
Abstract
Vaccination is, in theory, a safe and effective approach for controlling disseminated or metastatic cancer due to the specificity of the mammalian immune system, yet its success in the clinic has been hampered thus far by the problem of immune tolerance to tumor self-antigen. Here we describe a DNA vaccination strategy that is able to control cancer by overcoming immune tolerance to tumor self-antigen. We engineered a DNA construct encoding a dimeric form of a secreted single-chain trimer of major histocompatibility complex class I heavy chain, β2-microglobulin, and peptide antigen linked to immunoglobulin G (SCT-Ag/IgG). The chimeric protein was able to bind to antigen-specific CD8+ T cells with nearly 100% efficiency and strongly induce their activation and proliferation. In addition, the chimeric protein was able to coat professional antigen-presenting cells through the Fc receptor to activate antigen-specific CD8+ T cells. Furthermore, intradermal vaccination with DNA-encoding SCT-Ag/IgG could generate significant numbers of cytotoxic effector T cells against tumor self-antigen and leads to successful therapeutic outcomes in a preclinical model of metastatic melanoma. Our data suggest that the DNA vaccine strategy described in the current study is able to break immune tolerance against endogenous antigen from melanoma and result in potent therapeutic antitumor effects. Such strategy may be used in other antigenic systems for the control of infections and/or cancers.
Introduction
A particularly powerful strategy to increase the efficacy of DNA vaccines involves bypassing the antigen-processing pathway in DNA-transfected DCs by expressing a single chain trimer (SCT) composed of major histocompatibility complex (MHC) class I heavy chain, β2-microglobulin (β2-M), and peptide antigen (Primeau et al., 2005; Huang et al., 2007). The SCT localizes to the plasma membrane and stably displays peptide antigen to CD8+ cytotoxic T cells (CTLs). We have previously incorporated the SCT concept in several of our DNA vaccines. For example, we demonstrated that intradermal immunization with DNA encoding an SCT containing the H2-Kb-restricted immunodominant epitope of human papillomavirus (HPV) type-16 E6 protein linked to H2-Kb and β2-M generates more E6-specific CTLs than DNA encoding the wild-type E6 epitope (Huang et al., 2007). In addition, intradermal immunization with DNA encoding an SCT containing the human leukocyte antigen (HLA)-A2-restricted immunodominant epitope of the tumor-associated antigen mesothelin linked to HLA-A2 and β2-M generates human mesothelin-specific CTLs and protects against challenge with human mesothelin-expressing tumors in HLA-A2 transgenic mice (Hung et al., 2007a).
To further expand the SCT concept, we explored the immune response produced by a secreted form of the SCT. We reasoned that if such an SCT could be captured by DCs and remain functional, then it would have access to a much larger number of DCs in the draining lymph nodes compared to the membrane-bound form, greatly improving the activation of antigen-specific CTLs. To test this idea, we engineered an SCT containing the H2-Kb-restricted immunodominant epitope of chicken ovalbumin (Ova), a model antigen, linked to H2-Kb and β2-M and fused it with immunoglobulin G (IgG) (SCT-Ova/IgG). This construct contains the signal sequence for interleukin (IL)-2 and is thus secreted from transfected cells. In addition, the presence of IgG causes the SCT to form a dimer and to bind to the Fc receptor on DCs, rendering it available to be distributed among a large number of DCs in the lymph nodes. We found that the chimeric SCT-Ova/IgG protein was able to bind and to activate Ova-specific CD8+ T cells.
In addition, we demonstrate that vaccination with DNA encoding the chimeric SCT-Ova/IgG protein was able to generate a significant number of Ova-specific CD8+ T cells, leading to potent therapeutic antitumor effects against Ova-expressing tumors in vaccinated mice. Finally, We showed that intradermal vaccination with DNA encoding an SCT containing the H2-Kb-restricted immunodominant epitope of tyrosinase-related protein 2 (Trp2), a melanoma antigen, linked to H2-Kb and β2-M, and fused with IgG (SCT-Trp2/IgG) was capable of eliciting a Trp-2-specific CD8+ T-cell immune response and potent therapeutic antitumor effects against B-16 melanoma in treated mice. The clinical significance of the current studies is discussed.
Materials and Methods
Mice
We acquired 6- to 8-week-old female C57BL/6 mice from the National Cancer Institute (Frederick, MD). We performed all mice procedures in accordance with approved protocols and recommendations for the proper use and care of laboratory animals.
