
Abstract
Hepatic stimulator substance (HSS) has been suggested to protect liver cells from various toxins. However, the precise role of HSS in hepatic ischemia–reperfusion (I/R) injury remains unknown. This study aims to elucidate whether overexpression of HSS could attenuate hepatic ischemia–reperfusion injury and its possible mechanisms. Both in vivo hepatic I/R injury in mice and in vitro hypoxia–reoxygenation (H/R) in a cell model were used to evaluate the effect of HSS protection after adenoviral gene transfer. Moreover, a possible mitochondrial mechanism of HSS protection was investigated. Efficient transfer of the HSS gene into liver inhibited hepatic I/R injury in mice, as evidenced by improvement in liver function tests, the preservation of hepatic morphology, and a reduction in hepatocyte apoptosis. HSS overexpression also inhibited H/R-induced cell death, as detected by cell viability and cell apoptosis assays. The underlying mechanism of this hepatic protection might involve the attenuation of mitochondrial dysfunction and mitochondrial-dependent cell apoptosis, as shown by the good preservation of mitochondrial ultrastructure, mitochondrial membrane potential, and the inhibition of cytochrome c leakage and caspase activity. Moreover, the suppression of H/R-induced mitochondrial ROS production and the maintenance of mitochondrial respiratory chain complex activities may participate in this mechanism. This new function of HSS expands the possibility of its application for the prevention of I/R injury, such as hepatic resection and liver transplantation in clinical practice.
Introduction
Hepatic stimulator substance (HSS), a liver growth-promoting factor, was initially identified in the liver of weanling rats (LaBrecque and Pesch, 1975) and reported to stimulate remnant liver regeneration after partial hepatectomy (PH) (LaBrecque et al., 1987; Hagiya et al., 1995). HSS has also been referred to as augmenter of liver regeneration (ALR) or hepatopoietin, as it is able to enhance the growth of hepatocytes or hepatoma cells when combined with other hepatotrophic mitogens, such as epidermal growth factor (EGF) and transforming growth factor (TGF)-α, regardless of the animal species. Although HSS is synthesized in the liver parenchymal cells (He et al., 1993), it has multiple-tissue expression (Giorda et al., 1996), various subcellular localizations (Hofhaus et al., 1999), and diverse functions (Pawlowski and Jura, 2006). HSS exists in either a longer (with 205 amino acids) or a shorter form (with only 125 amino acids). In addition, together with Erv1p, a Saccharomyces cerevisiae homolog, HSS is a member of the new ALR/ERV1 protein family, which belongs to the sulfhydryl oxidase (SOX) enzymes that participate in disulfide bond formation (Wang et al., 2007; Daithankar et al., 2012). Structurally, HSS contains the conserved CXXC motif of SOX and a noncovalent flavin adenine dinucleotide (FAD) adjacent to CXXC, and these domains are vital to its catalytic activity (Wu et al., 2003).
Many studies have revealed that HSS protects the liver from injuries caused by various types of toxins, such as carbon tetrachloride (Mei et al., 1993),
It was found that ALR could protect the kidneys from ischemia–reperfusion injury in rats (Liao et al., 2010). However, it remains unknown whether HSS could play some role in defense against liver I/R injury. The present study aimed to evaluate the hepatoprotective effect of HSS both in in vivo hepatic I/R injury and in an in vitro H/R cell model. Moreover, the mitochondrial pathway of cell apoptosis or cell death was extensively investigated to elucidate potential mechanism of HSS protection. Here, we report that HSS exerts its protective effects mainly via attenuation of mitochondria-related cell apoptosis and preservation of mitochondrial function. Moreover, the suppression of H/R-induced mitochondrial ROS production and the preservation of mitochondrial energy production may be considered one of the protective mechanisms provided by HSS.
Materials and Methods
Animal model of hepatic ischemia–reperfusion injury
Male C57BL/6 mice weighing 18–22 g were purchased from the Academy of Military Medical Sciences (Beijing, China) and maintained at a constant room temperature (22–25°C) on a 12:12-hr light–dark cycle. All animals received humane care in compliance with the guidelines of the Capital Medical University (Beijing, China) Institutional Animal Care and Use Committee. Hepatic I/R injury was performed by surgical operation, using a procedure that was reported to produce 70% hepatic ischemia (Abe et al., 2009). In brief, the arterial and portal venous blood supply to the left lateral and median lobes was interrupted by an atraumatic clip for 90 min. The caudal lobes retained intact portal and arterial inflow and venous outflow, preventing intestinal venous congestion. After 90 min, the clip was removed, initiating hepatic reperfusion for 3 or 6 hr. The mice were then killed, and liver and serum samples were collected for analysis. Sham-operated mice underwent a similar procedure but without vascular occlusion and reperfusion.
HSS vector construction and gene delivery
A human HSS cDNA containing the entire coding sequence and tagged with FLAG was subcloned and constructed into a replication-deficient adenoviral vector, pAdxsi (Chinese National Human Genome Center, Beijing, China), as previously described (McConnell and Imperiale, 2004), and designated as Ad-HSS. For viral infection, mice were injected, at 1×109 plaque-forming units (PFU) per mouse, with viral particles diluted in pyrogen-free normal saline (NS) via the tail vein. An adenoviral vector without any gene insertion, named Ad-Null, was injected into mice and used as a control. At various times after injection, exogenous HSS expression was detected in the liver by immunohistofluorescence and Western blot using the anti-FLAG antibody.
The mice were randomly divided into four groups: mice that received the sham operation, mice that were subjected to I/R injury plus normal saline therapy, mice that were subjected to I/R injury plus Ad-HSS therapy, and mice that received I/R injury plus empty vector (Ad-Null) therapy. The sham group served as the control, and mice in the other groups received normal saline (NS), Ad-HSS (1×109 PFU), or Ad-Null (1×109 PFU) 4 days before I/R injury.
Biochemical measurement and histological observation
Serological activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH) were determined with an autoanalyzer (7600-020; Hitachi, Tokyo, Japan) in the clinical chemistry laboratory of Youan Hospital (Capital Medical University).
Liver specimens from the ischemic left lobes were fixed in 10% neutral-buffered formalin and embedded in paraffin. Tissue samples were sectioned into 5-μm-thick sections and stained with hematoxylin and eosin (H&E) for histological examination by light microscopy (DM5000 B; Leica Microsystems, Wetzlar, Germany).
