
Abstract
Adoptive therapy of cancer with genetically redirected T cells showed spectacular efficacy in recent trials. A body of preclinical and clinical data indicate that young effector and central memory T cells perform superior in a primary antitumor response; repetitive antigen engagement, however, drives T-cell maturation to terminally differentiated cells associated with the loss of CCR7, which enables T cells to persist in peripheral tissues. In this work, we explored the antitumor efficacy of CCR7− T cells when redirected in an antigen-dependent fashion by a chimeric antigen receptor (CAR) toward tumors in the periphery. CAR-engineered CCR7− T cells more efficiently accumulated at the tumor site, secreted more IFN-γ, expressed higher amounts of cytotoxic molecules, and showed superior tumor cell lysis compared to the younger CCR7+ cells. CCR7− T cells, however, were more prone to spontaneous and activation-induced cell death, which could be counteracted by simultaneous CD28 and OX40 (CD134) costimulation. Consequently, the combined CD28-ζ-OX40 signaling CAR rescued CCR7− T cells from apoptosis, which then produced more efficient antitumor efficacy than CCR7+ T cells redirected by the same CAR. Data suggest that T-cell therapy will benefit from combined CD28-ζ-OX40 stimulation in the long-term by rescuing continuously generated CCR7− T cells for an antitumor attack.
Introduction
Much work has focused on the need to identify the T-cell subset most suitable for a potent and sustained antitumor response. T cells with naive or central memorylike phenotype showed superior in this respect. Central memory T cells proliferate rapidly upon antigen encounter (Sallusto et al., 2004; Lefrançois, 2006) and express CCR7 and CD62L, enabling these cells to home into central lymphoid organs (Parish and Kaech, 2009). The latter process involves the interaction of CCR7 on T cells with CCL19 or CCL21 on lymph endothelial cells (Bromley et al., 2005; Debes et al., 2005). Upon repetitive antigen engagement, however, T cells acquire a late effector memory phenotype and cease CCR7 expression, which reduces their capability to reenter the lymph vessels and to recirculate. Adoptive cell therapy with any T-cell subset will finally produce those cells upon repetitive stimulation during an antitumor attack. For instance, central memory T cells, which have the benefit to establish a long-lasting antitumor response, migrate into the draining lymph node, reenter circulation through migration into lymph vessels, and further differentiate into CCR7− late effector T cells losing their recirculating capacities and allowing them to patrol peripheral tissues (Klebanoff et al., 2005). While these cells express higher amounts of effector molecules, their in vivo efficacy is paradoxically reduced (Gattinoni et al., 2005; Berger et al., 2008). Particularly cell therapy of cancer T-cell persistence in the periphery may be of advantage by promoting T-cell accumulation in the peripheral tumor lesion. We here revealed that CCR7− T cells, which are continuously produced from “young” T cells during an enduring immune response, need different prerequisites to sustain their activation than provided by the commonly used CARs. While CD28, 4-1BB (CD137), and OX40 (CD134) similarly improve proinflammatory cytokine secretion, OX40 most efficiently prevents activation-induced cell death (AICD) of CD62L- effector memory T cells (Hombach et al., 2011). Redirected by a combined CD28 and OX40 costimulatory CAR, CCR7− T cells can be rescued from activation-induced cell death and perform superior in antitumor activity compared to redirected CCR7+ T cells.
Materials and Methods
Cells and antibodies
All studies involving human blood cells have been approved by the Uniklinik Köln Institutional Review Board (reference no. 01-090). Peripheral blood mononuclear cells were isolated from healthy donors by density centrifugation and CD4+ and CD8+ T cells separated by magnetic activated cell sorting (MACS) (Miltenyi Biotec, Bergisch-Gladbach, Germany). CD3+ T cells were obtained by negative depletion during MACS procedures and separated into CCR7+ and CCR7− T cells using the mouse anti-human CCR7 IgM antibody 4H2 (BD Biosciences, Heidelberg, Germany) and anti-mouse IgM micro-beads (Miltenyi Biotec). Alternatively, cells were flow sorted using the FACS Aria III (Becton Dickenson, Mountain View, CA) and fluorochrome-conjugated anti-mouse IgM antibodies (Southern Biotech, Birmingham, AL) or fluorochrome-conjugated anti-human (clone RD12, BD Bioscience) and anti-mouse (clone 4B12, Biolegend, San Diego, CA) CCR7 antibodies, respectively.
