
Abstract
Inherited retinopathies (IRs) are common and untreatable blinding conditions inherited mostly as monogenic due to mutations in genes expressed in retinal photoreceptors (PRs) and in retinal pigment epithelium (RPE). Over the last two decades, the retina has emerged as one of the most favorable target tissues for gene therapy given its small size and its enclosed and immune-privileged environment. Different types of viral vectors have been developed, especially those based on the adeno-associated virus (AAV), which efficiently deliver therapeutic genes to PRs or RPE upon subretinal injections. Dozens of successful proofs of concept of the efficacy of gene therapy for recessive and dominant IRs have been generated in small and large models that have paved the way to the first clinical trials using AAV in patients with Leber congenital amaurosis, a severe form of childhood blindness. The results from these initial trials suggest that retinal gene therapy with AAV is safe in humans, that vision can be improved in patients that have suffered from severe impairment of visual function, in some cases for decades, and that readministration of AAV to the subretinal space is feasible, effective, and safe. However, none of the trials could match the levels of efficacy of gene therapy observed in a dog model of the disease, suggesting that there is room for improvement. In conclusion, these results bode well for further testing of AAV-mediated retinal gene therapy in patients with other monogenic and complex forms of blindness.
Introduction
Inherited retinopathies are untreatable blinding conditions and ideal targets for gene therapy
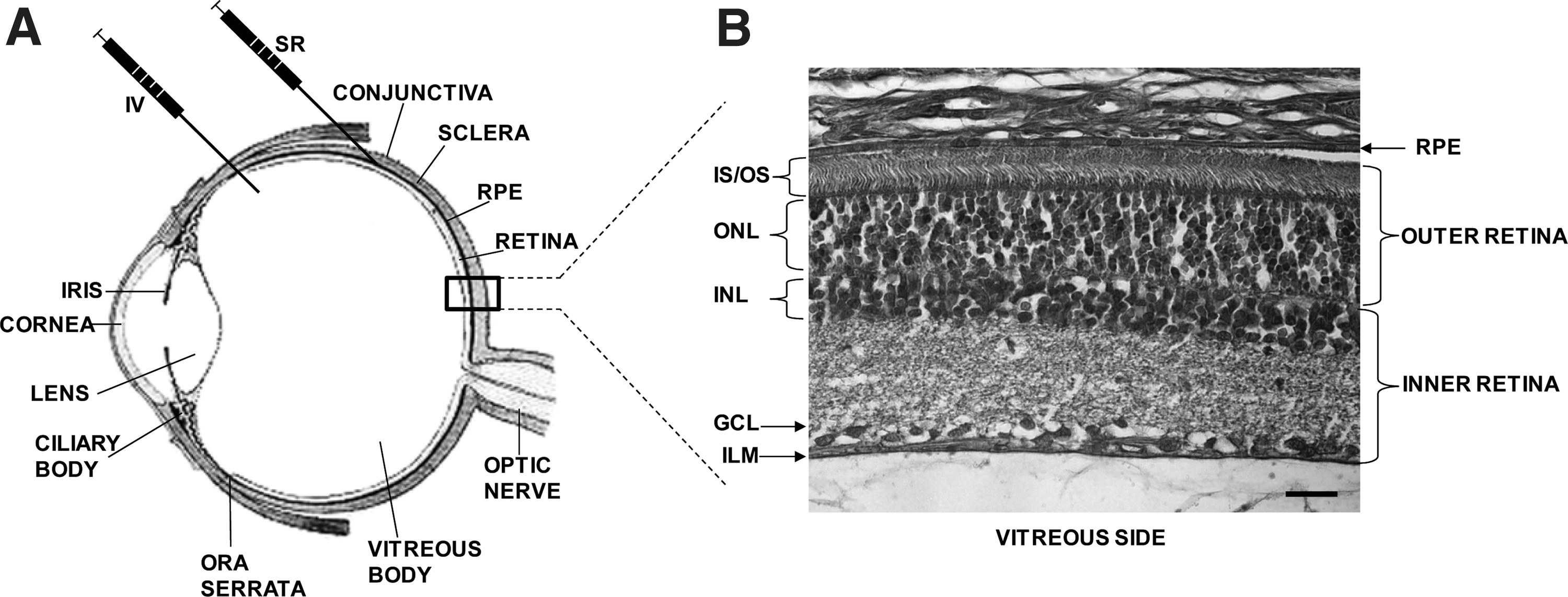
Inherited retinopathies (IRs) are common blinding diseases, mostly monogenic, that affect about one in 2,000 people worldwide and severely impair patients' living conditions (Berger et al., 2010). Vision loss in IR patients is due to the dysfunction/degeneration of retinal photoreceptor cells (PRs; rods and cones) and/or the retinal pigment epithelium (RPE) (Fig. 1). So far, IR causative mutations have been identified in more than 150 genes expressed mostly in PRs and to a lesser extent in the RPE (Daiger et al., 2011). IRs are both genetically and phenotypically heterogeneous. The different forms are commonly classified based on the genetic defect (when identified), the inheritance pattern (either autosomal dominant, recessive, or X-linked), the main cell type affected (rods, cones, or RPE), the onset and type of visual dysfunction, the disease progression rate, and the appearance of peculiar ocular fundus abnormalities (Berger et al., 2010). The most frequent and severe forms of IR are retinitis pigmentosa (RP) (Hartong et al., 2006), Leber congenital amaurosis (LCA) (den Hollander et al., 2008), Stargardt's disease (STGD) (Fishman, 2010), and achromatopsia (Berger et al., 2010). In addition, IR can be part of a syndrome, among which the most frequent is Usher syndrome (USH) that combines RP with hearing impairment (Millan et al., 2011). IRs are incurable, and no definite