The ultrastructural morphology of liver tissue samples was examined by electron microscopy. Briefly, liver specimens were fixed with 4% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 4 hr at 4°C. After fixation and an overnight wash in sodium cacodylate buffer, the specimens were postfixed with 1% osmium tetroxide in sodium cacodylate buffer at 4°C for 1 hr, dehydrated in alcohol, and embedded in Araldite resin. Semithin sections (1 μm) were removed for optical microscopy (H-800; Hitachi).
TUNEL staining and immunohistochemical staining for caspase-3
The terminal deoxynucleotidyltransferase (dUTP)-mediated nick end-labeling (TUNEL) assay was carried out according to the manufacturer's protocol with an in situ cell death/apoptosis detection kit (Roche Diagnostics, Mannheim, Germany). The results were scored semiquantitatively by averaging the numbers of TUNEL-positive cells per high-power field (HPF) for 10 fields per tissue sample.
For immunohistochemical analysis of caspase-3, 5-μm-thick tissue sections were deparaffinized and rehydrated, followed by microwave retrieval of antigen. Slides were treated sequentially with 3% H2O2 solution and diluted normal goat serum to block endogenous peroxidase activity and nonspecific antibody binding, respectively, and then incubated overnight at 4°C with cleaved caspase-3 antibody (Asp175, diluted 1:500; Cell Signaling Technology, Beverly, MA). After incubating with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA), a VECTASTAIN ABC kit (Vector Laboratories) was used according to the manufacturer's instructions. Sections were visualized with diaminobenzidine (DAB) and counterstained with hematoxylin. Caspase-3 activity was evaluated by counting the number of positive cells per HPF.
Western blot analysis
Liver tissues were homogenized, and total cell proteins were prepared. Protein concentrations of the lysates were determined according to the bicinchoninic acid (BCA) method, using a protein assay kit (Pierce Biotechnology, Rockford, IL). Samples containing 50 μg of protein were separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). For cell analysis, the cell cytosolic or mitochondrial proteins were extracted with a mitochondria/cytosolic fractionation kit (BioVision, Mountain View, CA), and 30 μg of protein was loaded and separated. After electrophoresis, proteins in the gel were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA) for Western blot analysis. Primary antibodies against FLAG-Tag (diluted 1:3000; Cell Signaling Technology), cytochrome c (diluted 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH, diluted 1:10,000; KangChen, Shanghai, China) or voltage-dependent anion channel (VDAC, diluted 1:1000; Cell Signaling Technology) were used. The membranes were washed and incubated with secondary antibodies conjugated to horseradish peroxidase. Immunoreactive proteins were detected by enhanced chemiluminescence (ECL). The relative density of the protein bands was quantitatively determined with ImageJ software (National Institutes of Health, Bethesda, MD).
Hepatic ATP levels
Hepatic mitochondria were isolated as previously described (Wu et al., 2010). Mitochondrial ATP content was measured with a CellTiter-Glo luminescent cell viability assay kit (Promega, Madison, WI) according to the manufacturer's instructions.
Measurement of mitochondrial respiratory activity
The activities of mitochondrial respiratory complexes were determined with Mito Complex I–IV activity assay kits (GenMed Scientific, Arlington, MA). Complex activities were expressed as the percentage of activity compared with that obtained from liver samples of mice without I/R injury.
Cell culture and H/R procedure
BEL-7402 cells, a human hepatoma cell line, were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO BRL, Paisley, UK) containing 10% fetal calf serum (FCS; HyClone, Rockford, IL), penicillin (100 U/ml), and streptomycin (100 U/ml). The cells were infected with adenoviral particles (Ad-HSS or Ad-Null) at a multiplicity of infection (MOI) of 100, 48 hr before H/R injury. The efficiency of HSS expression, based on immunofluorescence, was 70–80% by this method.
H/R injury was an ideal model for mimicking I/R injury in vitro (Li and Jackson, 2002). Cell hypoxia was manipulated by placing the cells into a specifically designed hypoxic incubator (HERACELL 150i; Thermo Scientific, Hanau, Germany) gassed with a mixture of 95% N2 and 5% CO2. The oxygen supply to the incubation chamber was extremely limited at a concentration of 1%. For hypoxia treatment, the cells received serum- and glucose-free deoxygenated DMEM and were incubated in the hypoxic incubator for 2 or 3 hr. After that, the medium was substituted with DMEM containing glucose and the cells were returned to a standard incubator (95% air and 5% CO2, 37°C). The reoxygenation time was set for 4 or 12 hr.
Cell viability and apoptotic analysis
After 3 hr of hypoxia and 12 hr of reoxygenation, cell viability was measured with a CellTiter96 AQueous One kit (Promega). To evaluate apoptosis, the cells were plated in 6-well plates and subjected to 3 hr of hypoxia followed by 12 hr of reoxygenation. Cell apoptosis was analyzed with an annexin V–FITC/PI apoptosis kit (Biosea, Beijing, China) according to the manufacturer's instructions by flow cytometry (FACScan; Becton Dickinson, Franklin Lakes, NJ) as previously described (Li et al., 2010). Data were analyzed with CellQuest software (Becton Dickinson). To further analyze cell apoptosis, the activations of caspase-3 and caspase-9 were monitored with CPP32/caspase-3 colorimetric kits (Millipore) and MCH6/caspase-9 colorimetric kits (Millipore), respectively, according to the manufacturer's instructions. Mitochondrial and cytosolic cytochrome c contents were also analyzed by Western blot. All data are presented as the mean of three determinations.
Mitochondrial ATP content and mitochondrial membrane potential
After 3 hr of hypoxia and 12 hr of reoxygenation, mitochondrial ATP content was analyzed with the CellTiter-Glo luminescent cell viability assay kit (Promega) according to the manufacturer's instructions. The mitochondrial membrane potential (ΔΨm) was measured after 2 hr of hypoxia and 4 hr of reoxygenation, using JC-1 (10 μg/ml; Sigma-Aldrich, St. Louis, MO) incorporation as previously described (Wu et al., 2007). JC-1 aggregates in the polarized mitochondrial matrix and forms J-aggregates, which emit red fluorescence at 595 nm when excited at 525 nm. However, JC-1 is hardly aggregated in the depolarized mitochondrial matrix and exists as JC-1 monomers, which emit green fluorescence at 525 nm when excited at 485 nm. Mitochondrial depolarization is indicated by a decrease in the red-to-green fluorescence intensity ratio.
