
Abstract
Gene transfer vectors derived from oncoretroviruses or lentiviruses are the most robust and reliable tools to stably integrate therapeutic transgenes in human cells for clinical applications. Integration of these vectors in the genome may, however, have undesired effects caused by insertional deregulation of gene expression at the transcriptional or post-transcriptional level. The occurrence of severe adverse events in several clinical trials involving the transplantation of stem cells genetically corrected with retroviral vectors showed that insertional mutagenesis is not just a theoretical event, and that retroviral transgenesis is associated with a finite risk of genotoxicity. In addressing these issues, the gene therapy community offered a spectacular example of how scientific knowledge and technology can be put to work to understand the causes of unpredicted side effects, design new vectors, and develop tools and models to predict their safety and efficacy. As an added benefit, these efforts brought new basic knowledge on virus–host interactions and on the biology and dynamics of human somatic stem cells. This review summarizes the current knowledge on the interactions between retroviruses and the human genome and addresses the impact of target site selection on the safety of retroviral vector–mediated gene therapy.
Introduction
When sequences of several vertebrate genomes became available, PCR-based methods were designed to clone and sequence the junctions between proviral and host-cell DNA (Schmidt et al., 2001; Schroder et al., 2002), and to analyze target site selection in a statistically significant fashion. These pioneering studies showed that retroviruses select their target in a sequence-independent manner, with preferences for transcribed genes in the case of the human immunodeficiency virus (HIV) (Schroder et al., 2002) or gene promoters in the case of the Moloney murine leukemia virus (MLV) (Wu et al., 2003). More recently, massive parallel sequencing technology has been adapted to retroviral integration studies, increasing exponentially the resolution of integration maps. Ligation-mediated (LM) or linear amplification-mediated (LAM) PCR coupled to pyrosequencing allowed to uncover genomic features systematically and specifically associated with retroviral insertions and revealed that each retrovirus has a unique, characteristic pattern of integration within mammalian genomes (reviewed in Bushman [2003] and Bushman et al. [2005]). Retroviral vectors, designed to integrate therapeutic transgenes in target cells, maintain the integration preferences of the viruses from which they derive, with significant implications for their biosafety.
Different Retroviruses Have Different Integration Preferences
Based on evolutionary relatedness, retroviruses are classified into seven genera (Alpha-, Beta-, Gamma-, Delta-, and Epsilon-retroviridae, Spumaviridae, and Lentiviridae). Except for epsilon retroviruses, integration preferences are known for at least one member of each family. The amount of available information reflects the clinical relevance of the virus, where the HIV and Moloney murine leukemia virus (MLV) integration profiles are the most extensively characterized. Integration studies revealed different patterns of favored and disfavored target sites for each retroviral family, suggesting differential involvement of viral and cellular factors in the integration process. Interestingly, an unsupervised clustering analysis of different retroviruses on the basis of their integration preferences overlap with phylogenetic trees based on the sequence similarity of their integrases, and both are in good agreement with traditional trees based on genomic sequences (Derse et al., 2007). This suggests a strong link between target site selection and evolution and shows that integration is part of the strategy by which retroviruses maximize survival and propagation.
Retroviral integrases have strict sequence requirements for the viral DNA ends: the dinucleotide CA is invariably located 2 base pairs from both viral ends, and certain nucleotides may recur up to 15 base pairs away from the CA. Conversely, sequences at the target site are very diverse. Early analysis of integration sites obtained from cells infected with HIV-1, MLV, the avian sarcoma-leukosis virus (ASLV), and the simian immunodeficiency virus (SIV) showed a statistically weak palindromic consensus centered on the virus-specific duplicated target site sequence (Wu et al., 2005). The consensus is weakly conserved but distinguishable between different retroviruses, as later confirmed by analysis of larger datasets (Berry et al., 2006; Wang et al., 2007). The same consensus was found around insertion sites in naked genomic DNA in vitro, suggesting that the nucleotide sequence preferences are determined by the integration machinery rather than by host-cell factors and most likely reflect spatial or energy requirements of the integration complex. In any case, the linear DNA sequence has little role in determining the integration preferences of each retroviral family, which depends on viral and cellular determinants that are poorly defined for most retroviruses.