treatment could be envisaged before the advent of gene therapy. Indeed, IRs are ideal candidates for gene therapy because: (i) they are conditions inherited mostly as monogenic traits (Daiger et al., 2011); (ii) many IR causative genes have been identified (Berger et al., 2010); (iii) the affected cells (PRs and RPE) can be approached by various surgical procedures used in the clinic to treat other ocular diseases (Liang et al., 2000); (iv) rodent and large animal models that mimic the human IRs are available (Chader, 2002; Lin et al., 2002; Baehr and Frederick, 2009); and (v) noninvasive analyses that are currently used in the clinic to diagnose and follow up patients with IRs (Maguire et al., 2008; Testa et al., 2011) have been successfully adapted to animal models (Aleman et al., 2004; Weymouth and Vingrys, 2008; Fischer et al., 2009). This has led, in the last 20 years, to many therapeutic proofs of principle of the efficacy of gene therapy in animal models of IR, and we are currently observing the successful translation of some of them from bench to bedside. In this article, we will review the landmark developments of gene therapy for IRs, the most recent breakthroughs, as well as the current challenges that the field is facing to expand the initial clinical successes to a larger number of IRs.
Review
Efficient viral vectors are key elements for successful retinal gene therapy: advantages and limitations of adenoviral and lentiviral vectors
The success of retinal gene therapy relies on vectors that can efficiently deliver nucleic acids to target cells (mainly PRs and RPE). One of the advantages of the eye as a gene therapy target organ is that it is easily accessible from the exterior. The retina and RPE can be targeted by two routes of injection: intravitreal (IV) (Liang et al., 2000) and subretinal (SR) (Liang et al., 2000) (Fig. 1). Whereas the IV injection releases the therapeutic agent in the vitreous, the SR injection releases it in the subretinal space that forms between the PRs and RPE as a consequence of a reversible, injection-induced retinal detachment (Liang et al., 2000). The IV injection is less invasive than the SR, but the diffusion of the therapeutic agent to the PRs and RPE is limited by several physical barriers, such as the vitreous, the inner limiting membrane (ILM), and the inner retina (Fig. 1). Thus, the SR is the preferred route used to date to target efficiently RPE and PRs. Many studies over the past years have provided evidence of the higher efficiency of viral versus nonviral vehicles (Andrieu-Soler et al., 2006) for retinal gene delivery; however, recent reports on nanoparticle-mediated retinal gene therapy showed an improvement compared with previous studies with nonviral agents (Cai et al., 2008). The vectors most studied and used for retinal gene transfer are those derived from adenoviruses (Kumar-Singh, 2008), lentiviruses (Balaggan and Ali, 2012), and adeno-associated viruses (Vandenberghe and Auricchio, 2012) which are capable of infecting and transducing nondividing cells such as PRs and RPE.