Cells
We acquired B16-F10 melanoma cells from the American Type Culture Collection (ATCC) (Rockville, MD). Fang et al. (2008) previously described the Ova-expressing B16 melanoma model (B16-Ova). We cultured these cells in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 50 units/ml of penicillin/streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate, and 2 mM nonessential amino acids at 37°C with 5% carbon dioxide. The generation of E7- (aa 49–57) and Ova- (aa 257–264) specific CD8+ T cells have been previously described (Martinez-Kinader et al., 1995; Wang et al., 2000). We acquired baby hamster kidney (BHK)-21 cells from the ATCC. Dr. Kenneth Rock (University of Massachusetts, Worcester, MA) kindly provided DC 2.4 cells (Shen et al., 1997). We cultured these cells in Roswell Park Memorial Institute-1640 medium supplemented with 10% fetal bovine serum, 50 units/ml of penicillin/streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate, and 2 mM nonessential amino acids at 37°C with 5% carbon dioxide.
Plasmid DNA constructs and preparation
Yu et al. (2002) previously described the SCT-Ova construct containing the immunodominant H2-Kb-restricted Ova epitope (SIINFEKL, aa 257–264), β2 microglobulin (β2M), and H2-Kb heavy chain in the pIRES vector (Yu et al., 2002). To generate SCT-Trp2, we synthesized an insert containing the immunodominant H2-Kb-restricted Trp2 epitope (VYDFFVWL, aa 181–188) (Bloom et al., 1997) with flanking AgeI/NheI restriction sites by annealing two single-stranded oligonucleotides (5’ – CCGGTTTGTATGCTGTGTATGACTTTTTTGTGTGGCTCGGAG GAGGTG – 3’ and 5’ – CTAGCACCTCCTCCGAGCCACACAAAAAAGTCATACACAGC ATACAAA – 3’). We cloned this insert into SCT-Ova using AgeI/NheI sites to replace the Ova epitope with Trp2. To generate SCT-Ova/IgG, we amplified the SIINFEKL-β2M-H2-Kb insert from the SCT-Ova construct by polymerase chain reaction (PCR) using the primers 5’ – TTTCGACTCTAGAAGCATGGCTCG – 3' and 5’ – AAAAGATCTAGTGGA TGGAGGAGGCTCCCA – 3’, and cloned it into the pFUSE-mIgG2A vector at EcoRI/BgIII sites (InvivoGen, San Diego, CA). To generate SCT-Trp2/IgG, we amplified the VYDFFVWL-β2M-H2-Kb insert from the SCT-Trp2 construct by PCR using the primers 5’ – TTTCGACTCTAGAAGCATGGCTCG – 3’ and 5’ – AAAAGATCTAGTGG ATGGAGGAGGCTCCCA – 3’) and cloned it into the pFUSE-mIgG2A vector (Invivogen) at AgeI/BgIII sites. We verified all plasmids by DNA sequencing.
Transfection and protein purification
We transfected 1×107 BHK-21 cells with 50 μg of plasmid-encoding SCT in T-150 flasks using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 3 days, we collected culture media, processed it with a 0.22 μm filter syringe (Millipore, Billerica, MA), and concentrated it with a 50 kDa cut-off Amicon® Ultra-15 filter tube (Millipore). We applied the concentrated media to a HiTrap Protein G HP column (GE Healthcare, Laurel, MD), washed with 20 mM sodium phosphate buffer (pH 7.0), and eluted with 0.1 M glycine-HCl buffer (pH 2.8). We measured protein concentration by Coomassie Plus assay (Pierce, Rockford, IL) and determined purity by reducing or nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis.
DNA vaccination
We performed intradermal DNA vaccination using a helium-driven gene gun (BioRad, Hercules, CA) as described previously (Chen et al., 2000). We administered SCT-Ova/IgG, SCT-Ova, SCT-Trp2/IgG, or SCT-Trp2 DNA-coated gold particles to the shaved abdominal region of mice with a discharge pressure of 400 psi. We immunized C57BL/6 mice with 2 μg of plasmid and boosted at the same dose and regimen 1 week later.
Intracellular cytokine staining and flow cytometry
We harvested splenocytes from mice 1 week after the last vaccination. We incubated 6×105 pooled splenocytes from each group with 1 μg/ml H2-Kb-restricted Ova peptide (SIINFEKL) or Trp2 peptide (VYDFFVWL) for 16 hr. We performed surface staining for CD8 with a phycoerythrin (PE)-conjugated rat anti-mouse CD8 monoclonal antibody (BD Biosciences, San Diego, CA) and intracellular cytokine staining for interferon (IFN)-γ with a fluorescein isothiocyanate-conjugated rat anti-mouse IFN-γ monoclonal antibody (BD Biosciences). We performed flow cytometry using a Becton Dickinson FACScan machine with CellQuest Pro software (BD Biosciences) as described previously (Chen et al., 2000).
T cell proliferation assay
We labeled 1×105 Ova-specific CD8+ T cells with 5 μM carboxyfluorescein succinimidyl ester (CFSE) and then incubated the cells with different amounts of purified protein (0, 0.1, 1, or 10 μg) for 3 days in the presence of 1,000 units of IL-2. We performed flow cytometry to detect CFSE dilution.