MLV-Derived Vectors Target Transcriptionally Active Regulatory Regions
MLV is part of the gamma-retrovirus family, historically known as Oncoretroviridae for their ability to induce tumors. Because they reach high expression levels in the hematopoietic system, MLV-based vectors carrying wild-type LTRs have been largely used in gene therapy for blood disorders, and their integration profile extensively analyzed in hematopoietic progenitors and other cell types. Early studies showed that MLV has a modest preference for active genes, and a peculiar distribution around transcription start sites (TSSs), with ∼20% of insertions landing 2.5 kb upstream or downstream from the +1 position of any gene (Wu et al., 2003; Hematti et al., 2004; Mitchell et al., 2004; Cattoglio et al., 2007). TSSs have been therefore considered as a major genomic determinant of Mo-MLV integration site selection. High-throughput sequencing studies showed, however, that the MLV bias for TSSs is simply one of the consequences of a more general preference of MLV PICs for genomic regions with a role in transcriptional regulation by RNA polymerase II (Pol II). In fact, regions flanking MLV integrations are enriched in promoters, CpG islands, DNase I hypersensitive sites, transcription factor binding sites (TFBSs), and phylogenetically conserved noncoding sequences, often predictive of cis-acting regulatory elements (Lewinski et al., 2006; Felice et al., 2009; Cattoglio et al., 2010a). Studies carried out in human CD34+ hematopoietic stem/progenitor cells (HSCs) and T lymphocytes showed that regions preferred by MLV are associated to histone modifications characteristic of active transcription, such as acetylations of histones H2A, H2B, H3, and H4, and binding of Pol II, CTCF, and the histone acetyl transferases p300 and CBP (Cattoglio et al., 2010a; Cattoglio et al., 2010b; Biasco et al., 2011). MLV integrations are strongly associated to methylations of H3 characteristic of promoters and enhancers, (i.e., H3K4me1, H3K4me2, and H3K4me3). In particular, H3K4me1, H3K27Ac and Pol II binding mark transcribed, activity-regulated enhancers (Djebali et al., 2012), often bound by CBP (Kim et al., 2010). Conversely, MLV integrations are under-represented in heterochromatic regions marked by H3K27me3 and H3K9me3 (Wang et al., 2007; Cattoglio et al., 2010a; Cattoglio et al., 2010b; Biasco et al., 2011). In more than one third of the cases, regions marked by H3K27me3 are also positive for the promoter-specific H3K4me3 modification, a “bivalent” chromatin signature characteristic of genes regulated during development and differentiation (Bernstein et al., 2006; Ernst et al., 2011). Most of the associations with chromatin epigenetic signatures are statistically significant for all MLV integrations, independently from their location with respect to promoters and in all analyzed cell types.
The obvious consequence of targeting active regulatory elements is that MLV integration patterns are strictly dependent on cell transcriptional programs and are therefore cell-specific. This is apparent when looking at the correlation between target genes and gene expression profiles (Schroder et al., 2002; Mitchell et al., 2004; Aiuti et al., 2007; Cattoglio et al., 2007; Cattoglio et al., 2010a; Cattoglio et al., 2010b) or at the functional characteristics of the targeted genes. Analysis of MLV insertion sites in human HSCs, T cells, or other cell types showed that genes targeted at high frequency are involved in cell-specific functions (Aiuti et al., 2007; Cattoglio et al., 2007; Cattoglio et al., 2010a; Biasco et al., 2011; Deichmann et al., 2011). Interestingly, developmentally regulated genes and genes involved in the control of cell growth, replication, and differentiation are targeted at a statistically higher frequency than housekeeping genes (Cattoglio et al., 2010b), suggesting that MLV PICs somehow discriminate between different types of promoter- and enhancer-binding complexes. The preference for highly regulated elements is suggested also by the strong association between MLV integration sites and binding of H2A.Z (Cattoglio et al., 2010b), a histone variant enriched at targets of the Polycomb complex and marking elements involved in the regulation of cell commitment and differentiation (Creyghton et al., 2008). An analysis of TFBSs around MLV integrations in HSCs, T cells, and HeLa cells identified cell-specific patterns and overrepresentation of binding sites for cell-specific families of factors (Felice et al., 2009; Cattoglio et al., 2010b). These preferences generate the typical MLV integration pattern, characterized by tight clustering in relatively small hot spots often arranged in higher-order clusters within complex loci or gene-dense regions (Cattoglio et al., 2010b; Ambrosi et al., 2011). High-definition integration maps show that regions bound by the Pol II basal transcriptional machinery, such as core promoters, are protected from the MLV insertion (Cattoglio et al., 2010a; Cattoglio et al., 2010b), confirming that integration is directed to occupied, transcriptionally active elements and not simply to open chromatin regions.
The simplest explanation of these preferences is that MLV PICs are tethered to genomic regions engaged by basal components of the enhancer-binding and/or RNA Pol II basal transcriptional machinery (Fig. 1). The viral determinants of these preferences have been relatively well defined by genetic experiments. An HIV-1 vector packaged with an MLV integrase acquires the MLV-specific bias for TSSs, CpG islands, and TFBS-rich regions (Lewinski et al., 2006; Felice et al., 2009), suggesting that the integrase plays a crucial role in targeting MLV to the genome. Other components of the PIC, such as glucosaminoglycan (GAG) polypeptides, play a minor but detectable role in the process (Lewinski et al., 2006). On the other hand, the cellular determinants of MLV target site selection are still undefined. Only one cellular factor, the protein BAF (barrier to autointegration factor), has been shown to date to be physically associated with the MLV PICs. BAF was originally identified as an inhibitor of suicide integration of the MLV provirus, which promotes efficient intermolecular DNA recombination (Lee and Craigie, 1994). Although essential for PIC integration activity, interaction with BAF alone does not obviously explain the MLV integration preferences. A yeast two-hybrid analysis of proteins potentially interacting with the MLV integrase provided a number of potential targets, many of which are component of chromatin and transcription complexes (Studamire and Goff, 2008). These targets await rigorous validation in a relevant cell system.

Retroviral vectors show different integration preferences.