Following SR delivery, adenoviral vectors (Ads) transduce the RPE efficiently, but PRs poorly (Bennett et al., 1994; Li et al., 1994). These vectors determine fast but short-lived transgene expression (Bennett et al., 1994; Reichel et al., 1998) due to the cytotoxic T lymphocyte–mediated removal of the transduced cells that express the Ad proteins encoded by the viral genome (Reichel et al., 1998). Despite this limitation, several studies reported that Ad-mediated gene transfer provided short-term improvements in IR animal models (Bennett et al., 1996, 1998; Kumar-Singh and Farber, 1998; Akimoto et al., 1999; Vollrath et al., 2001; Chen et al., 2006). Notably, the helper-dependent Ad vectors (Hd-Ads) that do not encode for viral proteins (Kochanek, 1999) evade the immune response and target the RPE stably (Kreppel et al., 2002). The thorough characterization of Hd-Ads in the retina of large animal models and the identification of new serotypes and/or the modification of known ones (Von Seggern et al., 2003; Cashman et al., 2007; Sweigard et al., 2010) to improve PR transduction will hopefully lead to the re-evaluation of Ad for the gene therapy of IRs. Indeed, due to their high DNA cargo capacity (up to 36 kb), the development of Hd-Ads that efficiently and stably transduce PRs and the RPE would be particularly useful to treat those IR forms caused by mutations in genes that would not fit in adeno-associated-viral vectors (AAVs) or lentiviral vectors (LVs), as well as to test large regulatory regions (i.e., endogenous enhancers and promoters) for a more physiological regulation of transgene expression.
LVs are integrating RNA retroviruses with a cargo capacity of about 8–10 kb (Balaggan and Ali, 2012). LVs are deleted of all viral genes and, thus, do not activate the immune system (Bennett, 2003; Pauwels et al., 2009), but the possibility of insertional mutagenesis, vector replication, and mobilization in human cells (Pauwels et al., 2009) raises significant concerns. Although no malignant transformation has been reported in the murine eye using high-titer integrating LVs (Balaggan et al., 2012), the issue of insertional mutagenesis is being addressed through the generation of nonintegrating LVs (Yanez-Munoz et al., 2006), whereas vector replication and mobilization are being tackled through the use of nonprimate LVs (Pauwels et al., 2009). Most studies showed that following SR delivery, LVs transduce the RPE efficiently in rodents (Auricchio et al., 2001; Bainbridge et al., 2001; Duisit et al., 2002; Kachi et al., 2009), but PRs variably and inadequately (Miyoshi et al., 1997; Bemelmans et al., 2005; Balaggan et al., 2006). Notably, PR transduction by LVs is inversely correlated to PR differentiation (Miyoshi et al., 1997; Bainbridge et al., 2001; Pang et al., 2006; Nicoud et al., 2007). Consistent with these observations, the SR delivery of LVs to the adult retina was effective in rodent models of IR caused by mutations in genes expressed in the RPE (Vollrath et al., 2001; Bemelmans et al., 2006), whereas perinatal (Takahashi et al., 1999; Hashimoto et al., 2007; Kong et al., 2008) or in utero (Williams et al., 2006) deliveries were required to treat rodent models that bear mutations in genes expressed in PRs. These preclinical studies served as the basis to test the nonprimate equine infectious anemia virus in ongoing clinical trials to treat exudative age-related macular degeneration (NCT01301443), STGD (NCT01367444), and USH type IB (USH1B) (NCT01505062). The results from these trials are not available yet. However, the transduction of mature PRs remains the fundamental limitation of LVs and Ads as vectors for the therapy of IRs. It is possible that the large size of Ad and LV particles (60–120 nm) prevents their efficient diffusion through the dense adult interphotoreceptor matrix (IPM) (Mieziewska, 1996). It is interesting that PR transduction by LVs has been reported to either positively (Balaggan et al., 2006; Pang et al., 2006) or negatively (Calame et al., 2011) correlate with disease-associated or induced PR damage and to be improved by loosening the IPM (Gruter et al., 2005). Therefore, it is possible that the degree of retinal degeneration impacts on PR transduction mediated by LVs or Ads in IR patients.
