
Abstract
Splicing is an essential cellular process to generate mature transcripts from pre-mRNA. It requires the splice factor U1 small nuclear ribonucleoprotein (U1), which promotes exon recognition by base-pairing interaction with the splice donor site (SD). After U1 dissociation, exon recognition is maintained by U6 small nuclear ribonucleoproteins (U6). It has been shown that SD mutations lower the binding affinity of U1 and cause splice defects in about 10% of patients with monogenetic diseases. U1 isoforms specifically designed to bind the mutated SD with increased affinity can correct these splice defects. We investigated the applicability of this gene therapeutic approach for different mutated SD positions. A minigene-based splicing assay was established to study a typical SD derived from the gene BBS1. We found that mutations at seven SD positions caused splice defects. In four cases, mutation-adapted U1 isoforms completely corrected these splice defects. Partial correction was found for splice defects induced by the mutation at SD position +5. The limited therapeutic efficacy at this position was alleviated by applying a combined treatment with mutation-adapted U1 and U6. The sequence complementarity between U6 and three SD positions (+4, +5,and +6) was relevant for the outcome of the therapy. Between 30 and 100% of the normal transcripts can be restored. The treatment significantly decreased both exon skipping and intron retention. Massive missplicing of off-target transcripts was not detected. Our study helps to assess the therapeutic efficacy of mutation-adapted U snRNAs in gene therapy and illustrates their strong potential to correct splice defects, which cause many different inherited conditions.
Introduction
At the initial steps of the splicing process, U1 is recruited to the splice donor site (SD). The U1 snRNA, which is part of the splice factor U1, binds to the SD through Watson-Crick base-pairing (Zhuang and Weiner, 1986). This involves the last three nucleotides of the exon (positions −3 to −1) and the first six nucleotides of the downstream intron (positions +1 to +6) (Lund and Kjems, 2002). In addition to these early interactions at the SD, other splicing factors including the U2 snRNP are recruited to the splice acceptor site, forming a complex with U1 across the same exon in vertebrates, a process called exon definition (Schneider et al., 2010). After initial formation of this cross-exon complex, a preassembled heterotrimeric complex containing U4, U5, and U6 snRNPs (U4/U6.U5 tri-snRNP) is recruited. This leads to the transition of the cross-exon complex into a complex that forms across the upstream intron, thus called cross-intron complex. It activates the spliceosome to accomplish the first catalytic steps of the splicing process, which subsequently leads to dissociation of U1 and U4 snRNPs (Wahl et al., 2009). Thereafter, correct recognition of the exon at 5’ splice sites is assured through the interaction of the U6 snRNA (U6) with nucleotides at positions +4 to +6 of the SD (Kandels-Lewis and Seraphin, 1993; Lesser and Guthrie, 1993). This interaction is maintained until the second catalytic step in the splicing process has been carried out and all U snRNPs are released from the mature transcript to be recycled for additional rounds of splicing (Wahl et al., 2009).
Gene therapeutic approaches have been successfully applied to several animal models of human diseases. An increasing number of clinical trials further document the need for treatment options in genetic diseases. Indeed, around 15% to 20% of all point mutations in patients affect splice sites and are a major cause of monogenetic diseases (Krawczak et al., 1992; Faustino and Cooper, 2003). Mutations of the SD have been found in many genes and were associated with the vast majority of inherited human disorders (for reference see the human gene mutation database online). Mutations in splice sites result in exon skipping, intron retention, or activation of cryptic splice sites, which leads to a reduction or absence of correctly spliced transcripts (Faustino and Cooper, 2003). The majority of mutations found in the SD affect positions −1, +1, +2, and +5. Nucleotides at these SD positions are more conserved than the others (Buratti et al., 2007; Krawczak et al., 2007).