An association of the MLV integrase with components of the Pol II transcriptional machinery, although supported only by indirect evidence at the moment, may be seen as an evolution of the mechanisms by which yeast retrotransposons, distantly related to retroviruses, target their integration to specific genomic regions (reviewed in Bushman, 2003). The Ty1 and Ty3 retrotransposons integrate at the 5’ end of genes transcribed by RNA Pol III, in regions that apparently tolerate insertions with no adverse consequences. The Ty3 element targets tRNA genes with extraordinary precision, inserting within few base pairs of the TSS, by tethering of PICs to the TFIIIB component of the Pol III basal transcription complex (Kirchner et al., 1995). The Ty1 element integrates less precisely, in a window of ∼750 base pairs upstream of TSSs. The histone deacetylase Hos2 and the Trithorax-group protein Set3, both components of the Set3 complex, have been proposed as the tethering factors of Ty1 (Mou et al., 2006). The domain of the Ty3 retrotransposase responsible for tethering to the Pol III complex is lacking in the evolutionarily related MLV integrase and may have been functionally replaced by domains mediating an association to Pol II–specific factors or to chromatin components associated to Pol II transcription. The structure of the integrase has interesting implications in terms of viral evolution. Oncoretroviruses may have developed a unique integration strategy that, by coupling target site selection to gene regulation, maximizes the chances of activating and maintaining proviral expression. In addition, as discussed in the following paragraphs, integration of the MLV provirus around promoters and regulatory elements of growth- and differentiation-controlling genes may increase the chances of inducing clonal expansion or transformation of the infected cells and ultimately favor viral propagation.
ASLV Vectors Integrate Almost Randomly in Mammalian Cells
ASLV is member of the alpha-retrovirus family, whose natural host is chicken. However, pseudotyped viral particles can be produced that are able to infect mammalian cells (Hu et al., 2008). Insertion site studies on this virus revealed only a weak, though still detectable, bias in favor of active genes, gene-dense regions, and genomic features associated with genes (e.g., slight enrichment compared to random sites for CpG islands, DNase I hypersensitive sites, and regulatory regions). Integrations are evenly distributed along the whole transcription unit, with no preference for TSSs (Moiani and Suerth, unpublished observations) (Fig. 1). The nearly random ASLV insertion profile in mammals is encouraging the development of optimized vectors as gene transfer tools for gene-therapy applications (Suerth et al., 2012).
Integration Preferences of Beta- and Delta-Retroviruses and Spumaviruses
The mouse mammary tumor virus (MMTV) is a representative of the beta-retrovirus family, for which only a single large-scale mapping study of integration sites has been performed, using murine and human mammary cell lines as target cells. MMTV displays the most random integration site distribution among retroviruses to date, with no preference for active genes, TSSs, gene-dense regions, CpG islands, or DNase hypersensitive sites (Faschinger et al., 2008).
The human T-cell leukemia virus type 1 (HTLV-1) is the only component of the delta-retroviral family for which an integration profile has been determined to date. The virus integrates into the human genome with little but significant preference for TSSs, transcription units, promoters, and gene-dense regions. No overrepresentation with respect to random sequences is observed near DNase I hypersensitive sites or CpG islands, and the guanine-cytosine (GC) content of surrounding genomic regions is comparable with that of controls. A role of the target cell transcriptional activity on target site selection by HTLV-1 has not been described (Derse et al., 2007).
Foamy viruses (FV), or spumaviruses, are complex exogenous retroviruses mainly prevalent in nonhuman primates. FV vectors have been developed that possess broad host range, large packaging capacity, and high transduction efficiency of human hematopoietic cells, making them promising alternatives to MLV vectors for gene therapy of hematological disorders. Low-resolution profiling of FV integration sites showed preferences similar to those of MLV, with overrepresentation of integrations in CpG islands and around transcription start sites (∼10%, compared to ∼20% of MLV) (Nowrouzi et al., 2006; Trobridge et al., 2006).
Lentiviral Vectors Target Transcribed Genes
HIV-1 is one of the several components of the Lentiviridae family. HIV-1 is the etiological agent of the acquired immunodeficiency syndrome (AIDS), and not surprisingly, its integration pattern was the first to be characterized, as soon as the ligation-mediated polymerase chain reaction (LM-PCR) technology became available (Schroder et al., 2002). The most evident characteristics of the integration profile of HIV-1, and of its simian counterpart SIV-1, is the preference for the transcribed portion of genes, with up to 80% of the proviruses, depending on the target cell, landing within a transcription unit (Schroder et al., 2002; Hematti et al., 2004; Mitchell et al., 2004). Two studies used deep-sequencing technology to map the integration profile of HIV-1-derived lentiviral vectors in T cells (Wang et al., 2007) and in primary HSCs (Cattoglio et al., 2010b) and allowed a better definition of the HIV-1 integration preferences. Differently from MLV, HIV-1 proviruses are evenly spread along the transcription body of active genes, with a tendency to avoid TSSs, CpG islands, G/C-rich sequences, DNase I hypersensitive sites, and TFBSs, denoting a negative preference for transcriptional regulatory regions (Cattoglio et al., 2010b). Again differently from MLV, genes controlling cell development and differentiation are not among the preferred target of HIV-1, which has instead a tendency to target a broad group of “housekeeping” genes, controlling cell cycle, metabolism, and replication. Integration hot spots are an obvious characteristic of the HIV integration profile, preferentially located in gene-dense regions of the genome and in highly expressed genes (Schroder et al., 2002; Wang et al., 2007; Cattoglio et al., 2010b). Interestingly, HIV hot spots are broader in size compared to the sharp and enhancer-centered MLV clusters and tend to accumulate in megabase-long regions of the genome (Ambrosi et al., 2011). The pattern of preferentially targeted genes, as well as the location of hot spots, changes with gene expression patterns and is therefore cell-type-specific.