In vivo tumor treatment experiments
For the B16-F10 tumor treatment, we challenged C57BL/6 mice (five per group) with 1×105 B16-F10 cells per animal in the tail vein to simulate hematogenous tumor spread (Ji et al., 1998). After 3 days, we immunized mice with 2 μg per animal of DNA-encoding SCT-Trp2 or SCT-Trp2/IgG by intradermal gene gun administration. After 1 week, we boosted mice at the same dose and regimen. We sacrificed mice 28 days after tumor injection. We counted the mean number of pulmonary nodules in the mice; investigators were blinded to sample identity. For the B16-Ova tumor treatment, we challenged C57BL/6 mice (five per group) with 1×105 B16-F10 cells per animal in the right hind leg. After 3 days, we immunized mice with 2 μg per animal of DNA-encoding SCT-Ova or SCT-Ova/IgG by intradermal gene gun administration. After 1 week, we boosted mice at the same dose and regimen. We monitored tumor growth by visual inspection and palpation twice each week.
Statistical analysis
Data presented in this study are from one out of two to three independent experiments performed and are expressed as mean+standard deviation. We performed all experiments at least in triplicate for each group. We evaluated the flow cytometry and tumor treatment data by analysis of variance and Tukey-Kramer multiple comparison test. We compared individual data points by Student's t-test. We analyzed the event-time distribution for mice by the Kaplan-Meier method and the log-rank test. We considered p values less than 0.05 to be significant.
Results
Generation and characterization of chimeric SCT-Ova/IgG protein
The diagram of our SCT construct is displayed in Figure 1A. We purified the recombinant SCT on a protein G column and confirmed its presence by sodium dodecyl sulfate polyacrylamide gel electrophoresis (Fig. 1B). SCT-Ova/IgG forms a dimer due to the presence of the IgG domain. As shown in Figure 1C, under nonreducing conditions, we are able to demonstrate the dimerization of the chimeric protein. Taken together, our data indicate that we have successfully generated a chimeric protein that is capable of forming a dimer under nonreducing conditions.
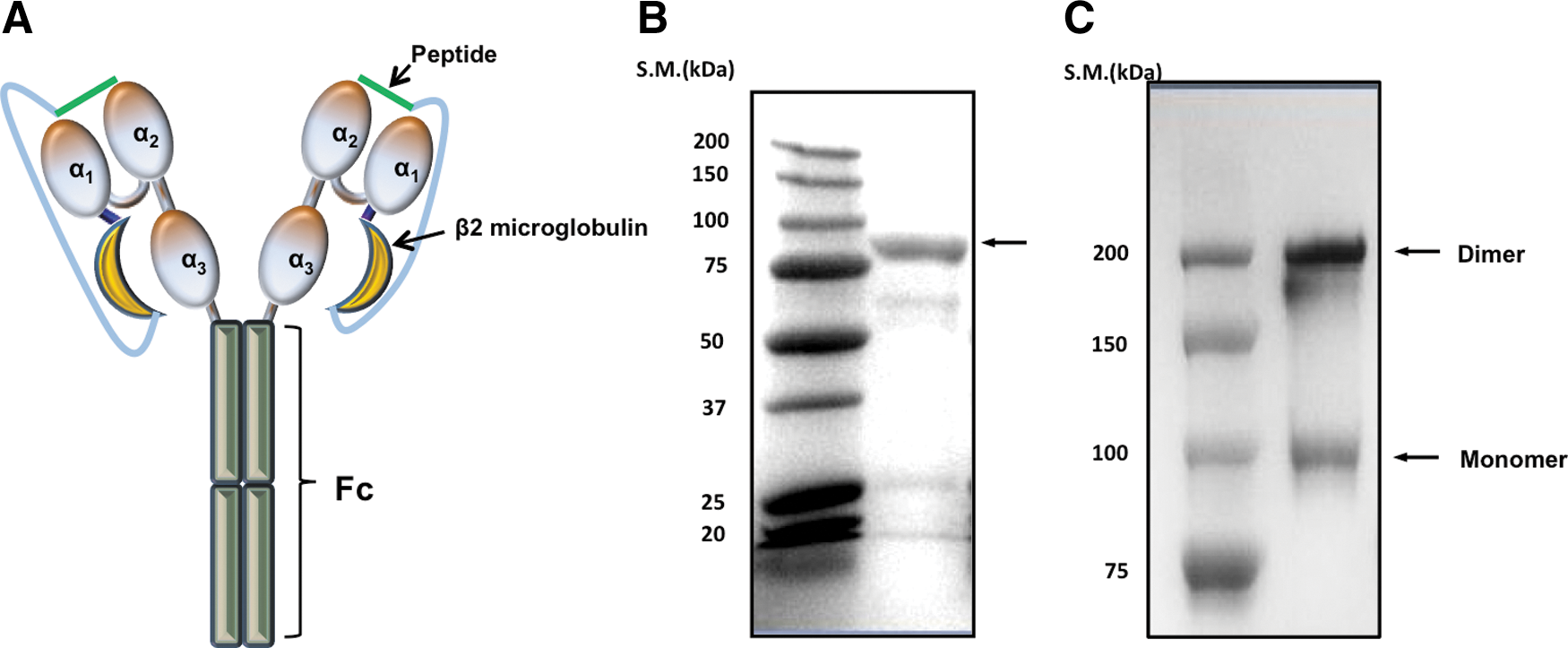
Characterization of the single-chain trimer-ovalbumin/immunoglobulin G (SCT-Ova/IgG) construct.