It has been documented that mutations reduce the interaction of U1 with the SD, subsequently causing aberrant splicing (Zhuang and Weiner, 1986). We and others showed that an increase in complementary of the U1 with the mutated SD corrects splice defects following virus-mediated treatment of patient-derived cell lines (Hartmann et al., 2010; Glaus et al., 2011; Schmid et al., 2011). The efficacy of this approach was tested for mutations at SD position −1, +1, or +3.
The study presented here uses a typical splice donor site to analyze the applicability of the U1 approach to treat SD mutations at several different positions. We further suggest that U6 is a promising candidate molecule in gene therapy and show that only a co-application of adapted U1 and U6 isoforms corrects the splice defect caused by the mutation at SD position +5.
Materials and Methods
Site-directed mutagenesis and cloning
Polymerase chain reactions (PCRs) were performed using Pfu-polymerase (Promega, Dübendorf, Switzerland). Sequences of primers used to produce mutated minigenes and different U snRNA constructs are shown in Supplementary Table 1 (Supplementary Material available online at
The minigene construct of the wild-type BBS1 gene includes the genomic region of intron 4 through 7. The wild-type BBS1 minigene was previously characterized (Schmid et al., 2011). Mutagenesis was performed as described by Tanner et al. 2009. Briefly, two PCR products were amplified from the minigene construct with primers containing the mutation to be analyzed. The two overlapping products were joined using primers pSPL3_MCS_F and pSPL3_MCS_R. PCR products containing the different SD mutations were subcloned into the pJet1 or pJet1.2 cloning vector (Fermentas, Le Mont sur Lausanne, Switzerland) and transferred into pSPL3 using XhoI and BamHI restriction sites.
The promoter and coding region of U6 snRNA was amplified from genomic DNA of human skin fibroblasts using primers U6_XbaI_fwd and U6_EcoRI_R. The XbaI- and EcoRI-digested PCR product was inserted into the cloning vector pGem3 using T4-Ligase (Promega). The pGem3 vector containing the genomic region encoding the U6 snRNA was used as PCR template to modify the U6 SD-binding sequence with primers carrying specific sequence alterations. Overlapping fragments were joined with primers binding to the T7 and SP6 promoter and cloned in the pGem3 vector. Constructs expressing different isoforms of U1 were produced as described previously (Tanner et al., 2009).
Cell culture and transfection
COS-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1.3% L-glutamine, and 1.1% penicillin/streptomycin (PAA; Chemie Brunschwig AG, Basel, Switzerland) at 37°C, 5% CO2. One to 4*105 cells were co-transfected with either 2.4 μg wt or mut minigene and 2.4 μg of a U1 expression construct using branched polyethyleneimine (PEI) (Sigma-Aldrich, Steinheim, Germany). Triple transfections with U6 snRNA isoforms were performed with 1.6 μg of each construct. Cells were harvested two days after transfection and each experiment was replicated three to eight times.
RNA extraction and RT-PCR analysis
Total RNA was extracted from COS-7 cells using the NucleoSpin® RNA II kit (Macherey-Nagel, Oensingen, Switzerland). To remove genomic DNA, RNA samples were treated with DNase using the DNA-free Kit™ (Applied Biosystems, Rotkreuz, Switzerland). RNA was randomly primed and reverse transcribed into cDNA using Superscript III reverse transcriptase following the manufacturer's protocol (Invitrogen, Basel, Switzerland). Minigene-derived splicing of BBS1 was analyzed by reverse transcriptase PCR (RT-PCR) as described previously (Schmid et al., 2011).
To search for potential side effects of the treatment, off-target transcripts were analyzed by RT-PCR. Potential binding sides of adapted U1 and U6 were identified by searching for perfect or almost-perfect sequence complementarity (at least seven base pairs [bp]) with the genomic reference sequences of various disease-associated transcripts. RNA extraction, cDNA construction, and RT-PCR from COS-7 cells transfected with different adapted U1 and U6 isoforms was performed as described above. Primer names and sequences are provided in Supplementary Table 1.