293T cells (ATCC CRL-11268) are human embryonic kidney cells that express the SV40 large T antigen. MC38 is a murine fibrosarcoma cell line (Schwegler et al., 2005); C15A3 cells were derived from MC38 cells by transfection with a plasmid-encoding carcinoembryonic antigen (CEA) (Robbins et al., 1991). (Both cell lines were kindly provided by Dr. M. Neumaier, Universität Mannheim-Heidelberg, Mannheim, Germany). C15A3 cells were engineered with click beetle luciferase and recorded by bioluminescence using D-luciferin as substrate. Panc02 is a murine pancreatic tumor cell line that was transfected to express CEA and click beetle luciferase (CBLuc). The anti-CD3 monoclonal antibody OKT3, the anti-CD28 antibody 15E8, and the antibody BW2064/36, which is an internal image anti-idiotypic antibody directed against the anti-CEA BW431/26 single chain antibody, were produced by the respective hybridoma cells and affinity purified from hybridoma supernatants utilizing goat anti-mouse IgG1 antibody (Southern Biotech) immobilized on N-hydroxy-succinimid-ester-(NHS)-activated sepharose (Amersham Biosciences, Freiburg, Germany).
All cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Invitrogen Life Technologies, Karlsruhe, Germany) containing 100 U/ml penicillin, 100 mg/ml streptomycin, 2 mM L-glutamine, and 10% (v/v) heat-inactivated fetal calf serum (FCS) (Invitrogen Life Technologies). Antibodies for CCR7 (clone 2H4 and clone 3D12), CD127 (clone HIL-7R-M21), CD28 (clone CD28.2), and CD45RO (clone UCHL-1) were purchased from BD Biosciences; CD45RA (clone ALB11), CD27 (clone 1A4CD27), and CD62L (clone DREG56) from Immunotech (Marseille, France); and CD25 (clone 4E3) from Miltenyi Biotec. CD57 (clone NC1) was purchased from Immunotools (Frisoythe, Germany), fluorescein isothiocyanate (FITC)-conjugated IgM from Beckmann Coulter (Krefeld, Germany), allophyocyanin (APC)-conjugated IgM from Southern Biotech, and PE/FITC conjugated anti-CD4 (clone MT310) and anti-CD8 (DK25) mAb from Dako (Hamburg, Germany). 7-AAD was purchased from BD Biosciences.
