with ethics committee approval no. 7/2009) were cultured at 37°C and 5% CO2 as described (Martoni et al., 2009). Cells were transfected for 4 hr with either AON (200 nM) plus AON ExGen 500 polyethylenimine (PEI) (3.3 μl/μg; MBI Fermentas/Thermo Fisher Scientific, Waltham, MA) or with PEI alone, as described (Evers et al., 2011).
RNA studies
Cells were harvested 24 hr post-transfection, and total RNA was isolated and retrotranscribed as reported (Martoni et al., 2009). RT-PCR was performed with COL6A primers within exons 2–10 and 4–10 (sequence available on request) to quantify exon 3 skipping, and to assess the proportions of wild-type (exon 9-plus) versus mutated (exon 9-minus) COL6A2 transcripts, calculated as the ratio between each type of transcript and the sum of all transcripts, on an Agilent high-sensitivity DNA chip (Agilent Technologies, Santa Clara, CA) (Supplementary Fig. S1B). TaqMan expression assays (Applied Biosystems) were used to quantify COL6A2 and COL6A1 transcripts (Hs00242484_m1 Ex27/28; Hs00242448_m1 Ex20/21) with respect to β-actin, the endogenous control. Real-time PCR was performed as described (Martoni et al., 2009). cDNAs from untreated fibroblasts served for calibration.
Western blot
Sixteen hours post-transfection, fibroblasts were supplemented with 0.25 mM sodium ascorbate for 24 hr. Medium was collected, and the cell layer was scraped and solubilized in radioimmunoprecipitation assay (RIPA) buffer. Protein content was quantified with a bicinchoninic acid (BCA) protein assay kit (Pierce/Thermo Fisher Scientific, Rockford, IL). Western blotting was performed as previously reported (Martoni et al., 2009). A loading check was performed with anti-β-catenin antibody (diluted 1:1000; BD Biosciences Transduction Laboratories, Lexington, KY) for cell layer extracts and Coomassie staining for culture medium.
Immunostaining
Sixteen hours post-transfection and after 24 hr in sodium ascorbate, cells were fixed with cold methanol and double-stained with anti-collagen VI (Fitzgerald, Concord, MA) and perlecan (Chemicon, Temecula, CA) antibodies, as previously reported (Sabatelli et al., 2001). After nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI), samples were mounted with antifade reagent (Molecular Probes/Invitrogen, Carlsbad, CA) and analyzed with an Eclipse 80i fluorescence microscope (Nikon Instruments, Melville, NY).
Immunoelectron microscopy
Cultures were incubated with 3C4 monoclonal antibody (Chemicon), diluted 1:50 in culture medium at 37°C for 3 hr, and revealed with anti-mouse IgG 5-nm gold-conjugated antibody (GE Healthcare, Piscataway, NJ), diluted 1:50 for 1 hr at 37°C. Rotary-shadowing electron microscopy was performed as described (Zhang et al., 2002). Samples were examined under a Philips EM 400 (FEI, Hillsboro, OR) or JEOL JEM 1011 electron microscope (JEOL, Tokyo, Japan) at 100 kV. For quantitative analysis of microfibrillar tetramer composition, 20 fields at ×17,000 magnification were evaluated for each condition.
Results
P221(T)AON induces exon 3 skipping, transcript out-of-framing, and COL6A2 transcript degradation
Patient-derived fibroblasts were transfected (in three independent experiments) with 200 nM P221(T)AON, and total RNA was isolated after 24 hr. RT-PCR and DNA chip analysis showed average exon 3 skipping of 30.3% (range, 21.3–39.3%), whereas none was observed in cells treated with control AON (against dystrophin exon 51) (Goemans et al., 2011). After P221(T)AON treatment, real-time PCR showed COL6A2 mRNA depletion to about 60% of baseline (0.62±0.10); COL6A1 expression was not significantly affected (1.07±0.20) (Fig. 1B).
P221(T)AON reduces collagen VI at cell layer and in medium
In parallel with transcript depletion, P221(T)AON treatment caused a decrease in the global amount of collagen VI in UCMD patient cell layer (Fig. 1D, left). P221(T)AON-treated samples showed even more pronounced reduction of collagen VI in the medium fraction (Fig. 1D, right) with respect to basal conditions. As medium extract is composed of proteins neither retained in cells nor deposited on ECM, the difference in collagen VI depletion between the two fractions seems to indicate more efficient post-treatment ECM deposition of the protein.