HIV integration is strongly associated with epigenetic signatures of transcriptionally active chromatin, such as mono-, di-, and tri-methylation of H3K4 and acetylation of H3 and H4, and negatively correlated with markers of heterochromatin such as H3K9me3 and H3K27me3 (Wang et al., 2007; Cattoglio et al., 2010a; Cattoglio et al., 2010b). The association is particularly evident with histone modifications marking the transcribed body of genes, such as H2BK5me1, H3K27me1, H3K36me3, and H4K20me1 (Wang et al., 2007; Wang et al., 2009; Cattoglio et al., 2010a). Although partially redundant with measures of gene density and chromatin structures, epigenetic modifications were shown to influence HIV integration independently of other genomic features (Figure 1).
HIV integration provides the best known model of target site selection through tethering of PICs to the host-cell chromatin. Several cellular proteins have been isolated as physically bound to lentiviral PICs, and for some of them, the association occurs via direct interaction with the integrase. These include members of the DNA repair machinery such as hRAD18 (Mulder et al., 2002), components of chromatin remodeling complexes such as INI1 (Kalpana et al., 1994) and EED (Violot et al., 2003), and the constitutive chromatin components HMGI(Y) (Li et al., 2000) and PSIP1/LEDGF/p75 (Engelman and Cherepanov, 2008). The lens epithelium-derived growth factor (LEDGF/p75) is a ubiquitously expressed nuclear protein, tightly associated with chromatin throughout the cell cycle, and is the most studied and best characterized interactor of the HIV-1 integrase. LEDGF/p75 was identified by its strong binding affinity to the HIV-1 integrase and was shown to stimulate its catalytic activity in vitro (Cherepanov et al., 2003; Emiliani et al., 2005; Turlure et al., 2006). It is characterized by a conserved N-terminal proline-tryptophan-tryptophan-proline (PWWP) domain, and a second, integrase-binding domain (IBD) at the C-terminus that allows its interaction with different lentiviral integrases (Cherepanov et al., 2004). The PWWP domain, together with a nuclear localization signal and a double copy of an AT-hook DNA-binding domain, mediates LEDGF/p75 association with chromatin, with no apparent sequence specificity except for a weak preference for AT-rich sequences (Llano et al., 2006; Turlure et al., 2006). Although the function of LEDGF/p75 remains largely unknown, its role in mediating HIV infectivity has been deeply investigated. Depletion of LEDGF/p75 by RNA interference knockdown (Llano et al., 2004a; Llano et al., 2004b; Ciuffi et al., 2005; Llano et al., 2006) or by homozygous gene-trap mutations (Sutherland et al., 2006; Marshall et al., 2007) leads to a re-localization of the HIV-1 integrase to the cell cytoplasm, with loss of chromosomal association and increased proteasomal degradation. The consequence is an overall reduction of infectivity due to a severe impairment in the integration process. Analysis of the residual integration sites showed significant detargeting of transcription units and increased insertion in nearby CpG islands and promoter regions, classical targets of other retroviruses. Integration did not become random, however, and transcribed genes were still favored, suggesting that cell factors other than LEDGF/p75 participate in tethering HIV-1 PICs to chromosomes. Recent reports indicate that the interactions between PICs and component of the nuclear import machinery may as well play a role in tethering HIV integration to transcribed genes (Matreyek and Engelman, 2011; Ocwieja et al., 2011).
Integration and Retroviral Evolution
The choice of the integration site has a deep impact on the fitness of a retrovirus, as it may influence the persistence and regulation of proviral gene expression. It is therefore reasonable that each retroviral family has evolved a molecular strategy to direct integration in order to maximize survival and propagation.
Gamma-retroviruses, and reasonably spumaviruses, may have evolved a mechanism coupling target site selection to gene regulation to take advantage of nearby cellular promoters and/or enhancers to activate their own expression. The other way around, integration of viral LTR enhancers in the proximity of cell-specific growth regulators increases the chance of clonal expansion or transformation by insertional gene activation, possibly resulting in expansion of infected cells and indefinite viral propagation.
Lentiviruses have apparently evolved a different strategy to target open chromatin regions while minimizing interference with the cell transcriptional machinery. This is expected to promote maximum production of daughter virions during the limited lifespan of infected cells in the phase of active replication. On the other hand, integration into active genes, but at a distance from promoters and regulatory regions, may be more permissive for the latent phase of the viral life cycle, at least if we consider latency as an HIV-deliberate survival strategy, which may not necessarily be the case (Persaud et al., 2003). In vitro latency models, in which silent HIV proviruses are reactivated by treatment with tumor-necrosis factor-α and then profiled for their integrations, suggest that transcriptional latency may also derive from integration in a “silencing” genomic environment (centromeric heterochromatin, long intergenic regions, or very highly expressed domains) (Lewinski et al., 2005). Relationship between HIV latency and integration sites remains, however, uncertain.
The nearly random integration pattern of alpha-, beta-, and delta-retroviruses is less obviously related to their chances of survival and propagation. In these cases, the host-virus interaction may have evolved to reduce damage to the host, or simply not evolved in any specific direction.