Chimeric SCT-Ova/IgG can bind and activate Ova-specific CD8+ T cells resulting in Ova-specific CD8+ T-cell proliferation
We furthered our study by carrying out additional experiments to characterize SCT-Ova/IgG. We observed that SCT-Ova/IgG attaches to Ova-specific T cells in an antigen-dependent manner, as well as a dose-dependent manner, by mixing various concentrations of the purified protein with either Ova-specific T cells or E7-specific T cells. We then stained the cells with PE-labeled anti-mouse IgG and performed flow cytometry to detect the binding of the SCT to the cells. As shown in Figure 2A, at all concentrations tested (i.e., 0.1, 1, and 10 μg/ml), Ova-specific T cells incubated with SCT-Ova/IgG displayed an about five-fold increase in fluorescence intensity compared to control. By contrast, E7-specific CD8+ T cells incubated with even 10 μg/ml SCT-Ova/IgG demonstrated a minimal difference in fluorescence intensity compared to control, proving that our SCT can interact with T cells in an antigen-specific manner. Since SCT-Ova/IgG attaches to Ova-specific T cells, we reasoned that it might be able to induce proliferation of these cells. To assess this, we labeled Ova-specific T cells with CFSE and mixed them with various concentrations of SCT-Ova/IgG together with recombinant IL-2. We performed flow cytometry 3 days later to detect CFSE dilution. As shown in Figure 2B, addition of SCT-Ova/IgG caused CFSE dilution in a dose-dependent manner, indicating proliferation of the T cells. We verified that these T cells were functional by using flow cytometry to detect surface CD8 and intracellular IFN-γ. As shown in Figure 2C and D, addition of 10 μg/ml SCT-Ova/IgG