Semi-quantitative analysis of spliced products
The amount of amplified RT-PCR products showing either exon 5 skipping, intron 5 retention, or correct splicing of BBS1 was determined for each treatment using an electrophoretic analysis system (2100 Bioanalyzer; Agilent, Basel, Switzerland). The level of each splice product was normalized to the amount found in control treatments (wild-type U6 in combination with either fully adapted U1 or the wild-type U1) and averaged from eight independent transfections. The error bars represent confidence intervals of 95%.
Results
Position-dependent effects of splice donor site mutations
We previously characterized the splice defects induced by a mutation in a typical splice donor site. The analyzed mutation causes retinal degeneration, effects exon 5 of the BBS1 gene, and leads to both exon skipping and intron retention (Schmid et al., 2011). Minigene assays closely resembled the splice pattern found in patient-derived and control cell lines. To evaluate the effect of different sequence alterations on splicing of this SD, minigenes were mutated at nine positions (Table 1). All of these positions are relevant to the interaction with U1 (Fig. 1) (Zhuang and Weiner, 1986; Lund and Kjems, 2002). The minigenes were co-transfected with constructs expressing either wild-type U1 or empty vector (Fig. 2). Splicing of transcripts was analyzed by RT-PCR (Fig. 2A and B). Minigenes mutated at positions −3 and +6 of the SD did not reveal obvious alterations in splicing. In contrast, mutations at positions −2 through +5 showed a reduction in correctly spliced transcripts and simultaneously caused an increase in exon 5 skipping (Fig. 2B, panels 1 and 2). Although less clear, retention of intron 5 might occasionally be increased.

Schematic drawing of base-pair interactions between U1 snRNA, U6 snRNAs, and the splice donor site (SD) in exon 5 of the BBS1 gene. The 5’ end of U1 interacts with nucleotides at positions −3 and −1 in the exon (uppercase letters) and with positions +1, +2, +4, and +5 in the intron (lowercase letters), whereas U6-wt base-pairs with the SD nucleotide at position +5 in the intron. Base-pairing is indicated by vertical lines. Pseudouridines are shown as italic letters.

Analysis and U1-based treatment of mutation-induced splice defects in BBS1.
Reference sequence NM_024649.
For comparability, the target sequence recognized by the adapted U1 snRNA is shown.
Bold letters indicate altered nucleotides; uppercase letters indicate exonic nucleotides; and lowercase letters indicate intronic nucleotides. U1 snRNA, U1 small nuclear RNA.
Therapeutic efficacies of U1 adaptations to correct the pathogenic effect of different SD mutations
The majority of the nine analyzed SD mutations caused splice defects (Fig. 1B). We tested whether increasing the complementarity between U1 and the mutated SD is an efficient therapeutic approach for all of these nine SD positions. In total, we analyzed the therapeutic efficacy of 18 different U1 isoforms to restore splice defects that were induced by nine single mutations at different positions in the splice donor site (Table1 and Fig. 1B).
Mutated minigenes were co-transfected with U1 constructs either adapted to the mutation only (U1-mut −/+ n) or with full complementarity to the mutated SD (U1-BBS1_mut −/+ n) (Table 1, Fig. 2B). Splice defects induced by mutations at positions −2, +3, and +4 were partially corrected by the corresponding single-adapted U1-mut isoforms (Fig. 2B, panel 3). In contrast, the different fully adapted U1-BBS1_mut isoforms completely restored normal splicing for mutations at positions −2, −1, +3, and +4 (Fig. 2B, panel 4; Table 2). In general, U1-BBS1_mut isoforms were more efficient to restore splice defects than U1-mut isoforms (Fig. 2B, panels 3 and 4). No therapeutic effect was detected for sequence alterations at SD positions +1 and +2 (Fig. 2B, panels 3 and 4; Table 2).
Interestingly, the mutation at position +5 only showed a partial correction of the splice defect, even after treatment with the fully adapted U1-BBS1_mut isoform (Fig. 2B, panel 4; Table 2). The single mutation-adapted U1 showed no detectable therapeutic effect. Mutations at +5 frequently cause splice defects (Buratti et al., 2007; Krawczak et al., 2007). Our findings suggest that the treatment with U1 is not sufficient to completely correct aberrant splicing induced by the +5 mutation. It seems likely that, under these circumstances, additional splice factors are required to efficiently recognize the SD.