Retroviral Integration and Mutagenesis
The covalent integration of viral DNA into the host-cell genome carries an intrinsic mutagenic potential, which is further exacerbated by the integration profile and/or some structural properties of certain retroviruses. This has an obvious impact in clinical gene therapy. Seminal clinical studies have shown the efficacy of retroviral gene transfer for the therapy of genetic diseases (Hacein-Bey-Abina et al., 2002; Mavilio et al., 2006; Aiuti et al., 2009; Cartier et al., 2009; Boztug et al., 2010) and of genetically modified T cells for the treatment of acquired disorders such as leukemia (Porter et al., 2011) or graft-versus-host disease (Bonini et al., 1997; Ciceri et al., 2007; Ciceri et al., 2009). Some of these studies also showed the genotoxic consequences of retroviral gene transfer technology: insertional activation of proto-oncogenes by MLV-derived vectors caused T-cell lymphoprolipherative disorders in patients undergoing gene therapy for X-linked severe combined immunodeficiency (SCID-X1) (Hacein-Bey-Abina et al., 2008; Howe et al., 2008) and Wiskott-Aldrich syndrome (WAS) (Avedillo Diez et al., 2011), as well as premalignant expansion of myeloid progenitors in patients treated for chronic granulomatous disease (CGD) (Ott et al., 2006; Stein et al., 2010). Insertion of a lentiviral vector in a proto-oncogene likewise caused clonal expansion in at least one patient undergoing gene therapy for beta-thalassemia (Cavazzana-Calvo et al., 2010). Understanding the causes of these events, and overcoming the genotoxic consequences of retroviral gene transfer, has been the objective of intense preclinical and clinical research in the last ten years.
MLV Integration Causes Insertional Gene Activation
Gamma-retroviruses often cause malignancy in their host by activating or deregulating proto-oncogenes, a mechanism called insertional oncogenesis (Coffin et al., 1997). Insertional activation of proto-oncogenes has always been considered a possible consequence of random insertion of vectors derived from gamma-retroviruses into the genome but, on statistical grounds, the probability of such an event was originally estimated to be less than one in ten million (Stocking et al., 1993). As it turned out, these calculations were based on a wrong assumption: retroviral integration into the human genome is all but random. The preference of MLV for transcriptional regulatory elements and for specific gene categories dramatically increases the probability of deregulating genes involved in crucial cell functions such as proliferation and differentiation, including proto-oncogenes (Aiuti et al., 2007; Cattoglio et al., 2007; Cattoglio et al., 2010a; Biasco et al., 2011; Deichmann et al., 2011). The combined effect of inserting the strong, constitutive enhancer of the MLV LTR and altering the physical integrity and spatial relationship of regulatory elements causes the “hijacking” of transcriptional regulation of cellular genes by the provirus. In human primary hematopoietic cells, the MLV LTR enhancer influences the expression of genes located far away from the insertion site (>100 kb), at a relatively high frequency, and independently from their location in the vector backbone and from the provirus orientation with respect to the target gene (Recchia et al., 2006; Cassani et al., 2009; Maruggi et al., 2009) (Figure 2).
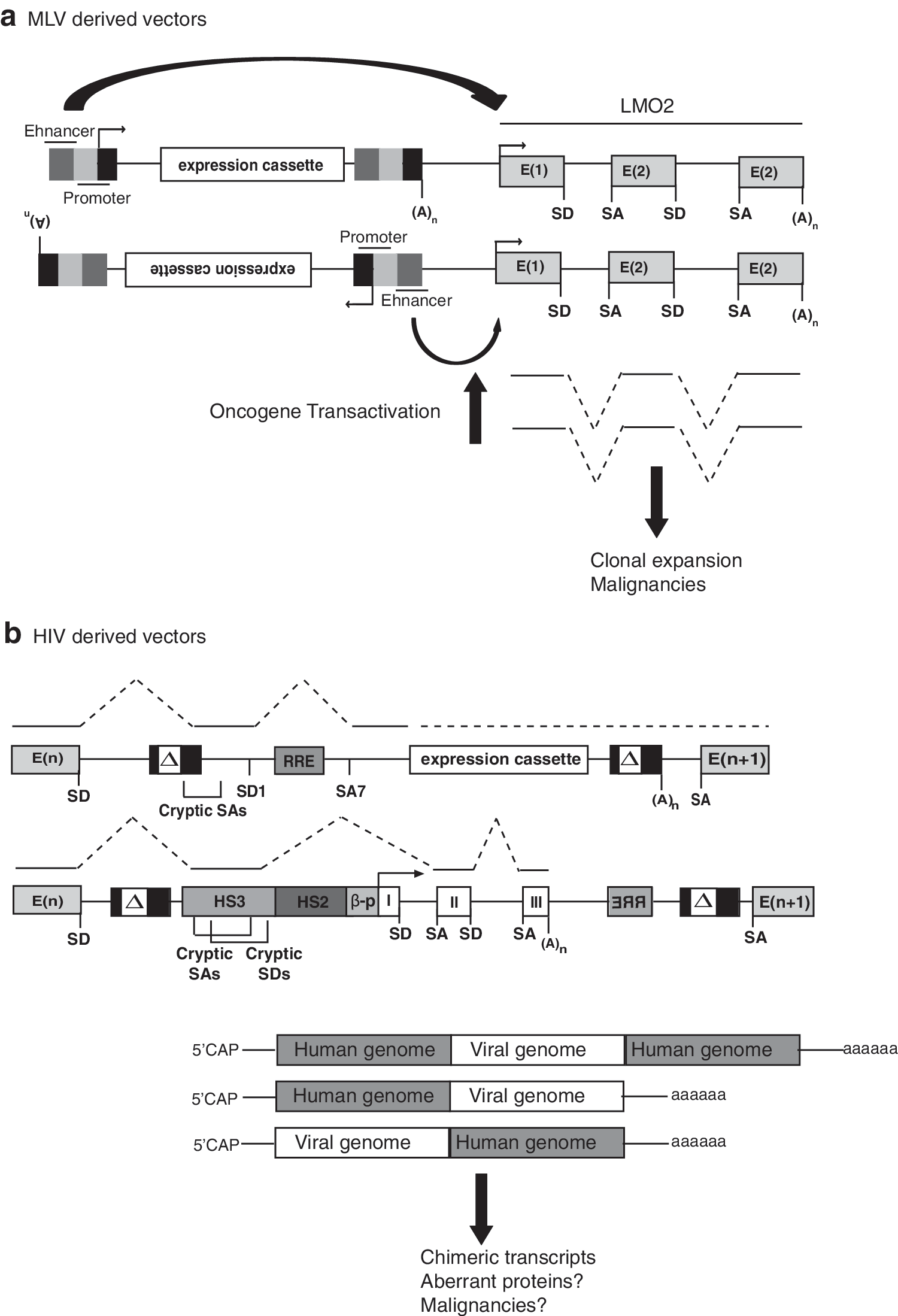