Measurement of mitochondrial ROS production
MitoSOX (Life Technologies, Grand Island, NY) was used to track mitochondria-specific ROS production. After 2 hr of hypoxia and 4 hr of reoxygenation, cells were incubated with MitoSOX (5 μM) for 15 min in a light-protected humidified chamber at 37°C and washed with phosphate-buffered saline (PBS) three times before analysis by confocal fluorescence microscopy. Flow cytometry was performed at excitation/emission wavelengths of 488/625 nm for MitoSOX.
Statistical analysis
The results of multiple observations are presented as the mean±SD of at least three independent experiments. Data were analyzed with statistics software SPSS 11.5 (IBM, Armonk, NY) and differences between various groups were analyzed by one-way analysis of variance. Differences were considered significant if the p value was less than 0.05.
Results
HSS gene transfer protects liver against ischemia–reperfusion injury
First, we examined the endogenous expression of HSS during in vivo hepatic I/R or in vitro H/R injury by real-time PCR and Western blot analysis. Results showed that hepatic endogenous expression of HSS was reduced in I/R injury at 3 or 6 hr after reperfusion in mice and in H/R of the cell model at 12 hr after reoxygenation as well (Supplementary Fig. S1; Supplementary Data are available online at
Adenoviral vector-mediated HSS gene delivery via tail vein injection effectively enhanced HSS expression in liver tissues. We had detected HSS expression in liver by immunofluorescence and Western blot using anti-FLAG antibody. As shown in Fig. 1A and B, injection via the vein tail of 1×109 PFU per mouse yielded higher HSS expression in liver tissues, and expression reached a maximum 4 days after gene transfer. In animals subjected to control vector gene transfer, HSS expression could not be detected. Therefore, these experimental parameters for adenoviral vector-mediated gene delivery were applied in the following in vivo experiments.
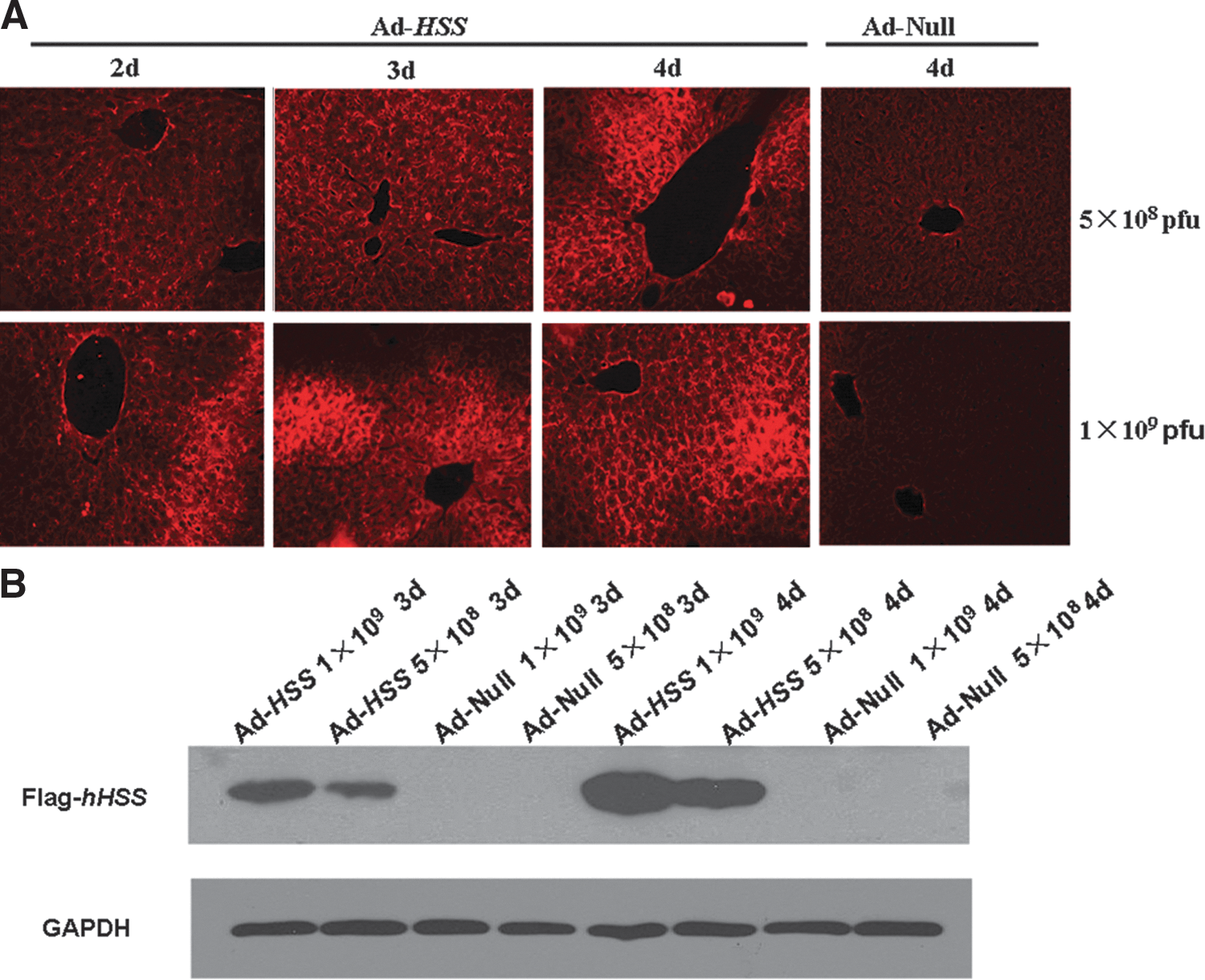
Hepatic stimulator substance (HSS) protein expression in liver tissue after transfection with adenoviral vector. C57BL/6 mice were injected intravenously with 1×109 or 5×108 PFU of Ad-HSS or empty adenoviral vector (Ad-Null) diluted in 0.5 ml of normal saline (NS). Expression of exogenous FLAG-tagged HSS in liver tissue was analyzed by immunofluorescence and Western blot, using the anti-FLAG antibody, 2, 3, and 4 days after infection.