U6 adaptations to correct splice defects induced by mutations at SD position +5
It has been described that U6 interacts with positions +4 to +6 in SDs (Fig. 1) (Kandels-Lewis and Seraphin, 1993; Lesser and Guthrie, 1993). Increasing the binding affinity of U6 to the mutated SD improved the correction of mutation-induced splice defects. We evaluated several adaptations of the U6 snRNA sequence with the aim to enhance the interaction of U6 with the studied SD (Fig. 3A). Cells were triple transfected with the minigene mutated at +5, the fully adapted U1, and an adapted U6 isoform. RT-PCR analysis revealed either partial or complete correction of the splice defect (Fig. 3B). The therapeutic efficacy was depending on the level of complementarity between U6 and the mutated SD. Compared to treatments with adapted U1 only, an increased amount of correctly spliced transcripts and a simultaneous reduction of intron 5 retention was detected by applying those U6 constructs that are complementary to either position +5 (U6+5) or +4 and +5 (U6+4/5). Moreover, the RT-PCR analysis suggested a nearly complete correction of the splice defect by adaptation of positions +5 and +6 (U6+5/6) or +4, +5, and +6 (U6+4/5/6). Other U1 and U6 combinations had no detectable effect on splicing (Fig. 3B).
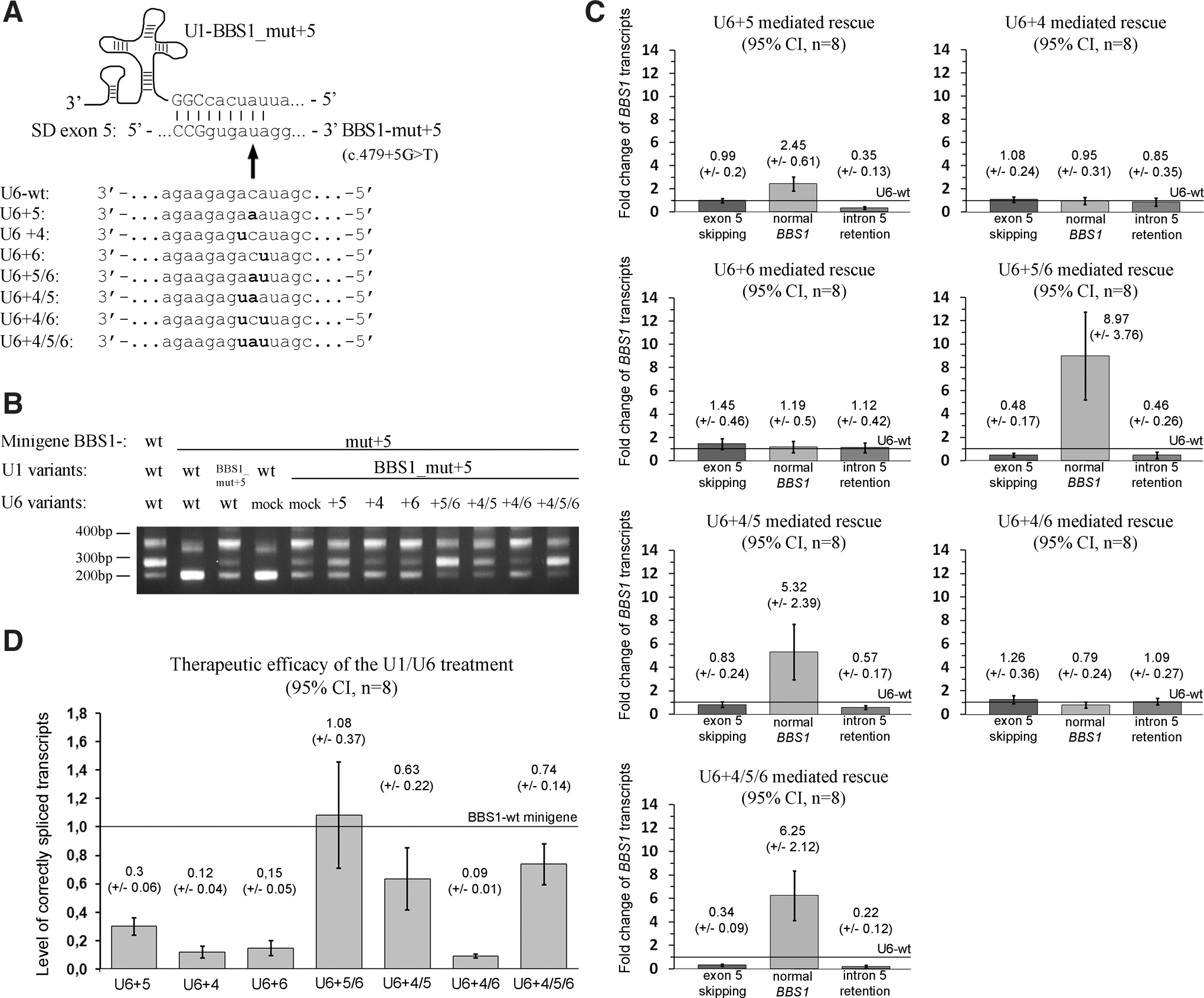
Combined treatment of mutation-adapted U1 and U6 to correct splice defects.
The changes in the splice pattern were quantitatively assessed (Fig. 3C and D). In reference to the wild type U6, the adapted U6+5 isoform resulted in two to three times higher levels of correctly spliced transcripts (Fig. 3C). Increasing the complementarity of U6 at positions +4/+5 and/or +5/+6 resulted in five to nine times higher levels of correct splicing (Fig. 3C). U6 isoforms without the complementary