stimulated close to 60% of CD8+ Ova-specific T cells. However, as expected, this construct failed to elicit IFN-γ production by E7-specific T cells. Together, these results demonstrate that SCT-Ova/IgG can directly interact with and activate antigen-specific T cells.
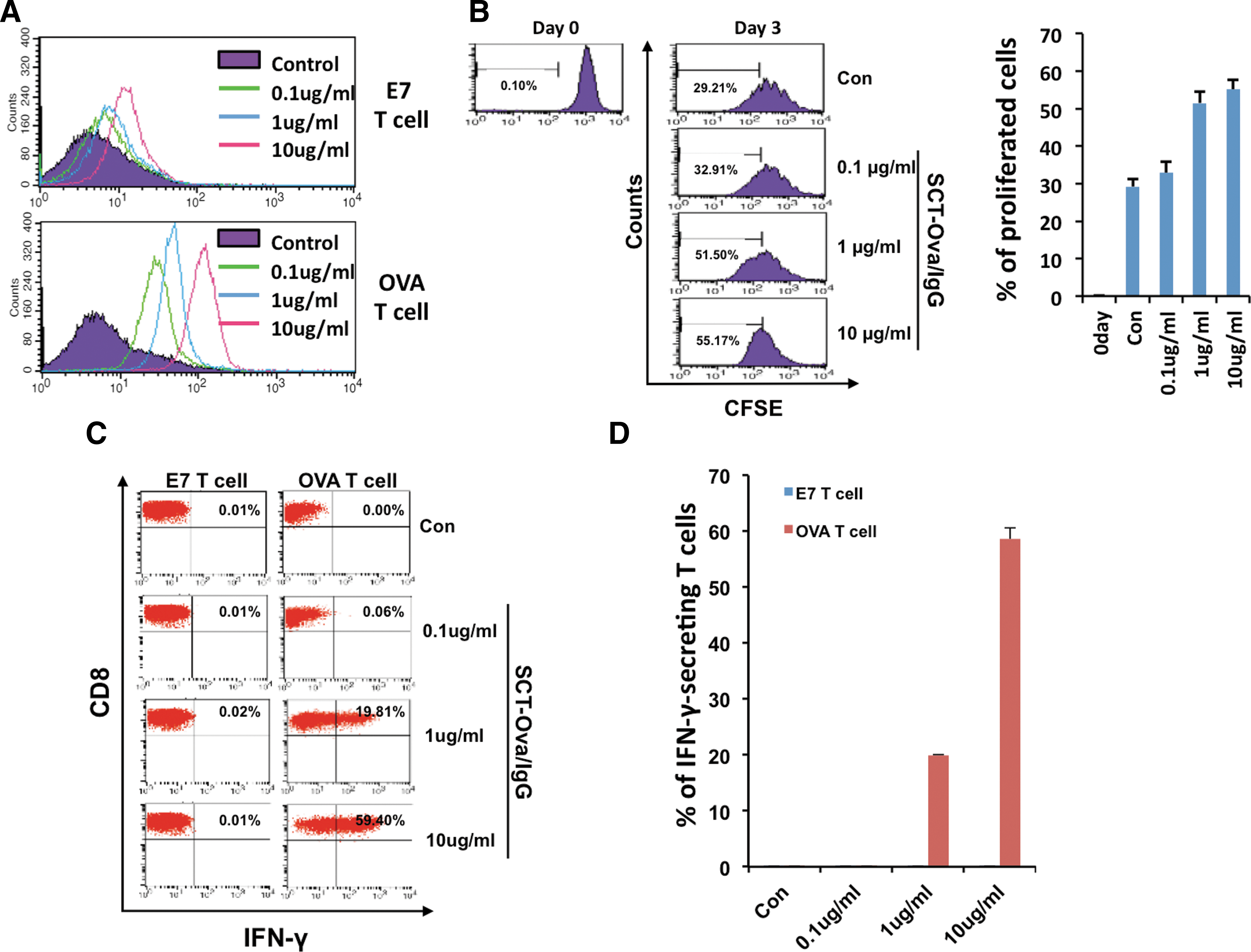
Characterization of Ova-specific T-cell activation and proliferation after stimulation with single chain trimer-ovalbumin/immunoglobulin G (SCT-Ova/IgG).
SCT-Ova/IgG is able to bind to Fc receptor-expressing DCs leading to activation of Ova-specific CD8+ T cells
Although we have demonstrated that SCT-Ova/IgG could directly activate T cells in vitro, our vaccination system hinges on the production and presentation of antigen by epidermal DCs that then migrate to the lymph nodes. In this context, it is unlikely that soluble SCT-Ova/IgG is present in the lymph nodes to activate T cells. Instead, a more likely scenario is that the DCs secrete SCT-Ova/IgG, which then attaches to those DCs and other surrounding cells through the Fc receptor. These SCT-Ova/IgG-bound DCs then migrate to the lymph nodes where they can prime antigen-specific T cells. To address this possibility, we mixed DCs with SCT-Ova/IgG either in the presence or absence of Fc blocker, which inhibits the Fc receptor. We washed away soluble SCT-Ova/IgG, incubated the DCs with Ova-specific T cells for 15 hours, and performed both intracellular cytokine staining and flow cytometry to detect IFN-γ expression by the T cells. As shown in Figure 3A and B, the presence Fc blocker reduced IFN-γ expression by Ova-specific T cells three- to four-fold. Therefore, our data are consistent with the theory that the Fc receptor on DCs plays an important role in the ability of the SCT to activate antigen-specific T cells in vivo.