The mice were randomly divided into four groups (see Materials and Methods), and each group contained at least six mice. Four days after gene transfer, animals were subjected to hepatic I/R, that is, 90 min of ischemia followed by 3 or 6 hr of reperfusion. For the assessment of hepatocellular damage, serum levels of ALT, AST, and LDH were measured after 90 min of ischemia and a subsequent 6 hr of reperfusion. As shown in Fig. 2A–C, remarkable increases in liver enzyme activities were observed in I/R-treated mice compared with sham-operated mice. However, the enhanced enzyme activities after ischemia–reperfusion injury were much improved in the Ad-HSS group compared with the Ad-Null group (for ALT, 1360.54±345.39 vs. 2554.00±455.74, p<0.05; for AST, 1969.53±460.55 vs. 4093.24±1015.94, p<0.05). The elevation of serum LDH level was also suppressed in the Ad-HSS group compared with the Ad-Null group (5799.60±1862.01 vs. 10,605.5±1548.96, p<0.05).

HSS transfection attenuates hepatic ischemia–reperfusion (I/R) injury. Mice were injected intravenously with Ad-HSS or empty adenoviral vector (Ad-Null) as a control 4 days before exposure to 90 min of hepatic ischemia (I) and 6 hr of reperfusion (R).
To further confirm the protective effect of HSS in hepatic I/R injury, liver tissues obtained from the ischemic left lobe 6 hr after reperfusion were sectioned for histopathology. I/R injury significantly induced hepatic damage compared with the sham-operated group. However, hepatic damage, which appeared as ballooning degeneration, cytoplasmic vacuolation, focal and confluent hepatocellular necrosis, endothelial swelling, and extensive hemorrhagic necrosis, was markedly reduced in HSS gene-treated mice compared with the Ad-Null group in Fig. 2D (panels c and d). The structure of the hepatic lobule was maintained well, the hepatic cords could be identified, and the degree of hepatocyte necrosis became more focal in the Ad-HSS group. The necrotic area in each group provided additional evidence of HSS liver protection (Fig. 2E).
HSS transfection prevents I/R-induced hepatocyte apoptosis
As another marker of liver injury, apoptosis was evaluated by TUNEL. After I/R injury, considerable numbers of apoptotic TUNEL-positive cells were identified in the liver tissue slides from the Ad-Null and NS groups compared with those from the sham group. In contrast, comparatively few TUNEL-positive hepatocytes were identified in the Ad-HSS group (Fig. 3A). The difference between the Ad-HSS and Ad-Null groups was found to be significant (Fig. 3B). Caspase-3 activation is reported to be a key mediator of the apoptotic cascade. Once activated, caspase-3 is cleaved and two fragments are released, which can be easily detected by an anti-cleaved caspase-3 antibody. To further analyze whether caspase-3 activation might be involved in I/R-induced hepatocyte apoptosis, we detected the activity of this apoptotic molecule in mouse liver after I/R, using immunohistochemical staining. As shown in Fig. 3C, the numbers of activated caspase-3-positive cells were significantly increased in the I/R-treated groups compared with the sham group. However, caspase-3-positive hepatocytes were decreased in the Ad-HSS group compared with the Ad-Null group, suggesting that HSS expression in liver inhibited cell apoptosis. Quantification of caspase-3-positive cells is shown in Fig. 3D.
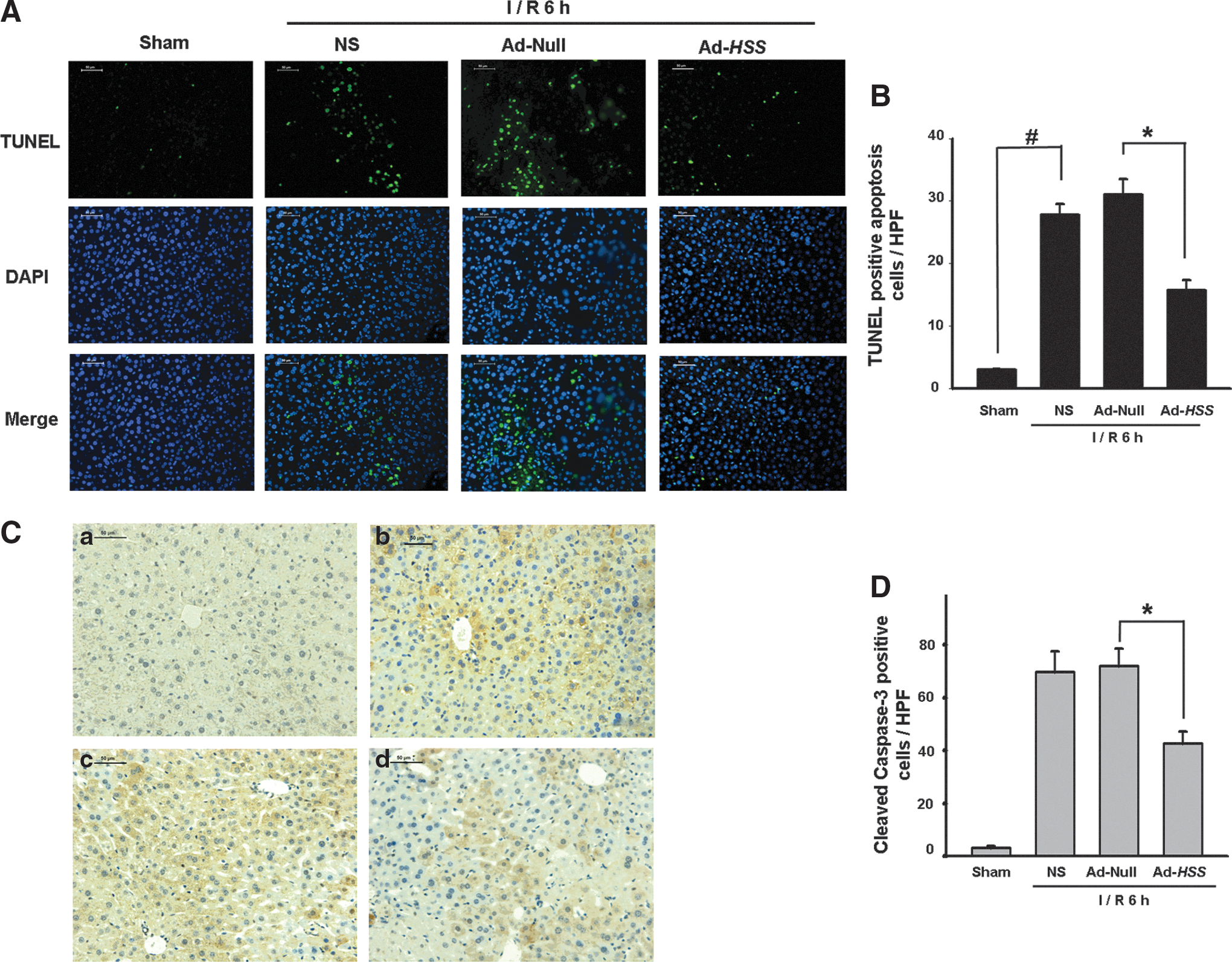
HSS transfection prevents I/R-induced liver cells apoptosis.