The consequences of deregulating cellular gene expression may, however, be very different depending on the species, the cell type, and even the individual genetic background. In T lymphocytes, insertional gene deregulation appears to have a negative influence on cell fitness, causing clonal loss rather than clonal expansion after transplantation in patients (Recchia et al., 2006; Cattoglio et al., 2010a). As a matter of fact, no malignancy or insertion-related clonal expansion has ever been observed in patients treated with genetically modified T cells in preclinical studies (Bonini et al., 2003; Newrzela et al., 2008) and throughout decade-long clinical trials (Bonini et al., 2003; Scholler et al., 2012). On the contrary, HSCs are apparently susceptible to insertion-mediated mutagenesis, and in particular, to the insertional activation of certain proto-oncogenes by MLV-based vectors. The murine Evi1 (for ecotropic viral integration 1) locus, originally identified as a common integration site in malignancies generated by oncogenic gamma-retroviruses, is targeted by MLV-derived vectors at a relatively high frequency. Activation of Evi1 leads to clonal expansion and eventually transformation of hematopoietic stem/progenitor cells in vivo (Li et al., 2002), and under certain conditions in clonal cultures in vitro (Modlich et al., 2006). Insertional deregulation of the homologous human MDS1/EVI1 locus by an MLV-derived vector carrying the potent spleen focus-forming virus (SFFV) enhancer, likewise led to pre-malignant clonal expansion of myeloid progenitors, as observed in a clinical trial of gene therapy for CGD (Ott et al., 2006; Stein et al., 2010). Clonal expansion of hematopoietic progenitors carrying a retroviral insertion in the Evi1 or EVI1/MDS1 loci has been observed in mice (Kustikova et al., 2005; Kustikova et al., 2007), nonhuman primates (Calmels et al., 2005), patients (Ott et al., 2006; Boztug et al., 2010) and even in culture (Sellers et al., 2010), indicating that HSCs are particularly susceptible to activation of this locus (reviewed in Metais and Dunbar, 2008). In fact, in vitro immortalization by Evi1 activation is a convenient read-out for testing alternative promoters or gene transfer vector designs (Modlich et al., 2009).
Analysis of the progeny of transduced HSCs in mice (Kustikova et al., 2005; Kustikova et al., 2007), nonhuman primates (Calmels et al., 2005), and humans (Ott et al., 2006; Deichmann et al., 2007, 2011; Schwarzwaelder et al., 2007; Boztug et al., 2010; Wang et al., 2010) identified “dominant” hematopoietic clones that hosted MLV insertions near a number of other proto-oncogenes or genes involved in signal transduction, cell growth, and proliferation. The conclusion of these studies was that vector-induced deregulation of certain categories of genes confers some growth and/or survival advantage to transduced progenitors, resulting in their in vivo amplification. High-resolution mapping of MLV integration sites in HSCs indicates, however, that many of the apparently dominant insertions are in fact over-represented also in nontransplanted, unselected cells as a consequence of the MLV preference for hot spots and certain categories of genes (Aiuti et al., 2007; Cattoglio et al., 2007; Cattoglio et al., 2010b; Biasco et al., 2011). In other cases, such as integrations in the MDS1-EVI1, PRDM16, or SETBP1 (Ott et al., 2006), the frequency by which integrations are retrieved in the progeny of repopulating stem cells is much higher than that observed in pretransplant cells (Cattoglio et al., 2010b), indicating true clonal amplification/selection in vivo. The availability of appropriate pretransplant controls is therefore crucial to assess the clonal dynamics of transplanted cells in clinical gene therapy trials and to distinguish the occurrence of dominant, potentially premalignant clones from the simple overrepresentation of naturally preferred retroviral integration sites.