base pair at position +5 resulted in a loss of the therapeutic effect (Fig. 3C). Interestingly, U6+5 and U6+4/5 significantly reduced the level of intron retention, whereas the exon skipping band was not influenced by these treatment options. In contrast, U6+5/6 resulted in both, significantly reduced exon skipping and decreased levels of intron retention (Fig. 3C). The strongest reduction in exon skipping and intron retention was detected with the fully adapted U6 (U6+4/5/6, Fig. 3C).
We further asked whether normal transcript levels can be restored by the combined treatment with mutation-adapted U1 and U6 (Fig. 3D). In comparison with the wild type minigene, the U6+5 adaptation was able to correct approximately 30% of the transcripts. Moreover, between 63 and 100% of the normal transcript levels were restored applying either U6+4/5, U6+5/6, or U6+4/5/6 (Fig. 3D). In summary, our results support that the combination of mutation-adapted U1 and U6 efficiently corrects splice defects caused by mutations at SD position +5.
We tested the possibility that other U6 positions than +4, +5, or +6 may be relevant for the correction of splice defects. We evaluated additional nucleotide changes at different U6 positions, but did not detect significantly altered levels of splice variants (Fig. 4). These observations confirm that increasing the U6 binding to positions +4, +5, and +6 has the highest therapeutic potential to correct splice defects.

The therapeutic potential of U6 is restricted to adaptations at positions +4, +5, and/or +6.
In combination with wild-type U1, none of the U6 isoforms showed clear correction of the splice defects (Fig. 5). This suggests that a treatment with adapted U6 alone is not sufficient to overcome the deleterious effects of SD mutations at intronic position +5.