Transfection of HSS attenuates mitochondrial dysfunction
It is well established that mitochondrial dysfunction plays an important role in controlling cell death. We next asked whether HSS-induced hepatic protection was accompanied by an attenuation of mitochondrial dysfunction. For this purpose, we examined the mitochondria ultrastructure by electron microscopy (EM). Intact liver cell mitochondria were regular in size, cristae-rich, and featured a typical folded intermembrane space and dense matrix (Fig. 4A). I/R injury elicited morphological abnormalities in the mitochondrial ultrastructure in the NS group and the Ad-Null group, including an increase in volume and deficient or swollen cristae. However, HSS gene therapy prominently prevented reduction in the numbers of cristae and dilation of the cristae structure. In addition, mitochondrial volume was apparently maintained within its normal range.
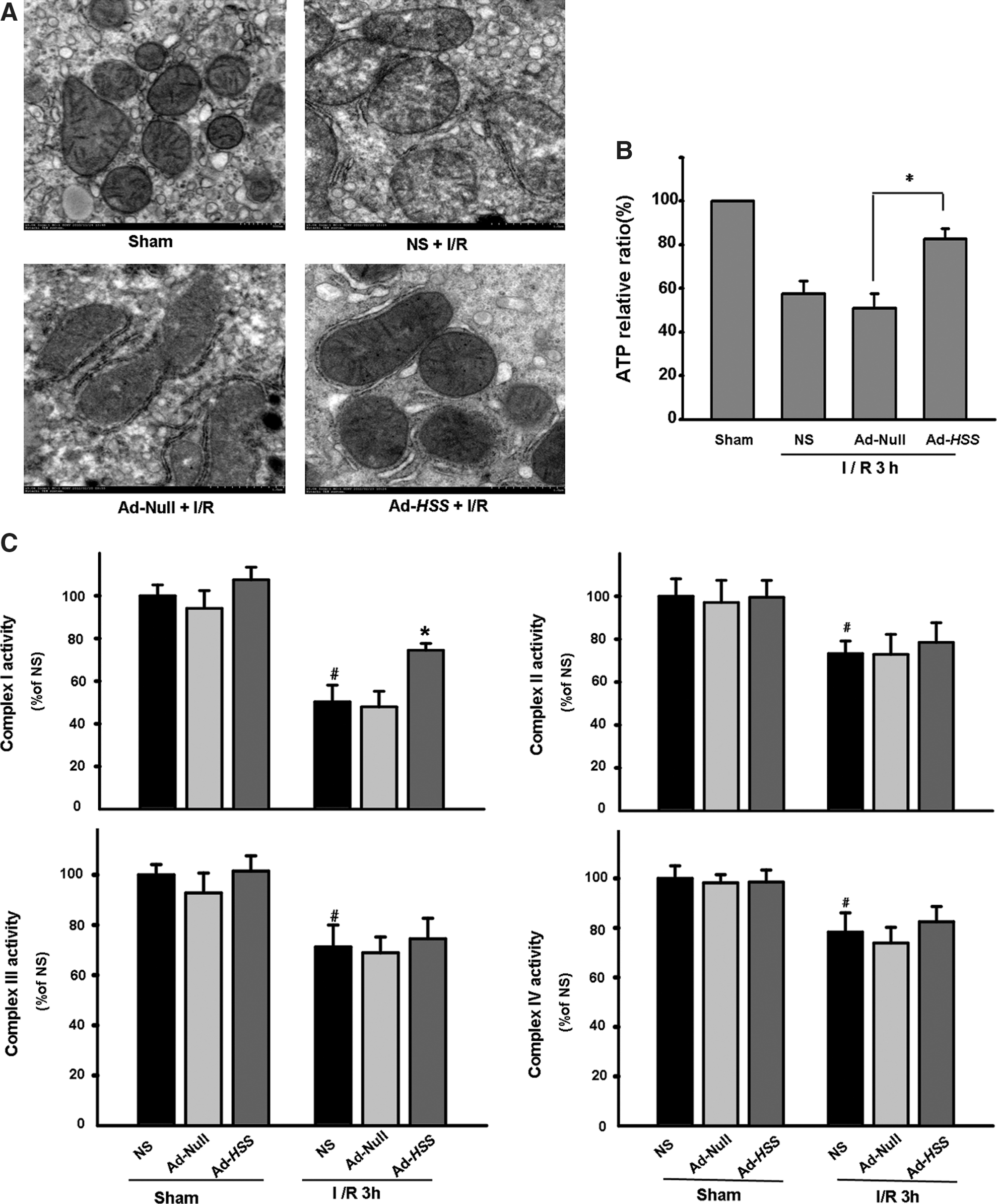
HSS transfection attenuates I/R-induced mitochondrial injury. Mice were injected intravenously with Ad-HSS or Ad-Null 4 days before exposure to 90 min of hepatic ischemia and 3 hr of reperfusion.
Hepatic mitochondrial damage in the setting of I/R is often the consequence of cellular ATP depletion, and the shortage in ATP supply worsens the efficiency of the Krebs cycle. To address whether mitochondrial protection by HSS was related to the preservation of ATP production, we measured mitochondrial ATP content. As indicated in Fig. 4B, the energy production by mitochondria was markedly impaired in mice in the NS and Ad-Null groups after 90 min of ischemia and a subsequent 3 hr of reperfusion. Although the relative amount of ATP was well preserved in the Ad-HSS group, it was approximately 34.2% higher than that in the Ad-Null group, suggesting protection of the mitochondria by restoring energy production. Preservation of hepatic mitochondrial ATP production as a result of HSS expression, in accordance with our mitochondrial EM results, revealed that HSS protection of mitochondria may be related to liver protection.
Last, we measured the activities of complexes I–IV of the mitochondrial electron transport chain during I/R injury. As shown in Fig. 4C, electron transport from mitochondrial complex I to complex IV was obviously impaired in the liver of mice subjected to I/R insult, in contrast with those in the sham-operated group. However, mitochondrial complex I activity was well preserved in the Ad-HSS group compared with the Ad-Null group, indicating that attenuation of mitochondrial dysfunction by HSS may underlie the protection and enhancement of mitochondrial respiratory chain complex I.