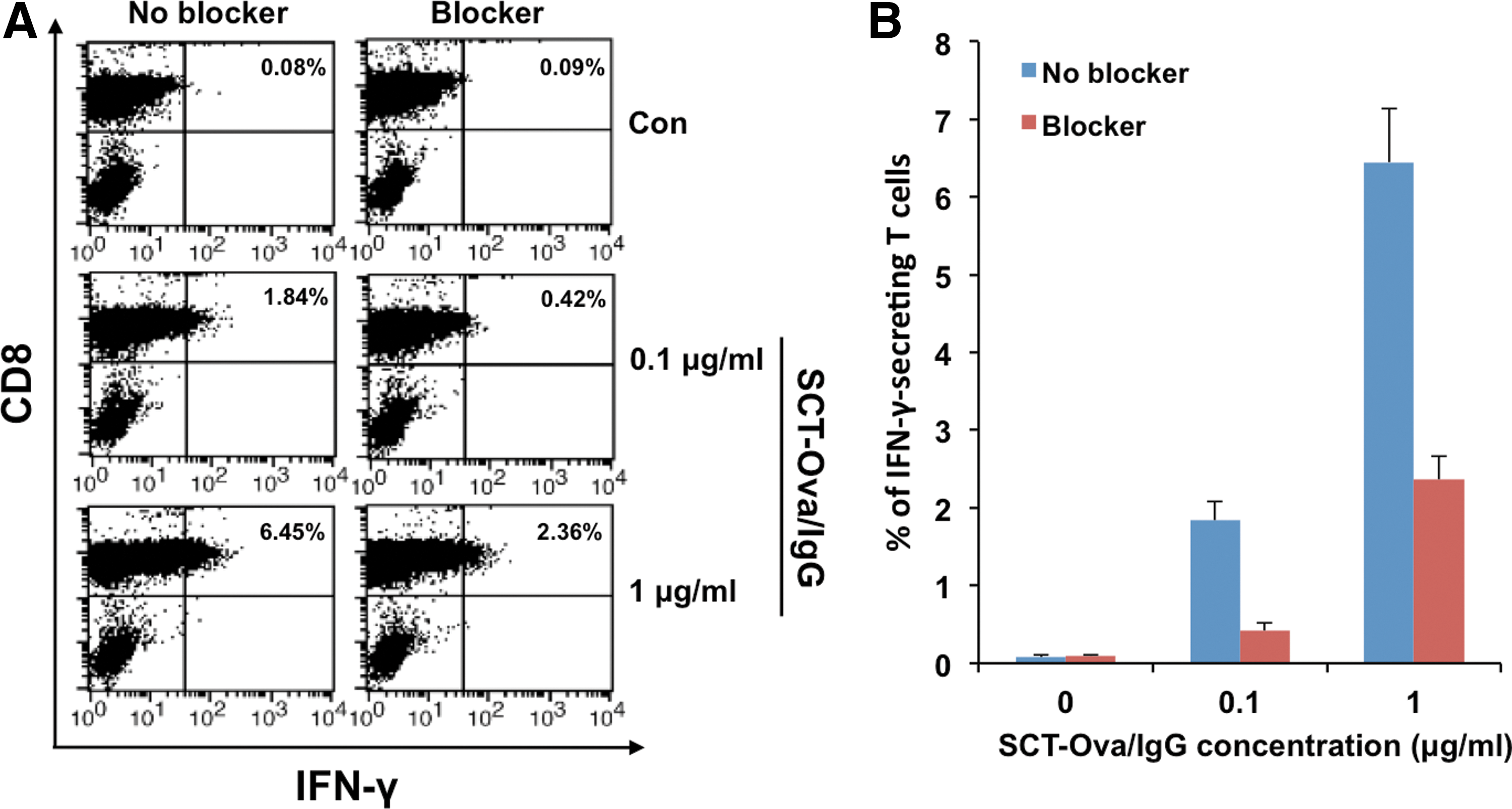
Characterization of the role of Fc receptor in mediating Ova-specific T-cell activation by single chain trimer-ovalbumin/immunoglobulin G (SCT-Ova/IgG). We treated dendritic 2.4 cells with or without Fc receptor blocker for 20 min and then added SCT-Ova/IgG protein for 20 min at 37°C.
Vaccination of DNA-encoding SCT-Ova/IgG generates a potent Ova-specific CD8+ T-cell response and therapeutic antitumor effect against Ova-expressing tumors
We have previously created a DNA vaccine-encoding SCT containing Ova peptide (pIRES-SCT-Ova). In order to compare the Ova-specific CD8+ T cell immune response generated by pIRES-SCT-Ova to that generated by DNA-encoding SCT-Ova/IgG (pFUSE-SCT-Ova/IgG) we vaccinated C57BL/6 mice with the DNA vaccines intradermally via gene gun. The splenocytes of the vaccinated mice were isolated for characterization of Ova-specific CD8+ T cells. As shown in Figure 4A and B, vaccination with pFUSE-SCT-Ova/IgG produced approximately three times more CD8+ IFN-γ+ Ova-specific CD8+ T cells than pIRES-SCT-Ova. These effects on the immune response correlated with the therapeutic outcome in mouse-bearing subcutaneous B16-Ova tumors. Animals administered with pFUSE-SCT-Ova/IgG had much smaller tumor sizes and survived significantly longer compared to animals administered with pIRES-SCT-Ova (p-value<0.01) (Fig. 4C and D). Thus, DNA-encoding SCT-Ova/IgG can generate significantly better Ova-specific CD8+ T-cell immune responses as well as a better therapeutic antitumor effect compared to DNA-encoding SCT-Ova.