MLV Integration Causes Insertional Oncogenesis
A number of preclinical and clinical studies clearly showed that integration of MLV-derived gene transfer vectors in certain genomic loci can lead to overt malignant transformation (reviewed in Nienhuis et al., 2006). Premalignant clonal expansion can predispose to subsequent accumulation of mutations or chromosomal aberrations, a classical model of neoplastic progression. In particular, deregulation of the MDS1/EVI1 locus led to chromosomal instability (monosomy 7) and eventually to a myeolodysplastic syndrome in patients treated for CGD (Stein et al., 2010). In other cases, malignant transformation was not preceded by clonal expansion and occurred abruptly a long time after the transplantation of HSCs genetically corrected with an MLV-derived vector-carrying wild-type LTRs. This is the case of T-cell myeloproliferative disorders that occurred in patients treated for SCID-X1 (Hacein-Bey-Abina et al., 2008; Howe et al., 2008) and WAS (Avedillo Diez et al., 2011). In these patients, T-cell malignancies arose in >30% of the patients years after treatment as the apparent consequence of insertion of the MLV vector in the LMO2 locus, a proto-oncogene previously known to cause childhood T-cell leukemia by a chromosomal translocation-mediated mechanism (McCormack and Rabbitts, 2004). Integrations in the LMO2 locus were common to all leukemic clones, although they were not the only genetic alteration observed in the clones (Hacein-Bey-Abina et al., 2008; Howe et al., 2008). Insertional activation of the Lmo2 and the common gamma cytokine receptor—the gene mutated in SCID-X1—was known to cause leukemia in mice infected by wild-type MLV (Dave et al., 2004)
Analysis of the T-cell clonal dynamics by high-throughput sequencing of retroviral integrations in patients from one of the SCID-X1 trials showed that clones carrying the LMO2 insertions did not expand before the occurrence of the leukemia (Wang et al., 2010), indicating a different oncogenic mechanism compared to that observed in the CGD trial. High-definition integration maps showed that MLV targets the LMO2 locus with a frequency of >1:500 in human CD34+ hematopoietic progenitors in hot spots that coincide with the LMO2 transcriptional enhancers (Cattoglio et al., 2010b) and co-map with the integrations found in the SCID-X1 leukemias. This suggests that neoplastic transformation occurs at an exceedingly rare frequency in cells carrying an MLV insertion in the LMO2 locus. Interestingly, integration at the same regions were found with fluctuating frequency in normal circulating T cells in several patients treated with an MLV vector for ADA-deficient SCID (Aiuti et al., 2007). No neoplastic event was observed in >20 patients treated with gene therapy for ADA-SCID in two different clinical trials for as long as 14 years after treatment (Aiuti et al., 2009; Gaspar et al., 2011). The history of the SCID-X1, ADA-deficient SCID and WAS trials shows that MLV insertion in the LMO2 locus is not sufficient to transform a T-cell progenitor and that establishment and progression of malignancy are influenced by yet unknown factors that include the disease context, the patient's individual genetic background, the vector design and copy number, the therapeutic gene expression, the bone marrow conditioning regimen, and the dose of genetically corrected cells.
Retroviral Vectors Cause Post-Transcriptional Deregulation of Gene Expression
Insertion of proviral sequences in cell transcription units may cause deregulation of gene expression also at a post-transcriptional level, for example, upon insertion of functional splicing and polyadenylation signals of retroviral origin or present within the transgene expression cassette. Splice donor and acceptor sites enhance titers and transgene expression and are therefore maintained in retroviral vector backbones. However, upon integration within a transcription unit, the viral splicing signals may function as alternative donors or acceptor sites for those of the target gene. The result may be aberrant splicing, leading to truncated or otherwise mutated products, with potentially altered function (Fig. 2). Insertion of an active polyadenylation signal can have similar consequences, producing either premature transcript termination of a host gene (strong polyA signals) or read-through transcription from a proviral promoter (weak polyA signals). The mutagenic potential of viral post-transcriptional regulatory elements is exemplified by the genomic distribution of human endogenous retroviruses (HERVs), extinct retroviral elements that account for ∼8% of the entire human genome. HERVs are mainly located outside transcription units and away from gene-rich regions and regulatory elements. When inside a transcription unit, they are found in opposite transcriptional orientation with respect to the host gene, so that their splicing and/or polyadenylation signals cannot interfere with gene transcription. Integration profiling of an experimentally “resurrected” HERV showed, instead, a preference for genomic regions involved in active transcription, with no bias for sense or antisense orientation with respect to target genes (Brady et al., 2009). This observation indicates that integrations leading to the insertion of splicing and polyadenylation signals inside transcription units are deleterious and, in the case of HERVs accumulating in the human germ line, strongly counterselected. Evidence for negative selection of cells harboring same-orientation integrations within genes has emerged also in the follow-up of clinical gene therapy studies (Recchia et al., 2006; Cattoglio et al., 2010a).
The propensity of lentiviral vectors to integrate into the body of transcribed genes increases the probability of post-transcriptional gene deregulation compared to MLV-derived vectors. A number of preclinical studies indeed showed that lentiviral vector–mediated insertion of splicing and polyadenylation signals within transcription units may cause post-transcriptional deregulation of gene expression by inducing aberrant splicing, premature transcript termination, and the generation of chimeric, read-through transcripts originating from internal promoters (Almarza et al., 2011; Cesana et al., 2012; Moiani et al., 2012), a classical cause of insertional oncogenesis (Nilsen et al., 1985). In addition, the deletion of the U3 region typical of the most commonly used self-inactivating (SIN) vector design decreases transcriptional termination and increases the generation of read-through transcripts (Yang et al., 2007). A recent report showed that downregulation of the expression of the Ebf1 transcription factor caused by insertion of a lentiviral vector can cause haploinsufficiency and the insurgence of leukemia in mice (Heckl et al., 2012). In a clinical context, insertion of a lentiviral vector caused post-transcriptional activation of a truncated form of the HMGA2 proto-oncogene in hematopoietic cells of a patient treated with gene therapy for beta-thalassemia, resulting in benign clonal expansion of the affected cells (Cavazzana-Calvo et al., 2010).