Exogenously delivered HSS in vitro is located mainly in mitochondria
An in vitro I/R model was further used to more clearly elucidate the possible mechanism of HSS liver protection. Adenovirus-mediated HSS gene transfer to hepatoma cell line BEL-7402 was achieved 2 days before hypoxia–reoxygenation (H/R) insult. The efficiency of HSS gene transfer was relatively high, and approximately 70–80% of the cells were positive (data not shown). Because the transfected HSS was labeled with FLAG-tag, the exogenously delivered HSS gene could be stained and visualized by confocal microscopy. Our results showed that transfected HSS was localized mainly in the mitochondria (Supplementary Fig. S2A). Western blot analysis showed that HSS expression, displayed as a 15-kDa band, could be clearly observed both in the cellular extract and the mitochondrial extract (Supplementary Fig. S2B).
HSS transfection reduces H/R-induced cell apoptosis
To elucidate whether HSS protects hepatocytes from H/R injury, cell viability was detected. H/R treatment increased cell mortality to 30% and 29% in untransfected and the adenoviral vector-transfected cells, respectively (Fig. 5A). However, cell mortality was only 12.5% in HSS-transfected cells (p<0.05 compared with Ad-Null). Next, H/R-induced apoptosis was analyzed by annexin V–FITC/PI flow cytometry (Fig. 5B). The statistical calculation of apoptotic rates is shown in Fig. 5C. After H/R injury, the apoptotic rates in untransfected cells and empty vector-transfected cells were obviously increased with ratios of 32.9±1.8 and 29.4±1.9%, respectively. However, apoptosis in HSS-transfected cells was reduced to 21.2±1.5% (p<0.05). Furthermore, we examine whether cell apoptosis was related to the mitochondrial signaling pathway. Figure 5D and E indicates that the activities of caspase-3 and caspase-9 were increased after H/R damage. However, the elevated activities of caspase-3 and caspase-9 were significantly inhibited in HSS-transfected cells compared with vector-transfected cells, suggesting that HSS plays a prominent role in protecting hepatocytes from mitochondria-dependent apoptosis.
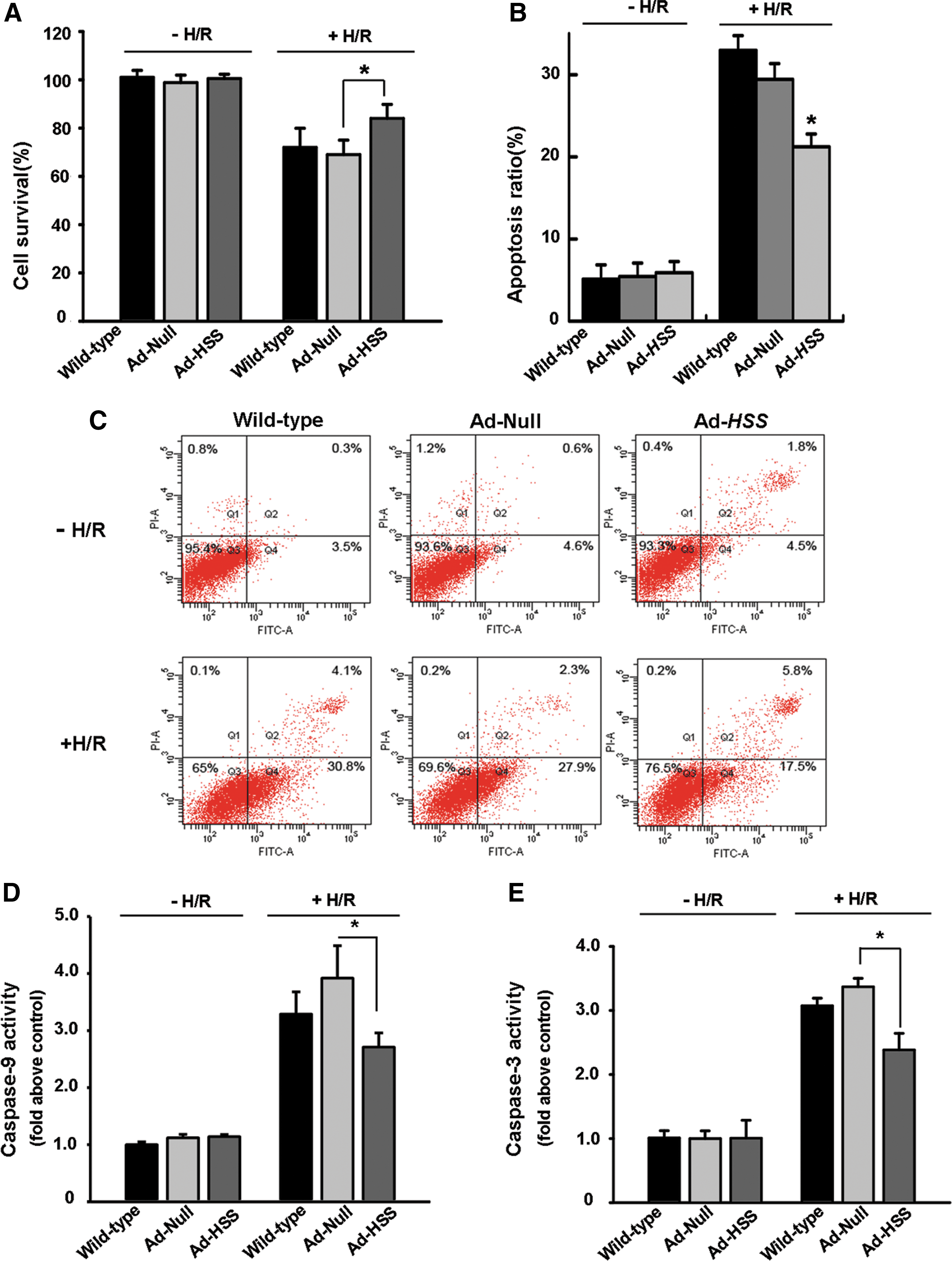
HSS transfection inhibits hypoxia–reoxygenation (H/R)-induced cell death. HSS-transfected and vector-transfected cells were transfected 2 days before undergoing hypoxia (3 hr)–reoxygenation (12 hr) injury.