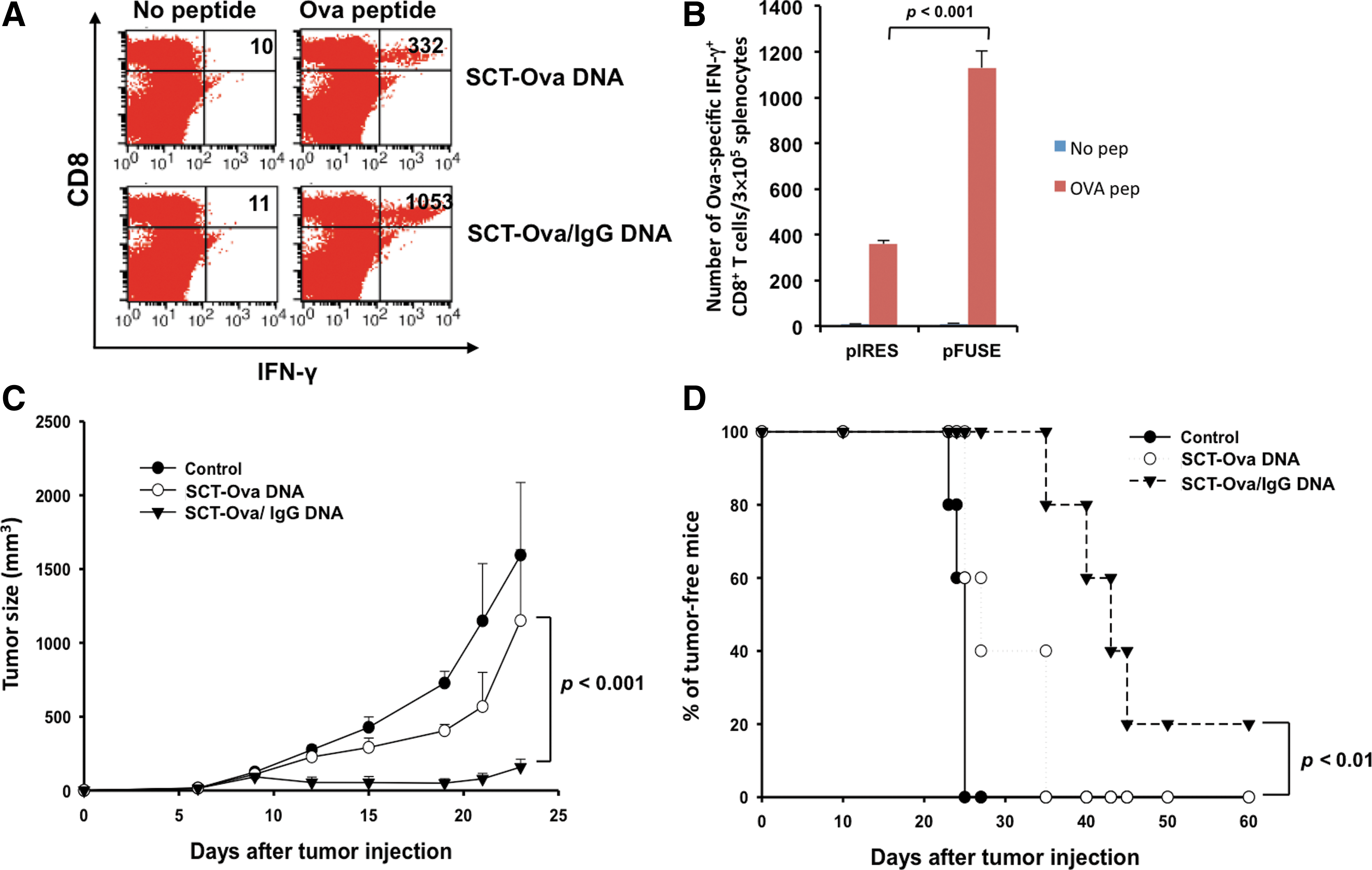
Comparison of the Ova-specific immune response and antitumor effect generated by vaccination with DNA-encoding SCT-Ova/IgG or SCT-Ova.
Vaccination of DNA-encoding SCT-Trp2/IgG generates a potent Trp2-specific CD8+ T-cell response and therapeutic antitumor effect against Trp2-expressing tumors
Because we have developed a potent vaccination strategy employing SCT technology and characterized its mechanism of action, we sought to extend this strategy to a clinically relevant setting. We synthesized an SCT-containing H2-Kb linked to the immunodominant epitope of the melanoma Trp2 antigen and IgG (pFUSE-SCT-Trp2/IgG). For comparison, we also synthesized DNA-encoding SCT containing Trp2 peptide (pIRES-SCT-Trp2). We vaccinated mice with pFUSE-SCT-Trp2/IgG or pIRES-SCT-Trp2 intradermally via gene gun and measured the frequency of IFN-γ-secreting Trp2-specific CD8+ T cells in the spleen after 2 weeks. As shown in Figure 5A and B, vaccination with pFUSE-SCT-Trp2/IgG produces a significant number of Trp2-specific IFN-γ+ CD8+ T cells, whereas vaccination with pIRES-SCT-Trp2 produces only a minimal number of these T cells. Administration of pFUSE-SCT-Trp2/IgG also led to a reduced number of pulmonary nodules and increased survival in mice challenged intravenously with melanoma cells compared to administration of pIRES-SCT-Trp2 or control vector (Fig. 5C and D). Together, these results demonstrate that the SCT strategy can be successfully used to break immune tolerance against a clinically important tumor-associated antigen.

Characterization of the tyrosinase-related protein 2 (Trp2)-specific immune response and antitumor effect generated by DNA-encoding SCT-Trp2/IgG or SCT-Trp2.