P221(T)AON affects mutated/wild-type COL6A2 allele ratio
To assess the sequence specificity and efficacy of P221(T)AON, the proportions of mutated versus wild-type COL6A2 transcripts were compared in treated versus untreated cells. In untreated cells, the mutated COL6A2 allele (missing exon 9) comprised 82.72±1.86% of the wild-type. After P221(T)AON transfection, this pathological messenger was reduced by 10% to 72.87±1.18%. Control AON did not significantly affect the allele ratio (80.70±1.00%) (Fig. 1C).
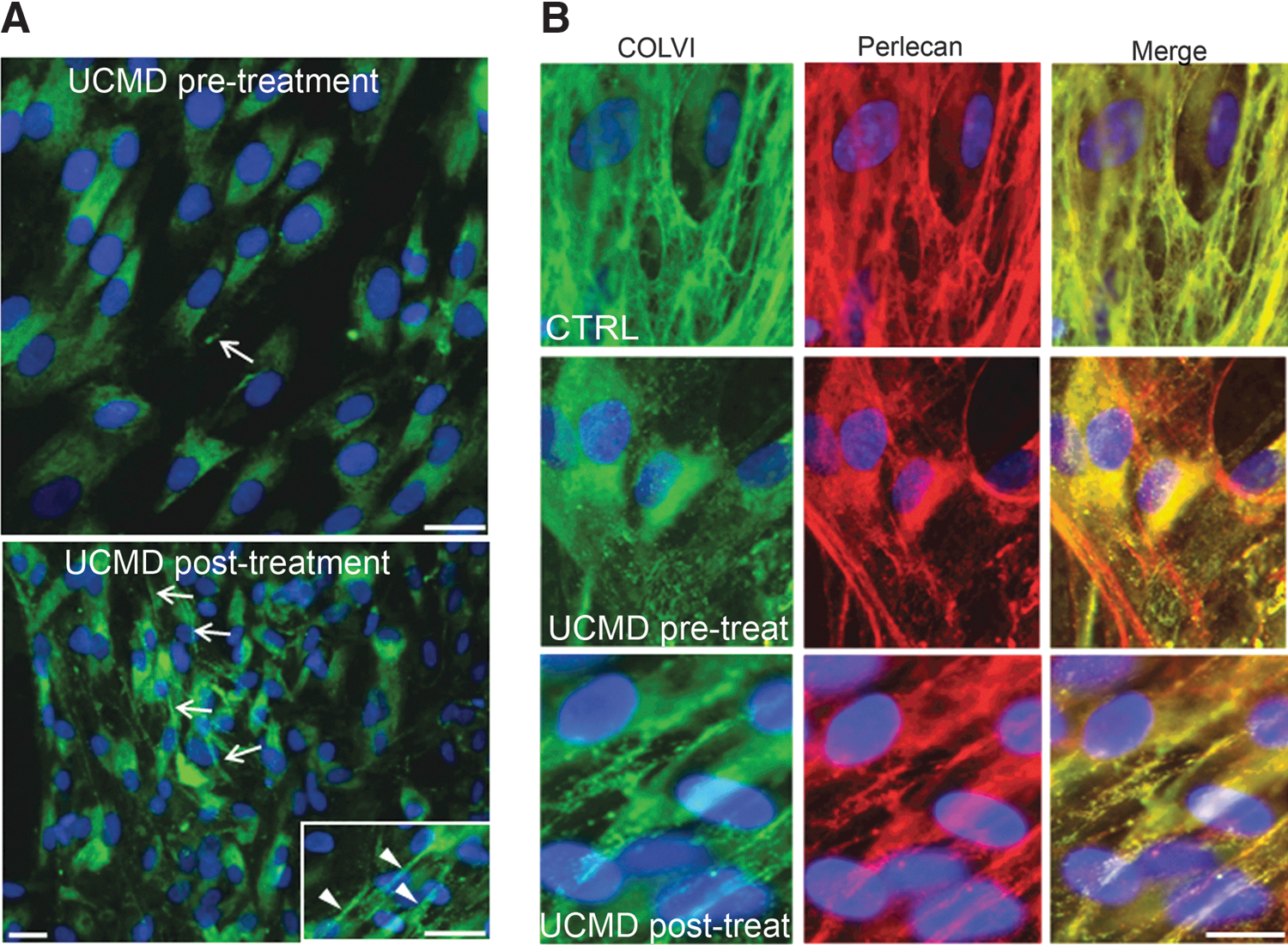
P221(T)AON partially recovers collagen VI-based ECM
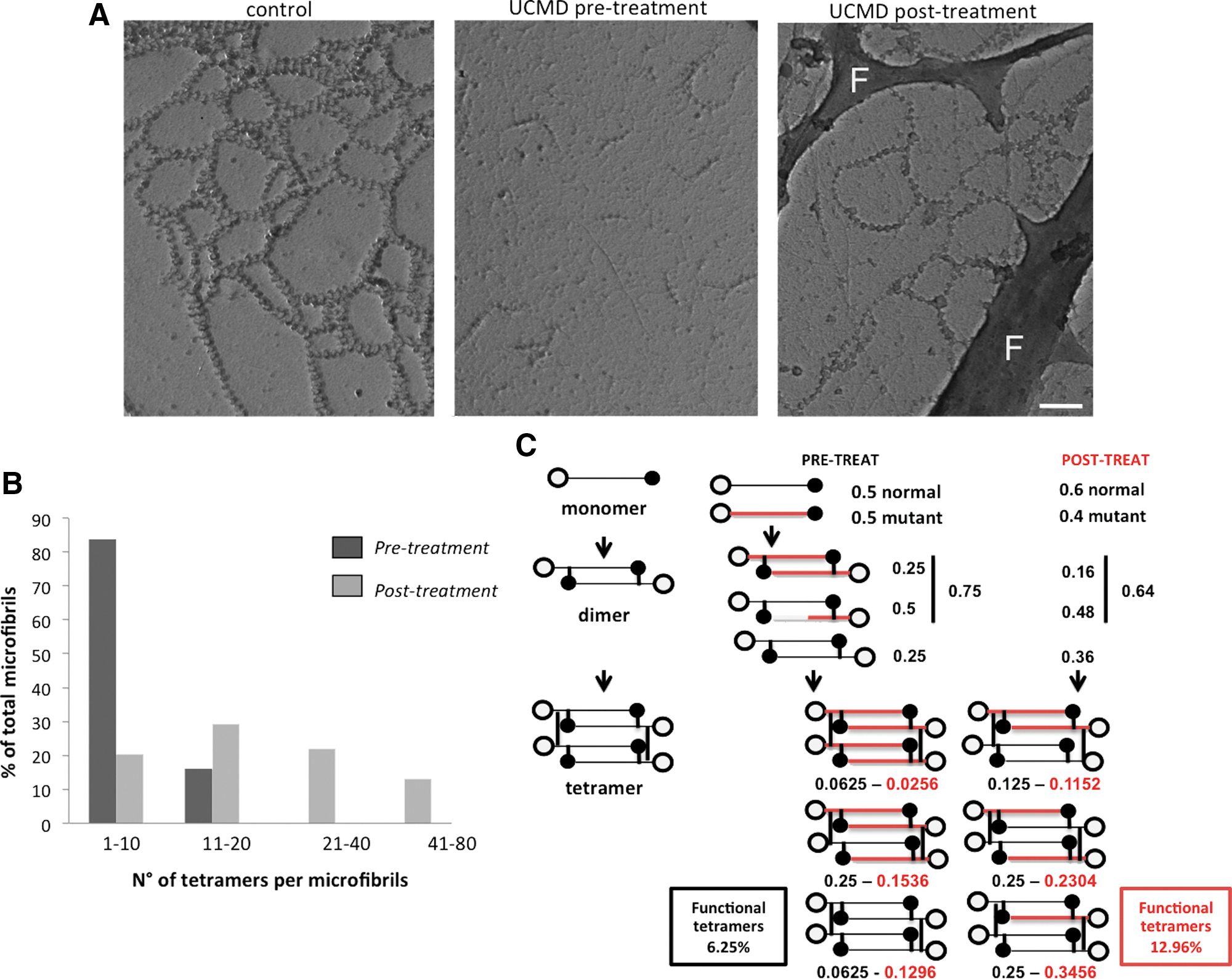
In untreated UCMD patient fibroblasts, collagen VI secretion appeared severely affected, with little protein deposited in the ECM in a spotlike arrangement, indicating a decreased ability of secreted mutant tetramers to associate into microfibrils. P221(T)AON treatment appeared to increase the amount of collagen VI associated with the ECM (Fig. 2A); the secreted protein was organized in a microfibrillar network and colocalized with perlecan labeling, as per control (Fig. 2B). Electron microscopy (EM) analysis showed improved tetramer–tetramer association and enhanced ability to form a microfilament network (Fig. 3A). In untreated UCMD culture, 83% of microfibrils were constituted by 2 to 10 tetramers, whereas microfibrils with more than 20 tetramers were absent. After treatment, about 80% of fibrils contained more than 20 tetramers (Fig. 3B). No effects were seen in fibroblasts treated using AON directed toward dystrophin exon 51 (data not shown).