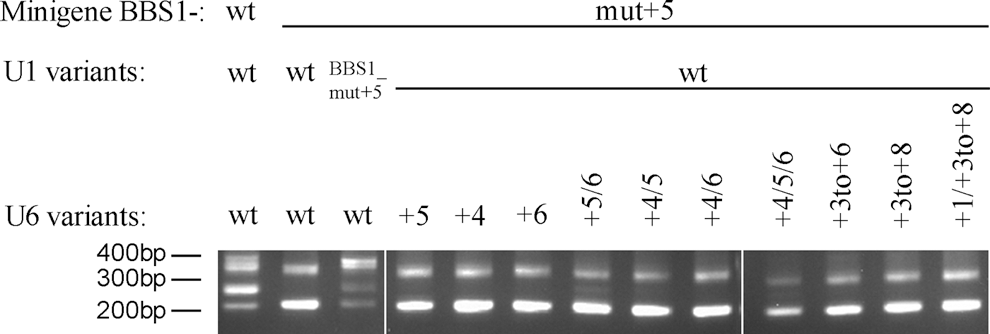
Treatment with U6 isoforms alone shows no significant therapeutic effect. Without a co-transfection with therapeutic U1, U6 isoforms did not lead to significant correction of splice defects. Treatment of the BBS1-wt minigene in combination with U1-wt and U6-wt, as well as a treatment of the BBS1-mut +5 minigene with U1-BBS1_mut +5 and U6-wt, were used as positive controls. The mutated minigene treated with U1-wt and U6-wt served as a negative control. DNA sizes are given in base pairs (bp).
Side effects of the U1/U6 treatment
The adaptation of binding sites in U1 and U6 may interfere with splicing of off-target pre-mRNAs. Using RT-PCR, we analyzed six disease-associated off-target transcripts that contained strong binding sites for the adapted U1 and/or U6 described herein (Supplementary Fig. 1). We did not detect splice alterations in these transcripts, suggesting that massive missplicing is not induced by the treatment.
Discussion
Defects in the splicing have been associated with many inherited human diseases. Independent of the disease-causing gene, 15% to 20% of all mutations are usually found in splice sites and affect splicing of the pre-mRNA (Krawczak et al., 1992; Krawczak et al., 2007). Several therapeutic interventions targeting aberrant transcripts have been investigated in order to restore correct splicing (for reviews, see Cooper et al., 2009, and Wang and Cooper, 2007). This involves application of pharmacological reagents or antisense oligonucleotides that block or increase exon inclusion. Furthermore, the usage of specific siRNAs and antibodies to reduce the amount of aberrantly spliced products has been reported. We have recently demonstrated that increasing the complementarity of U1 to the mutated SD is an efficient therapeutic approach to correct splice defects in primary human skin fibroblasts derived from patients with eye diseases (Glaus et al., 2011; Schmid et al., 2011).
SD consensus sequences are identical between mice and human and show perfect sequence complementarity to the U1 binding sequence at nine bp (Ast, 2004; Yeo et al., 2004). Previous studies showed that five to six matching bp between U1 and the SD are sufficient for correct splicing (Ketterling et al., 1999). In the wild-type SD of BBS1 exon 5, U1 matches to the highly conserved nucleotides at positions +1, +2, and also to positions −3, −1, +4, and +5. This indicates that the correct recognition of exon 5 in BBS1 requires six Watson-Crick bp with U1.
Not all positions of the SD are equally important to enable the recognition by U1 and to ensure correct splicing. Furthermore, various bp combinations within the SD show increased binding to U1, indicating mutual relationships between specific nucleotides of the SD. In our study, defects in splicing were not induced by mutations at positions −3 and +6. Indeed, +6 is among the less conserved positions in the SD (Carmel et al., 2004), indicating that mutations at +6 frequently have no effect on splicing. Nucleotide position −3 appears to be more conserved. Nevertheless, nucleotides at −3 are frequently not conserved if the SD matches U1 at positions −1 and +5, but does not show sequence complementarity at position −2 (Burge and Karlin, 1997). The SD of BBS1 exon 5 studied here exhibits exactly this configuration, which might explain why the mutation at position −3 caused no obvious defect in transcript splicing. In contrast, all other mutations at the SD caused various degrees of exon skipping. The mutation at −2 may be considered a mild mutation since it produced a detectable amount of normal transcripts in addition to exon skipping. Furthermore, published data are in agreement with our findings, demonstrating a higher degree of aberrant splicing caused by the mutation at −1 compared to the mutation at −2 (Carmel et al., 2004). In summary, the studied exon–intron border can be considered a typical splice donor site.