HSS transfection attenuates H/R-induced mitochondrial dysfunction
It is well known that mitochondria are highly susceptible to hypoxic damage, leading to severe mitochondrial impairment, including the loss of mitochondrial membrane potential (MMP) and subsequent collapse of the permeability transition pore (PTP), triggering downstream pathways that promote cell apoptosis. Alteration of the MMP (ΔΨm) is known to be an early event in the apoptotic signaling cascade. JC-1 is a mitochondrion-specific dye used to evaluate the mitochondrial membrane potential. At low membrane potentials, JC-1 continues to exist as a monomer and produces green fluorescence (emission at 525 nm). At high membrane potentials or concentrations, JC-1 forms J aggregates (emission at 595 nm) and produces red fluorescence. As shown in Fig. 6A, on H/R injury, all three cell groups (untransfected, vector-transfected, and HSS-transfected) had more green fluorescence, that is, depolarized mitochondria (25.7, 23, and 10.5%, respectively) compared with untreated cells (7.25, 5.5, and 5.6%, respectively). The ΔΨm in Ad-HSS cells appeared to be relatively high with smaller alternations (Fig. 6B), indicating that HSS might protect the MMP from H/R injury. As a consequence, leakage of cytochrome c from the mitochondrial intermembrane space into the cytosol was also inhibited. As shown in Fig. 6C and D, H/R treatment induced an increase in the leakage of mitochondrial cytochrome c in untransfected cells and vector-transfected cells. In contrast, the release of cytochrome c was markedly reduced in HSS-transfected cells. Densitometric analysis demonstrated that the level of mitochondrial cytochrome c within HSS-transfected cells was approximately twice as high as that in untransfected and vector-transfected cells (Fig. 6C). Furthermore, the ATP content was also observed as an index of mitochondrial function (Fig. 7). H/R treatment severely impaired mitochondrial ATP production because the ATP content significantly decreased. The ATP content in HSS-transfected cells was still maintained at higher levels compared with control cells. The relative ratio was 72.6±2.1% in HSS-transfected cells compared with 50.8±2.5% in untransfected cells and 52.0±2.7% in vector-transfected cells, suggesting that HSS gene therapy could prevent energy depletion induced by hypoxia in hepatocytes (Fig. 7).

Transfection of HSS protects mitochondria from H/R injury.

Alteration of cellular ATP content after H/R treatment. *p<0.05 versus Ad-Null plus H/R treatment.
HSS transfection decreases H/R-induced mitochondrial ROS production
The mitochondria are an important source of reactive oxygen species (ROS) implicated in ischemia–reperfusion injury (Jaeschke, 2003b). H/R-induced cell damage is believed to be associated with an excess of mitochondrial ROS generation. Hence, we applied confocal laser scanning microscopy and a MitoSOX red fluorescence assay to examine whether HSS transfection could contribute to overwhelming mitochondrial ROS production. As shown in Fig. 8A, the level of red fluorescence (ROS production) was significantly increased in Ad-Null cells (as a control) after H/R treatment; however, the fluorescence intensity was diminished in Ad-HSS cells. Furthermore, the number of MitoSOX red-positive cells detected by flow cytometry was lower, indicating that positively stained cells decreased in the Ad-HSS group compared with the Ad-Null group after H/R (Fig. 8B). These results suggested that HSS has an important role in suppressing the production of mitochondrion-specific ROS.
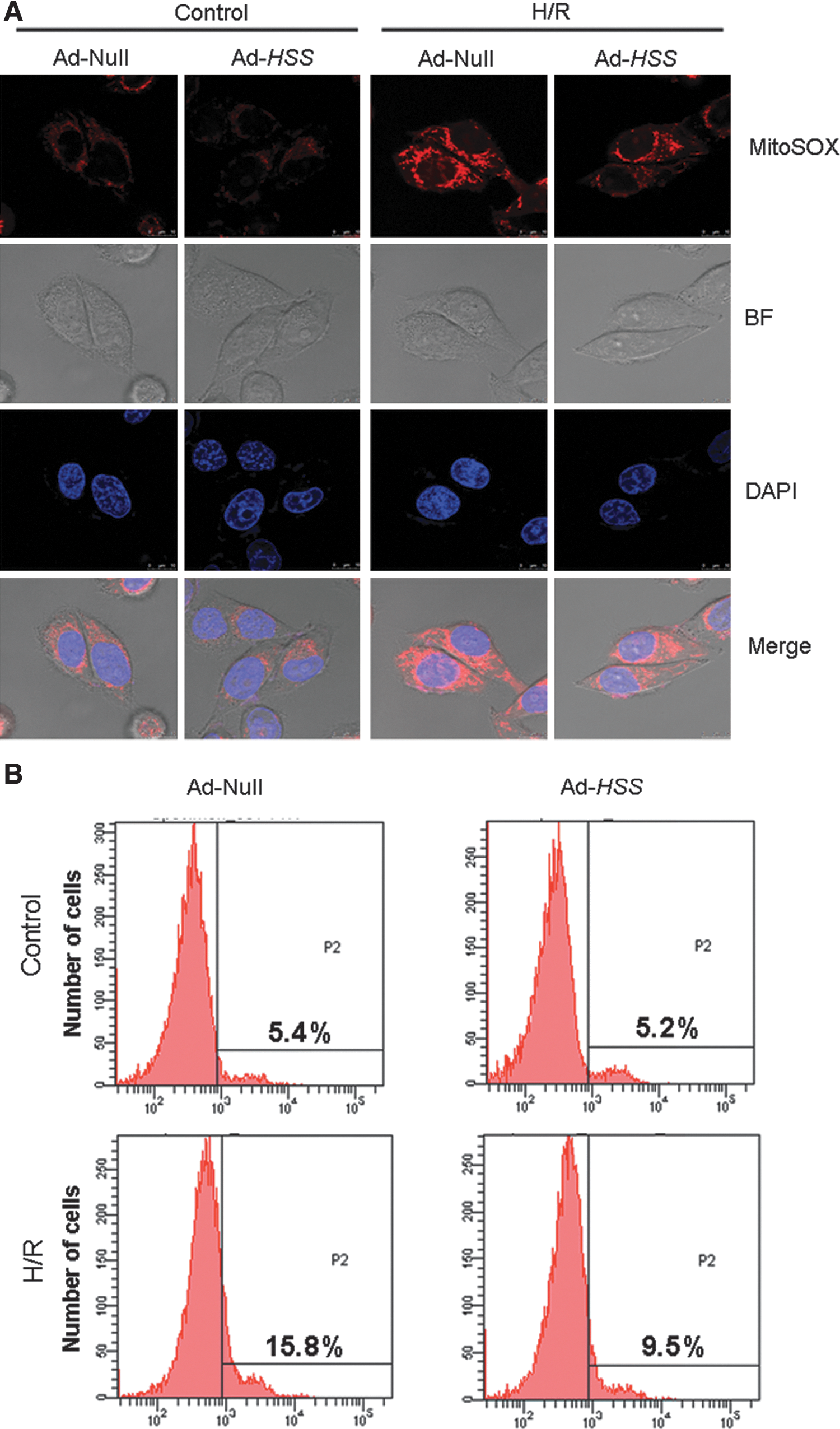
Transfection of HSS inhibits mitochondrial reactive oxygen species (ROS) production. Mitochondrial ROS production was evaluated by MitoSOX, using confocal microscopy and flow cytometry (FCM) after H/R injury.