Discussion
In our study, we have successfully established an SCT-antigen/IgG dimeric protein that binds to T cells with high antigen specificity. The SCT-Ova/IgG dimeric protein has the ability to specifically bind Ova-specific CD8+ T cells and induce the proliferation and activation of Ova-specific T cells in a concentration-dependent manner in vitro. In addition, SCT-Ova/IgG can activate T cells through an Fc receptor-mediated pathway (Fig. 3). Furthermore, we demonstrated that vaccination with DNA-encoding SCT-antigen/IgG was able to trigger potent antigen-specific CD8+ T-cell immune responses against tumor self-antigen, resulting in a potent antitumor effect. Thus, we have created a vaccination platform technology that is able to break immune tolerance. Other vaccination technologies have been discovered that are also able to break immune tolerance, and it will be of interest to compare them with the technology employed in the current study (O et al., 2003; Yamano et al., 2006; Tanaka et al., 2012; Vasievich et al., 2012)
We compared the DNA construct encoding SCT-Ova/IgG with the DNA construct encoding SCT-Ova using an Ova-expressing murine tumor models. We observed a three- to four-fold better T-cell immune response and better tumor control in response to the DNA construct encoding SCT-Ova/IgG compared to the DNA construct encoding SCT-Ova (Fig. 4B and C). The observed improvements of the DNA vaccine effects are likely due to several factors. Firstly, the dimerization of chimeric SCT-Ova/IgG may increase the avidity of the chimeric molecule for binding to T-cell receptors of Ova-specific CD8+ T cells resulting in increased T-cell activation (Lebowitz et al., 1999). Secondly, the chimeric SCT-Ova/IgG has an Fc domain that can bind to the Fc receptor expressed in many antigen-presenting cells including macrophages and DCs. The bound macrophage or DC will contribute to the priming of the Ova-specific CD8+ T cells. Taken together, these factors account for the observed enhancement of the Ova-specific CD8+ T-cell immune response and antitumor effect mediated by the SCT-Ova/IgG.
The successful application of our DNA vaccine in generating the potent Trp2-specific CD8+ T-cell immune response and antitumor effect against the Trp2-expressing melanoma represents a platform technology for breaking immune tolerance. It is now clear that many tumors are associated with antigens that are endogenous. The major problem in generating potent immune responses against these antigens is immune tolerance. Since Trp2 is also an endogenous tumor antigen, the successful application of this technology suggests that such technology may be applied to other endogenous antigens. Therefore, it is important to further explore our strategy using other endogenous antigens in order for this to become a useful platform for breaking immune tolerance.
Our vaccination technology can potentially be used in conjunction with immune-modulating agents to further enhance the vaccine potency. Previously, Tanaka et al. (2012) demonstrates an immune-modulating agent, Lentinula edodes mycelia, mitigating immune suppression by T-regulatory cells can be used to improve TRP2 peptide-based vaccine potency. They demonstrate that TRP2 peptide-based vaccination in conjunction with immune-modulating agent Lentinula edodes mycelia can further enhance the antigen-specific immune response as well as the therapeutic antitumor effect against TRP2-expressing tumors. Their data suggests that the immune-modulating agent could potentially be used in conjunction with our vaccination technology to further enhance the therapeutic antitumor effect of our DNA construct.
Several significant limitations of the DNA vaccine using the current approach are identified. For example, it is essential to determine the immunodominant CTL epitope of the antigen of interest in order to be used for the generation of DNA vaccines. However, the immunodominant CTL epitopes of antigens of interest have successfully been determined by many investigators, including us (Peng et al., 2004; Hung et al., 2007b). Another limitation is that such vaccines can only benefit individuals that express the specific MHC class I molecule used in the DNA vaccine. However, with the increasing identification or characterization of immunodominant CTL epitopes of the antigen of interest, which are restricted by different MHC class I molecules, we have now generated multiple different DNA constructs employing CTL epitopes corresponding to the MHC class I molecules expressed by a particular individual. It is expected that such technology will be particularly useful for personalized medicine.
In conclusion, we have demonstrated vaccination with DNA-encoding SCT-Ag/IgG generates robust cytotoxic T-cell responses against tumor self-antigen and leads to successful therapeutic outcomes in a preclinical model of metastatic melanoma. With the advancement in gene sequencing and personalized medicine, we would further the translational value of this treatment by overcoming the MHC restriction and maximizing the efficacy of this therapeutic vaccine.
Footnotes
Acknowledgments
We thank Dr. Shiwen Peng for helpful discussion, as well as Ms. Barbara Ma for critical review of this manuscript. We would like to thank Jayne Knoff for preparation of this manuscript. This work was funded by the National Institutes of Health Cervical Cancer SPORE and Head and Neck Cancer SPORE (P50 CA098252 and P50 CA96784-06), RO-1 grant (CA114425-01), and Melanoma Research Alliance.
Author Disclosure Statement
No competing financial interests exist.