The efficacy of the U1-based therapeutic approach to restore the splice defects seems to reflect the pathogenic potential of the SD mutation to affect splicing. Defects in splicing caused by mutations at positions −2, −1, +3, and +4 were completely corrected by fully adapted U1 isoforms, whereas single adapted U1 partially restored aberrant splicing. These results suggest that high binding affinities between U1 and the mutated SD are beneficial to achieve complete correction of the splice defect.
Aberrant splicing activated by mutations at +1 and +2 could not be restored. Almost all human introns contain the GT dinucleotide at position +1 to +2, two residues that are essential for correct splicing of transcripts (Sheth et al., 2006). Hartmann et al. (2010) reported restoration of normal splicing using U1, which was only adapted to a single mutation at SD position +1. In this case, U1-wt already shows eight bp complementary to the SD of the affected exon indicating a strong interaction. However, further studies will be required to evaluate whether splice defects derived from mutations at +1 and +2 are a suitable target for U1-based therapies.
Only partial correction of the splice defect caused by the mutation at position +5 was found upon treatment with fully adapted U1. Similar as the GT nucleotides at the beginning of introns, the G nucleotide at position +5 in the SD is conserved (Carmel et al., 2004). Mutations at position +5 have frequently been found to cause aberrant splicing (Krawczak et al., 2007), e.g., can cause Stickler syndrome (Richards et al., 2010; Richards et al., 2012) or autosomal dominant polycystic kidney disease (Wang et al., 2009). The human gene mutation database lists +5 SD mutations in the majority of disease-associated genes. These findings substantiate the importance of developing treatment options for diseases caused by +5 mutations in SD. Our findings indicate that the U1 treatment only partially corrects splice defects induced by these mutations and suggest that additional factors need to be modified to achieve complete correction of the splice defect. Indeed, it has been reported previously that U6 is essential for accurate performance of the splicing process mediated by its interaction with SD positions +4, +5, and +6 (Kandels-Lewis and Seraphin, 1993; Lesser and Guthrie, 1993; Hwang and Cohen, 1996). Clearly improved exon recognition upon co-treatment with both adapted U1 and U6 isoforms was found. Interestingly, only the combination of adapted U1 and U6 resulted in an almost complete correction of the splice defect, whereas a treatment with U6 isoforms alone, i.e., without the help of fully adapted U1, had no significant effect. It is unclear whether these two splice factors collaborate on a molecular basis or whether changes in the kinetics of the splice mechanism lead to the observed therapeutic effects.
Additional studies are required to substantiate the search for possible side effects of the U1 and U6 treatment. We did not detect that the adapted splice factors U1 and U6 interfere with splicing of off-target pre-mRNAs, suggesting that massive missplicing is not induced by the treatment described herein. This finding is also confirmed by studies that successfully applied the U1-based treatment to patient-derived cell lines (Hartmann et al., 2010; Schmid et al., 2011). Nevertheless, further data are required to evaluate splice alterations in off-target transcripts on a broader basis. Applicability and safety of the therapeutic approach should be tested also in in vivo models affected by SD mutations. A global survey of pre-mRNA splicing in treated tissues or organs will help to determine the balance between therapeutic benefits and side effects.
In summary, the treatment with U1 and U6 splice factors shows strong potentials in the gene therapy of mutations at SDs. Our results will have implications on the development of therapeutic approaches applicable to many different inherited diseases.
Footnotes
Acknowledgments
We thank Silke Feil for help with cell culture and Barbara Kloeckener-Gruissem for helpful discussions. This work was supported by the Velux foundation and the Schweizerischer Fonds zur Verhütung und Bekämpfung der Blindheit.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.