Two recent studies identified the significant potential of HIV-derived vectors to generate abnormally spliced transcripts upon integration in human genes. In the first one, clonal analysis of cell lines and primary T cells identified fusion transcripts between viral and cellular sequences in the majority of in-gene integrations. Chimeric transcripts were generated through the use of constitutive and cryptic splice sites in the HIV 5’ LTR and gag gene, and in a beta-globin minilocus inserted in opposite transcriptional orientation as an example of a therapeutic transgene carrying cellular introns and polyadenylation sites. Compared to constitutively spliced transcripts, most aberrant transcripts accumulated at low level, at least in part as a consequence of nonsense-mediated mRNA degradation (Moiani et al., 2012). The second study used high-throughput RNA sequencing technology to map transcripts generated by aberrant splicing and read-through transcription and identified essentially the same critical signals (Cesana et al., 2012). A limited set of cryptic splice sites therefore causes the majority of aberrant transcripts, providing a strategy for recoding lentiviral vector backbones and transgenes to reduce their potential post-transcriptional genotoxicity. Interestingly, cryptic sites located in either vector orientation generated fusion transcripts at higher frequency compared to the constitutive sites located in the HIV gag or in the beta-globin gene. This indicates that the cell-splicing machinery removes canonical introns efficiently by using their native donor and acceptor sites, and that most of the aberrant splicing events are caused by uncoupled, cryptic splice signals.
The relatively low efficiency by which proviruses induce aberrant splicing has important implications in terms of vector genotoxicity, since it predicts a low frequency of gene downregulation or true monoallelic knock-out. However, aberrant splicing caused by cryptic proviral signals may occasionally lead to gain-of-function mutations, as observed for the HMGA2 proto-oncogene in the beta-thalassemia trial (Cavazzana-Calvo et al., 2010). The fact that constitutive introns appear to interfere only marginally with cellular gene splicing suggests that intron-containing genes may still be incorporated in a recoded vector backbone if necessary for a specific therapeutic application.
Overcoming Insertional Genotoxicity
The current clinical applications of retroviral gene transfer technology, either ex vivo or in vivo, involve the transduction of billions of cells and the generation of a very high number of potentially mutagenic insertion events. The integration preferences of each vector type makes some of these events more or less likely to happen, but given the numbers involved, even a completely random integration machinery would have only a few-fold lower probability of inducing a potentially oncogenic or otherwise dangerous mutation than an MLV- or an HIV-based vector. Many approaches have been proposed in the last few years to replace transgenesis based on viral integrases with more “intelligent” machineries achieving site-directed rather than quasi-random integration and gene correction rather than gene addition (Urnov et al., 2010). Although extremely promising, these techniques will probably take years before matching the unsurpassed transduction efficiency of retroviral vectors and becoming practically applicable in a clinical context. In the meantime, all we can do is improve the safety profile of retroviral vectors and make our best efforts in terms of evaluating risks and benefits of each specific application.
Many years of preclinical and clinical studies have identified vector elements and features most critical in terms of potential genotoxicity. The development of robust in vitro and in vivo models of genotoxicity has dramatically improved our understanding of the factors involved in insertional oncogenesis and our capacity to predict the potential risk of any given vector design in a comparative fashion (Modlich et al., 2006; Montini et al., 2006; Modlich et al., 2009; Montini et al., 2009). These studies showed that the nature of the sequences borne by a retroviral vector may have as much impact as the vector itself in terms of overall genotoxic potential. Strong viral enhancers, like those carried by the MLV and SFFV LTR U3 regions, induce transcriptional activation of cellular genes at high frequency and at long distance in vitro, induce cell transformation in vitro and in vivo, and caused most of the severe side effects seen in clinical trials. The oncogenic potential of viral enhancers is increased in the context of MLV vectors, and when they are carried by the viral LTRs, but they activate transcription and induce cell transformation also in the context of lentiviral vectors, although with reduced frequency (Maruggi et al., 2009; Modlich et al., 2009). The development of U3-deleted SIN vectors, the use of cellular rather than viral enhancer/promoter elements, and of lentiviral rather than gamma-retroviral integration machineries, reduces dramatically the potential genotoxicity of clinical vectors. In general, cellular enhancers scored better than viral enhancers and SIN-HIV vectors better than SIN-MLV vectors in preclinical models, indicating that cis-acting transcriptional activation and integration preferences are independent factors with additive effects on the overall genotoxic potential of a retroviral vector (Modlich et al., 2009; Montini et al., 2009). However, even cellular regulatory elements may have cis-acting activity when they have long-range regulatory potential (Hargrove et al., 2008), while the use of short-range regulatory elements such as the PGK or the EF1-alpha have a very low effect, if any, on neighboring genes. Finally, recoding of cryptic splice sites in vector backbones and transgenes will probably add an additional safety measure to vectors that are anyway performing much better in terms of biosafety compared to those used in the first clinical trials of gene therapy for immunodeficiency. The first data on the clonal dynamics of hematopoietic progenitors in patients treated with stem cells transduced with lentiviral vectors in preclinical and clinical studies are so far confirming the data predicted by the genotoxicity tests (Cartier et al., 2009; Biffi et al., 2011). These data are very encouraging and indicate that the risk–benefit balance of gene therapy with last-generation retroviral vectors has become more favorable and more manageable, and justifies a new wave of clinical trials and new therapeutic applications.
Footnotes
Author Disclosure Statement
No competing financial interests exist.