Discussion
Correcting dominant mutations by splicing modulation is appealing, because the only way to intervene in these cases is by specifically silencing the mutated allele, a feat that has been achieved using various genetic methodologies (Wood et al., 2007; Le Roy et al., 2009). In particular, the exquisite sequence specificity of siRNAs has been used with encouraging results for allele-specific knockdown in several nonmuscular diseases in vitro and in animal models (Wood et al., 2007), and a phase 1b trial based on mutation-targeted siRNA, locally injected within skin lesions of pachyonychia congenita, is underway (Leachman et al., 2010). Nevertheless, concerns regarding delivery, interference with endogenous RNAi processing, and nonspecific off-target effects have thus far hampered a wider clinical exploitation of siRNA-based therapies (Wood et al., 2007; Wang et al., 2010). In contrast, AON-based treatment has reached phase 1–2a clinical trials in neuromuscular diseases. In patients with Duchenne muscular dystrophy (DMD), 2′OMePS-AONs and phosphorodiamidate morpholino oligomer (PMO) against dystrophin exon 51 have been systemically used to induce in-frame exon skipping, which can restore dystrophin production, apparently without overt toxic effects, paving the way to extended treatment phases (Cirak et al., 2011; Goemans et al., 2011).
We adopted exon skipping, exploring the efficacy of an SNP-specific 2′OMePS-AON to induce out-of-frame skipping and consequent allele degradation, as a corrective approach for a COL6A2 gene dominant negative mutation. This strategy was based on the following evidence: (1) 2′OMePS-AON sequence specificity and efficacy in inducing exon removal have been largely demonstrated in humans (Spitali et al., 2009); (2) COL6 gene transcripts with nonsense mutations almost invariably undergo RNA nonsense-mediated degradation, and quantification of transcripts has been proposed as a tool for pinpointing the mutated gene (Allamand et al., 2011); (3) COL6 genes are highly polymorphic and several common SNPs are known, which, if cistronic with pathogenic mutations, can be specifically targeted by AONs to induce transcript depletion in large patient populations; (4) dominant de novo mutations represent the predominant type, being the 67% of the COL6 genes missense mutations, 57% of small deletions, and 44% of splice site mutations leading to in-frame exon exclusion (Allamand et al., 2011). By AON-targeting a common SNP, we were able to target the causative mutation and deplete the mutated transcript, partially correcting the associated severe cellular phenotype. Treatment was not completely specific and caused a global reduction in COL6A2 mRNA level, preferentially involving the mutated allele. We observed that a relatively low efficiency of mutated allele depletion (10% variation in mutated/wild-type allele ratio) was nevertheless able to significantly improve extracellular collagen VI assembly, and restore its ability to develop a microfilament network, in single-dose and short-term treatment. The collagen VI complex assembly process, which explains the dominant negative behavior of mutations leading to aberrant chains, also accounts for this “magnification effect” of protein correction (Fig. 3C).
Progress in unraveling the pathogenic mechanisms of collagen VI-related myopathies (Grumati et al., 2010) has opened intriguing therapeutic perspectives; nevertheless, RNA-splicing modulation is appealing, because it is based on safe and highly reliable drugs in terms of systemic administration and tissue targeting (Cirak et al., 2011; Goemans et al., 2011). Furthermore, exon skipping can be personalized, treating specific mutations using dedicated AONs.
We provide proof of principle that out-of-frame exon skipping by AONs induces allele targeting and corrects COL6 gene dominant mutations, highlighting molecular splicing as a potential treatment for collagen VI-related myopathies. In addition, because our AON design targeted a common SNP linked to the mutation, we envisage a novel AON therapy that may facilitate personalized treatments, targeting “common variations embracing rare mutations.”
Footnotes
Acknowledgments
This work was supported by the Italian Ministry of Education, Universities, and Research (grant PRIN 2008PB5S89 to A.F., P.B., and S.S.) and by the Telethon Foundation Italy (contract grant GGP07004 to F.G.). The authors thank Dr. Marina Fabris for technical assistance in cell transfection experiments.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.