AAV as safe and most efficient vector for therapy of IRs
AAVs are currently the most favored vectors for gene therapy of IRs given their: (i) small (25 nm) size and ability to efficiently target various retinal layers; (ii) low immunogenicity, allowing the possibility to readminister the vectors to the subretinal space; (iii) excellent safety profile; and (iv) availability of over 100 different forms, the AAV serotypes. This last feature is one of the major strengths of the AAV vector platform; the versatility of the AAV production system allows the easy exchange of capsids among various AAV variants, thus creating hybrid vectors that contain a genome with the same AAV inverted terminal repeats (ITRs) (i.e., those from AAV2) and the capsid from a different variant (Auricchio, 2003). AAVs obtained through this transcapsidation system are named AAV2/n, where the first number refers to the ITRs and the second to the capsid (Surace and Auricchio, 2008). Over the past years, AAV vectors derived from naturally-occurring serotypes (Auricchio et al., 2001; Gao et al., 2002, 2005) or with rationally-modified (Zhong et al., 2008b) or in vitro evolved capsids (Perabo et al., 2008) have been generated, significantly expanding the potential of AAVs for the treatment of IRs. The most relevant findings have been: (i) the identification of AAV2/8 as the most efficient naturally-occurring serotype for PR transduction in mice (Allocca et al., 2007; Auricchio, 2011), pigs (Mussolino et al., 2011a), and nonhuman primates (NHPs) (Vandenberghe et al., 2011); (ii) the availability of AAV2/7, 2/9, and 2/5 as alternatives to AAV2/8 for PR transduction (Auricchio et al., 2001; Lotery et al., 2003; Allocca et al., 2007; Mussolino et al., 2011a); and (iii) the generation of tyrosine-mutated AAV capsids (Zhong et al., 2008b) that avoid targeting of AAVs to the proteasome, thus increasing the vector load that reaches the nucleus, resulting in increased transgene expression (Zhong et al., 2008a). In particular: (i) the single and triple mutants from AAV2/2 (Y444F and Y444,500,730F) and 2/8-Y733F determine stronger and more widespread transduction than their wild type (wt) counterparts when delivered SR in mice (Petrs-Silva et al., 2009); and (ii) quadruple and pentuple tyrosine-mutant AAV2/2 vectors are capable of transducing murine PRs and RPE when injected IV (Petrs-Silva et al., 2011). PR and RPE transduction from the vitreous is particularly attractive for IR treatment, because it would allow a more widespread and homogeneous transgene expression and reduces the risk of surgical morbidity. However, the presence of a thick (especially in large animals) ILM limits the diffusion of vectors to the outer retina after IV administration. Indeed, the IV delivery of wt AAV2/2 in mice and NHPs allows transduction of the inner retina (Auricchio et al., 2001; Yin et al., 2011) and only a few scattered foveal cones (Wadsworth, 2012). However, if the retinal architecture is altered by a degenerative process, the diffusion of AAV viral particles to the outer retina from the vitreous side is enhanced (Park et al., 2009; Kolstad et al., 2010).
Two major limitations of AAVs in the retina are the slow onset of transgene expression due to the time-consuming conversion of the single-stranded AAV genome into a double-strand, and the vector limited cargo capacity. The first limitation can be circumvented by using self-complementary (sc) AAV vectors that package double-stranded AAV genomes (McCarty, 2008). scAAV2/2, 2/8, and 2/5 drive faster onset and stronger levels of transgene expression than their single-stranded counterparts in the retina of mice (Yokoi et al., 2007; Natkunarajah et al., 2008; Kong et al., 2010) and dogs (Petersen-Jones et al., 2009), theoretically allowing the delivery of lower vector doses to obtain similar levels of transgene expression. However, in some cases, it is not clear whether this is due to the inherent properties of scAAVs or to a systematic erroneous titration (Fagone et al., 2012) that results in the administration of higher doses of scAAVs than expected. The major limitation of AAVs remains their packaging capacity, which is considered to be restricted to the size of the parental genome (4.7 kb) and thus hampers the treatment of certain forms of IR caused by mutations in genes whose cDNA exceeds 5 kb (i.e., Abca4, Myo7a, or CEP290, mutated in STGD, USH1B, and LCA10, to name a few). Strategies to overcome AAV size restriction are based either on the generation of dual AAV vectors that reconstitute the full-length genome in transduced cells (referred to as trans-splicing, overlapping, or hybrid dual-vector strategies) (Duan et al., 2001; Ghosh et al., 2008) or on packaging of oversized genomes (Grieger and Samulski, 2005; Allocca et al., 2008; Hirsch et al., 2010). Notably, oversized AAVs allow the expression of full-length proteins in vitro and in the murine retina (Allocca et al., 2008), but contain highly heterogeneous genomes (Dong et al., 2010; Hirsch et al., 2010; Lai et al., 2010; Wu et al., 2010), which limit their use in clinical settings. On the other hand, the efficiency of the dual AAV systems for gene delivery to the retina still remains to be thoroughly exploited, because few studies have been performed to date (Reich et al., 2003).
To silence, to replace, or to supply?