Discussion
In the present study, we demonstrated that efficient gene transfer of HSS protected liver against I/R injury, as evidenced by decreased liver enzyme activities, improved hepatic morphology, and reduced hepatocyte apoptosis. The underlying mechanism of this hepatoprotective effect might be involved in the attenuation of mitochondrial dysfunction and mitochondrion-dependent apoptosis, as verified in both the in vivo animal I/R model and the in vitro cellular H/R model. More importantly, this study has identified that the scavenging of H/R-induced mitochondrial ROS production, restoration of the mitochondrial ATP level, and preservation of the mitochondrial respiratory chain complex activities may be considered possible mechanisms of HSS protection. Although HSS hepatic protection has been studied previously in our laboratory and other laboratories, this is the first report of its protective effects against I/R damage, a pathological process that is more frequently encountered in clinical situations, such as liver transplantation and hepatic resection.
Although the pathophysiological roles of apoptosis and oncotic necrosis in hepatic I/R injury have not been fully elucidated yet, they are undoubtedly prominent features (Georgiev et al., 2006). Many investigations have shown that interventions targeting apoptosis are therapeutically efficient in the inhibition of hepatic I/R injury. To evaluate the dynamics of apoptosis by I/R treatment, we examined apoptotic cells by TUNEL assay and analyzed the activation of caspase-3, a component of the enzymatic cascades that cause apoptotic cell death. As a result, we found a significant downregulation of the activation of caspase-3 as well as a reduction in TUNEL-positive cells on HSS gene transfer into liver. These data further demonstrated that the inhibition of apoptosis might be responsible for the protective effect of HSS against hepatic I/R injury.
Next, we tried to reveal the underlying mechanisms of the beneficial effect of HSS on hepatic I/R injury. It is well established that mitochondrial dysfunction is both a direct and indirect consequence of I/R-induced cell death (Kim et al., 2003). For example, I/R treatment causes ATP depletion, inhibits mitochondrial oxidative phosphorylation, and impairs the electron transport chain, which leads to the loss of mitochondrial membrane potential (MMP) and opening of the permeability transition pore (PTP), triggering downstream pathways that promote cell death. In addition, mitochondrial functional impairment was considered one of the major sources of cellular ROS generation immediately after H/R (Haga et al., 2008; Mukhopadhyay et al., 2012). Previous studies have also suggested that the protection of hepatocytes by HSS is related to mitochondrial preservation and blockade of the mitochondrial permeability transition (Wu et al., 2010). To clarify the role of mitochondrial protection during HSS gene therapy in H/R injury, we analyzed both mitochondrial morphology and its function. Although mitochondrial morphology and production of energy are severely destroyed by I/R treatment, transfection with HSS preserved mitochondrial morphology, preventing the defects observed in Figs. 3 and 4. Furthermore, the activity of respiratory chain complex I was somewhat maintained after H/R damage, suggesting that protection of the mitochondria by HSS may be the result of promotion of the expression and activity of the respiratory chain complex, which is in agreement with previous findings that treatment with the HSS protein enhances mitochondrial gene expression and the oxidative phosphorylation capacity of liver mitochondria (Polimeno et al., 2000).
Hypoxia–reoxygenation (H/R) is a reliable in vitro model and is widely used to study I/R injury. The H/R model allows for exploration of the mechanisms of HSS protection. Adenoviral vector-mediated HSS transfer in hepatoma cells improved cell viability after H/R treatment (Fig. 5A). An antiapoptotic effect could also be observed (Fig. 5B and C). Exogenously delivered HSS was located mainly in the mitochondria (Supplementary Fig. S2). HSS transfection attenuated mitochondrial dysfunction and protected the hepatocytes from mitochondria-dependent apoptosis by H/R treatment, as shown by suppression of the depolarization of the mitochondrial membrane potential, inhibition of cytochrome c leakage, deactivation of caspase-9 and caspase-3 (Fig. 6), and preservation of ATP content (Fig. 7).
H/R-induced apoptosis is considered to result mainly from ROS generation. Excessive ROS production not only causes damage to cells through lipid peroxidation, protein degradation, and DNA damage, but it also causes indirect damage by regulating redox-sensitive signals that may affect cell fate and inflammation (Mukhopadhyay et al., 2012). A number of reports have shown that reduced ROS generation suppressed injury after reoxygenation by catalytic antioxidants or antioxidative enzymes (Serviddio et al., 2010). One of the major sources of cellular ROS generated immediately after H/R may be functionally impaired mitochondria (Weinberg et al., 2000). In the present study, ROS generation by mitochondria after H/R was suppressed by HSS gene therapy (Fig. 8), demonstrating the antioxidative effect of HSS. Although the mechanism of free radical scavenging by HSS remains unclear, the promotion of antioxidative enzyme activity such as by glutathione peroxidase (GPx) (Cao et al., 2009) and the abundance of thiol and disulfide bonds within the HSS crystal structure may account for this process.
In conclusion, we have demonstrated for the first time the protective effects of HSS during the process of hepatic I/R injury. The underlying mechanisms may be associated with mitochondrial protection, including the attenuation of mitochondrial dysfunction and the inhibition of mitochondria-dependent apoptosis. More importantly, improvement of mitochondrial function as a result of the alleviation of ROS production and enhancement of oxidative phosphorylation capacity could also be considered as a possible protective mechanism of HSS. Although further studies are needed to clarify the mechanisms of HSS, it may be considered be a potential candidate as a liver-protecting agent to treat hepatic injury in the clinic.
Footnotes
Acknowledgments
This work was supported by the National Key Basic Project (grant 2010CB5344903) and National Natural Science Foundation of China (grant 81100310).
Author Disclosure Statement
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.