Around 40% of IR patients that present a recognizable pattern of inheritance are affected by dominant forms of the disease (Berger et al., 2010). In several cases, the mutation underlying the disease exerts a toxic, gain-of-function effect. More than 150 different mutations associated with dominant RP have been described in the rhodopsin (RHO) gene alone. Dominant forms of the disease pose two main challenges: (i) gain-of-function mutations require silencing rather than replacement; and (ii) mutation-independent silencing that may be desirable in the presence of high allelic heterogeneity requires efficient replacement, as both the mutant and wt alleles will be silenced. Allele-dependent approaches have used AAV-mediated delivery of either mutation-specific ribozymes (Lewin et al., 1998; LaVail et al., 2000) or short-hairpin (sh) RNAs (Tessitore et al., 2006) directed to the common P23H-RHO mutation. This provided partial (Lewin et al., 1998; LaVail et al., 2000) to no (Tessitore et al., 2006) amelioration of PR degeneration, presumably due to the high levels of RHO expression in rods. Notably, mutation-independent approaches whose design is not restricted by the position of the mutation provided more potent silencing whether based on ribozymes (Gorbatyuk et al., 2005, 2007b) or on shRNAs (Gorbatyuk et al., 2007a; Chadderton et al., 2009). Recently, Mussolino et al. (2011b) showed that it is possible to effectively repress the expression of the RHO gene in a mutation-independent way by targeting engineered zinc-finger–based DNA binding proteins to the RHO promoter. This is an interesting approach, as promoter repression targets the locus of a gene that is present in two copies per diploid cell, as opposed to the transcript, which can be very abundant and challenging to silence, as in the case of RHO. To date, the most advanced proof of concept of the “suppression–replacement” strategy for dominant RHO mutations has been provided by O'Reilly et al. (2008), Mao et al. (2012), and Millington-Ward et al. (2011), who used a single AAV2/8 vector that codelivers an shRNA directed to RHO and a silencing-resistant RHO transgene. This strategy resulted in significant delay of PR cell loss in treated RHO-P23H mice (O'Reilly et al., 2007; Mao et al., 2012) and in increased rod function (Mao et al., 2012). The use of two separate AAVs, one that delivers the shRNA and the other the RHO transgene, allows RHO suppression and replacement to be modulated and optimized by varying the two vector doses (Millington-Ward et al., 2011). The results achieved so far using the suppression–replacement strategy in animal models with RHO dominant mutations are encouraging. However, as both high and low levels of RHO have been shown to be deleterious for rods (Gorbatyuk et al., 2005; Mao et al., 2011), further studies are required to optimize this complex approach in order to consistently achieve therapeutic efficacy and avoid toxicity. For those dominant IR forms due to mutations that exert a dominant-negative effect, this can be reduced by gene replacement, i.e., by overexpressing the wt protein that competes with the mutant. Indeed, AAV-mediated delivery of wt RHO has been recently reported to ameliorate the phenotype of heterozygous RHO-P23H mice in a Rho +/+ background (Mao et al., 2011), raising the possibility that P23H exerts a dominant-negative rather than gain-of-function effect.
Most forms of IR are determined by loss-of-function mutations (Daiger et al., 2011), which can be treated by gene replacement, that is achieved by the delivery of the correct cDNA copy of the defective gene in the affected cells (mainly PRs and RPE) (Colella et al., 2009). Historically, AAV-mediated gene replacement has been reported to be more effective in forms of IR due to mutations in genes expressed in the RPE (Smith et al., 2003; Dejneka et al., 2004; Surace et al., 2005; Gargiulo et al., 2009) than in PRs (Bennett et al., 1996; Schlichtenbrede et al., 2003a; Allocca et al., 2011). This can be attributed to the features of the RPE (i.e., transducibility), the need for fine-regulated transgene expression in PRs, or the different rates of retinal degeneration, which is usually faster in PR- than RPE-specific diseases (Smith et al., 2009). To date, the most successful preclinical gene therapy study for a recessive RPE defect is represented by the AAV-mediated delivery of the 65-kDa RPE-specific isomerase (RPE65) in a dog model of LCA2 (Acland et al., 2001; Narfstrom et al., 2003; Le Meur et al., 2007; Annear et al., 2011), which has led to three independent human clinical trials in LCA2 patients, as discussed later. Gene replacement has been successfully evaluated in additional rodent models of IR caused by mutations in genes expressed in the RPE, such as MER tyrosine kinase (Mertk) (Vollrath et al., 2001; Smith et al., 2003; Tschernutter et al., 2005) and lecithin-retinol acyltransferase (LRAT) (Batten et al., 2005). Gene transfer to PRs has been enormously advanced in recent years by the development of more efficient AAV vectors; before then, attempts had been made using LV, Ad, or AAV vectors with poor PR transduction ability. Indeed, the first evidence of the benefits as well as the limitations of gene replacement was provided for two forms of RP caused by mutations in genes expressed in PRs: the beta cGMP phosphodieserase 6 (PDE6-β), which is required for rod phototransdution (Bennett et al., 1996), and the peripherin 2 (Prph2), which is essential for proper formation and activity of cones and rods (Ali et al., 2000). Very limited improvement of rod function was achieved in the murine models of PDE6-β deficiency following the delivery of the PDE6-β by Ad (Bennett et al., 1996), AAV2/2 (Jomary et al., 1997), and LV (Takahashi et al., 1999) vectors that inefficiently transduce PRs, whereas transient improvements (due to onward rod degeneration) were observed by using either AAV2/5 (Pang et al., 2006) or AAV2/8 (Allocca et al., 2011). Recently, the delivery of PDE6-β by AAV2/8-Y733F in rd10 mice (which carry homozygous PDE6-β missense mutations) restored retinal function and preserved rods up to 6 months (Pang et al., 2011), indicating that the faster onset and more widespread transgene expression provided by the tyrosine-mutant serotype 8 can significantly slow PR loss in this severe model of PR degeneration. The recent report of successful AAV-mediated gene transfer in rcd1 dogs (Monnier et al., 2012) with PDE6-β deficiency suggests that the time is ripe for the clinical translation of gene therapy for this common form of recessive RP (Hartong et al., 2006). Other successful proofs of concept of AAV-mediated gene replacement in models of inherited PR disease include the rescue of the aryl hydrocarbon receptor interacting protein like-1 (Aipl1)–deficient mouse (Tan et al., 2009; Sun et al., 2010; Ku et al., 2011) and the retinitis pigmentosa GTPase regulator interacting protein (RPGRIP) −/− mouse (Pawlyk et al., 2005). These experiments show that AAV2/8 and its tyrosine-mutant version are the most effective gene transfer vectors for rod-cone dystrophies. Both AAV2/5 and 2/8 have been successfully applied to the retina of animal models of cone-specific or cone–rod dystrophies (Michaelides et al., 2004; Thiadens et al., 2010; Thomas et al., 2011). Mice (Alexander et al., 2007; Michalakis et al., 2010; Carvalho et al., 2011; Pang et al., 2012), dogs (Komaromy et al., 2010), and squirrel monkeys (Mancuso et al., 2009) with achromatopsia or color blindness have been successfully treated long-term after single SR administrations of AAVs. Similarly, in a mouse model of the common LCA1 due to mutations in the guanylate cyclase-1 gene (the Gucy2e−/− mouse), a single SR administration of AAV2/8 resulted in long-term cone functional rescue that was more pronounced than that observed with AAV2/5 (Boye et al., 2010, 2011; Mihelec et al., 2011). Overall, these data suggest that gene therapy of IRs due to mutations in genes that are expressed in rods and cones is feasible with AAV2/5 and AAV2/8, and soon we should see the clinical translation of these proofs of concept.
As PR apoptosis is the final outcome and hallmark of IRs, independent of the specific causative gene (Wright et al., 2010), therapies based on “supply” of growth, neurotrophic, anti-apoptotic, anti-oxidative, and anti-inflammatory molecules have been widely exploited to sustain PR survival (Colella and Auricchio, 2010). The list of such molecules delivered by intraocular gene transfer with AAV vectors, which allow safe and sustained expression, include the fibroblast and lens-epithelium–derived growth factors (FGF and LEDGF, respectively), the ciliary and brain-derived neurotrophic factors (CNTF and BDNF, respectively), the glial-cell–derived neurotrophic factor (GDNF), the pigment epithelium–derived factor (PEDF), erythropoietin (EPO), the X-linked inhibitor of apoptosis (XIAP), and the heme oxygenase 1 (HO-1) (Colella and Auricchio, 2010). The data collected so far by using protective factors showed that it is possible to delay PR loss in a mutation-independent way; however, it is still difficult to predict which IR form would be responsive to a specific factor and to evaluate the risks and benefits related to the nonphysiological expression of factors with growth and anti-apoptotic properties (Colella and Auricchio, 2010). The safety concerns are even more critical in view of the fact that gene supply does not directly target the causative gene and does not represent a definitive cure for IRs. A paradigmatic example of gene supply is CNTF, which has been tested in a phase I safety trial (NCT00063765) by intraocular release in IR patients using encapsulated-cell implants (Sieving et al., 2006). Despite minor improvements in visual acuity in the absence of severe adverse effects, significant reductions in electroretinogram (ERG) were reported (Sieving et al., 2006), confirming what was previously observed in preclinical studies (Bok et al., 2002; Schlichtenbrede et al., 2003b; Wen et al., 2006). The detrimental effects observed following the intraocular delivery of high CNTF doses highlight the importance of regulating the expression of molecules with growth/neurotrophic/anti-apoptotic activities, which may have tight therapeutic ranges. Intraocular delivery of EPO offers an additional example of the risks and benefits associated with the use of neurotrophic molecules: EPO is a potent neuroprotective molecule (Brines and Cerami, 2005; Grimm et al., 2005), but it is endowed with marked erythropoietic activity, an undesirable feature when treating IRs. The recent discovery of EPO mutants that lack erythropoietic activity while retaining neuronal protective effects (Leist et al., 2004) has allowed the successful testing of AAV-mediated intraocular delivery of these derivatives in IR animal models (Colella et al., 2011; Sullivan et al., 2011). Another strategy that is independent of the mutation underlying the disease relies on gene delivery of light-activated ion channels or pumps to retinal neurons (either PRs, bipolar, or ganglion cells) to stimulate retinal activity (Busskamp et al., 2012). As this optogenetic strategy has the potential to render light-sensitive cells that are normally not, it can be applied to end-stage retinal diseases in which PRs are lost at the time of intervention. So far, AAV-mediated delivery of microbial opsins (channelrhodopsin-2 and halorhodopsin) or of the light-gated ionotropic glutamate receptor (LiGluR) restored light sensitivity in various murine models of IRs and human postmortem retinas (Busskamp et al., 2012).
Gene therapy clinical trials for LCA2
The first IR for which retinal gene therapy has been developed up to a phase I/II clinical trial is LCA2 due to mutations in RPE65. This is because of the excellent and consistent preclinical results obtained in small and large animal models, as well as of the LCA2 phenotype, which is ideal to test the efficacy of gene therapy: LCA2 patients have severe visual dysfunction (negligible ERGs, reduced light sensitivity, and progressive visual acuity decline), slow PR degeneration, and an adequate RPE preservation (Cideciyan, 2010). Therefore, restored expression of RPE65 in RPE cells is predicted to result in production of 11-cis-retinal and in reactivation of PR function. To date, 30 LCA2 patients have been treated in three independent phase I/II clinical trials (NCT00643747, NCT00516477, NCT00481546) by SR delivery of AAV2/2 encoding for the human RPE65 gene (Bainbridge et al., 2008; Cideciyan et al., 2008, 2009a,b; Hauswirth et al., 2008; Maguire et al., 2008, 2009; Simonelli et al., 2010; Ashtari et al., 2011; Bennett et al., 2012; Jacobson et al., 2012) (Table 1). The preliminary results from a fourth trial (NCT004815469) that is being conducted using the same AAV2-RPE65 vector as NCT00481546 have been recently published; however, we will focus on the trials with a longer follow-up. Notably, all trials showed significant improvements of both retinal and visual function (Table 1), despite the fact that: (i) the vectors used contained slightly different regulatory sequences and were independently produced, purified, and characterized; (ii) the LCA2 patients were independently selected, characterized, and followed-up at each site; and (iii) the surgical delivery was performed independently at each site. The data collected so far showed: (i) improvement of retinal light sensitivity in most treated eyes; (ii) increase of visual acuity in 30% of treated patients; (iii) reduction of nystagmus in 30% of patients; (iv) increase of visual fields; (v) reactivation of the visual cortex; and (vi) significant mobility improvement in navigation tasks (Table 1). A longer follow-up will be required to assess whether PR cell loss is halted and/or slowed in the AAV2/2-RPE65–treated eyes. Importantly, Bennett et al. (2012) have successfully readministered AAV2/2-RPE65 to the contralateral eye of three LCA2 patients previously treated with the same vector and have observed that this is safe and effective (Table 1), confirming the immune-privileged status of the subretinal space (Anand et al., 2002).
CBA, CMV enhancer/chicken beta actin promoter; fMRI, functional magnetic resonance imaging; FST, full-field stimulus testing; mfERG, multifocal electroretinogram; PLR, pupillary light reflex; RPE65, 65-kDa RPE-specific isomerase; VA, visual acuity.
However, unlike what was observed in the preclinical studies (Acland et al., 2001; Narfstrom et al., 2003; Le Meur et al., 2007; Annear et al., 2011), full-field Ganzfeld ERGs remained negligible in all treated LCA2 eyes. In two cases, the reduced nystagmus allowed the measurement of electrical responses from the treated area by the sensitive multifocal ERG technique (Maguire et al., 2009) (Table 1). The lack of full-field ERG responses in the trials suggests that either the levels of RPE65 transduction achieved are insufficient or that most of the surviving LCA2 PRs are irreversibly compromised due to the 11-cis-retinal deficiency. To better understand the effect of 11-cis-retinal deprivation on PR physiology, it would be worth testing the effect of substitute chromophores in the LCA2 retina. In fact, a clinical trial (NCT01014052) that evaluates the efficacy of systemic administration of substitute chromophores to LCA2 patients has been recently initiated, and the preliminary efficacy results are encouraging (Cideciyan et al., 2012). The substitute chromophores could thus be exploited to counterbalance PR demise before RPE65 gene delivery (Batten et al., 2005). On the other hand, AAV serotypes, other than AAV2, which efficiently transduce the RPE [i.e., AAV2/4 (Le Meur et al., 2007) and AAV2/1 (Auricchio et al., 2001; Dejneka et al., 2004; Jacobson et al., 2005)], used in combination with improved RPE65 expression cassettes could provide higher levels of RPE65 transgene expression, thus resulting in ERG improvement.
The LCA2 trials also raised some concerns on the invasiveness of the SR vector delivery to a diseased retina, in particular regarding the foveal region (Jacobson et al., 2005; Maguire et al., 2008). Delivery of the vector to extrafoveal PR-rich regions (Jacobson et al., 2005, 2008) should still achieve significant visual improvement, reducing the risks of damaging the remaining foveal cones in the LCA2 retina.
Conclusions and Future Prospects
The excellent safety and efficacy results from the LCA2 trials strongly argue for testing AAV-mediated gene therapy in other rare and common blinding conditions. The best candidates are retinal diseases with severe functional defects but slow PR degeneration, such as achromatopsia or LCA1. However, several IRs have a mild phenotype with a slowly-progressing retinal degeneration. The design of clinical trials for these conditions is much more challenging, as long-term observations and a detailed knowledge of the natural history of each specific form will be required to establish efficacy.
Another challenge to be overcome in the next years will be gene therapy of those IRs caused by mutation in large genes expressed in PRs, such as Abca4 mutated in STGD. As these genes are not efficiently accommodated in single AAVs, to date the most efficient viral vector for PR transduction, the identification of Ads or LVs able to efficiently target PRs upon SR administration or improvements in dual or oversized AAV technology may satisfy this yet unmet need.
An additional major hurtle in the development of gene therapy of IRs is the extremely high genetic heterogeneity of this group of diseases, which renders particularly challenging the identification of the mutations underlying the disease in each given patient. Considering that more than 50 genes have been associated with RP (Daiger et al., 2011) and more than 15 are involved in LCA (Daiger et al., 2011), a classic Sanger sequencing-based method would be unfeasible to screen the DNA sample of an RP or LCA patient for mutations in each of these genes. The recent development of high-throughput sequence technologies referred to as next-generation sequencing (NGS) (Metzker, 2010) will most likely allow the sequencing of the whole exome or genome of a patient with IR in a very limited time and at very low costs.
In vivo, noninvasive retinal imaging is also dramatically improving thanks to the development of optical coherence tomography (van Velthoven et al., 2007), which allows the precise evaluation of the structure and thickness of the various human retinal layers over time. The combination of information coming from NGS and optical coherence tomography will allow to: (i) overcome the poor genotype–phenotype correlation of IR patients; (ii) select the most appropriate window of time for therapeutic intervention; and (iii) deliver the therapeutic vector to the most preserved and responsive retinal areas.
In conclusion, the time is ripe for the clinical development of gene therapy for IRs. From now on, the pace will be dictated mainly by the availability of sufficient funding supporting the translation of many proofs of concept from bench to bedside. U.S. and E.U. funding agencies are increasing the budget allocated to support the development of new therapies for rare diseases, such as IRs. Biotechs and pharmaceutical companies are viewing rare diseases as important targets to test the safety and efficacy of novel therapeutic platforms, like gene therapy, before moving to more common conditions. The future looks bright for gene therapy of blindness.
Footnotes
Acknowledgments
We thank Dr. Graciana Diez-Roux for the critical reading of the manuscript. A.A. acknowledges funding from the National Institutes of Health (grant R24 EY019861-01A1), the European Union (ERC Starting Grant “RetGeneTx” and the FP7 “TREATRUSH” grant), the Italian Telethon Foundation (grant TAAMT1), and the Italian Ministry of the University and Scientific Research (grant PRIN no. 2008SR7557_003).
Author Disclosure Statement
The authors have nothing to disclose.