CAR's and retroviral modification of T cells
The retroviral expression cassette for the CEA-specific CAR BW431/26scFv-Fc-CD28-ζ has been described (Hombach et al., 2001). The CEA-specific scFv antibody BW431/26 was used as binding domain and linked through the human IgG1 CH2CH3 (Fc) to the transmembrane and intracellular domain of CD28, which is either fused to the intracellular CD3ζ or CD3ζ-OX40. To generate the cDNA coding for the chimeric CD28-ζ-OX40 and ζ-OX40 signaling domain, the cDNAs of OX40 and CD28/CD3ζ were amplified by polymerase chain reaction (PCR) utilizing the following oligonucleotides: 5zetaOX40 (sense): 5′-CACATGCAGGCCCTGCCCCCTCGCAGGGACCAGAGGCTGCCCCCCGATGCC-3′; OX40AS (antisense): 5′-TCAGTCGACCTCGAGTCAGATCTTGGCCAGGGTGGAGTG-3′; TMcd28bam BamHI (sense): 5′-CTGGATCCCAAATTTTGGGTGCTGGTGGTGGTTG-3′; 3zetaOX40 (antisense): 5′-GGCATCGGGGGGCAGCCTCTGGTCCCTGCGAGGGGGCAGGGCCTGCATGTG-3′. The amplified DNAs were flanked with overlapping sequences and assembled by PCR. The assembled PCR product was reamplified utilizing oligonucleotides TMcd28bam BamHI and OX40AS, thereby flanked by BamHI and XhoI restriction sites, respectively, and inserted into the retroviral expression vector pBULLET. The intracellular amino acid sequence of the CD28-ζ-OX40 CAR is as follows: CD28[DPKFWVLVVVGGVLACYSLLVTVAFIIFWVRSKRSRLLHSDYMNMTPRRPGPTRKHYQPYAPPRDFAAYRSL]-ζ[RVKFSRSADAPAYQQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEAYSEIGMKGERRGKGHDGLYQGLSTATKDTYDALHMQALPPR]-OX40[RDQRLPPDAHKPPGGGSFRTPIQEEQADAHSTLAKI]. Human T cells were transduced to express the CARs as previously described in detail (Weijtens et al., 1998). CAR expression was monitored by flow cytometry utilizing a PE-conjugated F(ab’)2 anti-human IgG1 (Southern Biotech) and a fluorescein isothiocyanate (FITC)-conjugated anti-CD3 antibody (UCHT-1; Dako, Glostrup, Denmark) and analyzed utilizing a fluorescent-activated cell sorter (FACS) Canto cytofluorometer equipped with the FACS DIVA software (Becton Dickinson). Mouse CD8+ T cells were cultured in RPMI 1640 medium, Dutch modification, supplemented with 20 mM HEPES, 10% (v/v) heat-inactivated FCS (Biochrom, Cambridge, UK), 2 mM L-Glutamine, 2 mM sodium-pyruvate, 0.2 mM nonessential amino acids, and 55 μM β-mercaptoethanol (all Gibco/BRL). Mouse spleen T cells were retrovirally modified as follows: T cells were activated with 500 IU/ml IL-2 (Chiron GmbH, Ratingen, Germany), 0.5 μg/ml anti-CD3 mAb clone 145-2C11 (Becton Dickinson), and 0.1 μg/ml anti-CD28 mAb clone 37.51 (Becton Dickinson) for 24 hr. Retroviruses were produced in 293T cells using the helper plasmids pVpack-Eco (Stratagene, La Jolla, CA) coding for Moloney murine leukemia virus (MMLV)-based ecotropic env gene and pVpack-GP (Stratagene) coding for MMLV-based gag and pol genes. Activated CD3+ T cells were infected for 24 hr in fresh culture medium supplemented with 200 IU/ml IL-2 and 5 μg/ml IL-15. For bioluminescence imaging, T cells were marked by an optimized version of a membrane-anchored, recombinant form of the Gaussia princeps luciferase (G-luc). Briefly, G-luc cDNA was modified by PCR according to Welsh et al. (2009) to increase duration of luminescence. Membrane-anchored G-luc was obtained by fusion of the G-luc cDNA to the CD8 transmembrane cDNA (Santos et al., 2009). G-luc was expressed by a retroviral vector together with green fluorescent protein (GFP) to allow identification of marked cells by flow cytometry. C15A3 tumor cells were marked by the click beetle luciferase (CB-luc). In vivo imaging was performed upon intraperitoneal injection of D-luciferin (1,5 mg/mouse) or benzyl-coelenterazine (100 μg/mouse) (PJK GmbH, Kleinblittersdorf, Germany) as substrate for the CB and G luciferase, respectively, using Photon Imager (Biospace Lab, Paris, France).
Redirected activation of T cells
Specific cytotoxicity of CAR-engineered T cells was monitored by an XTT (2,3-bis(2-methoxy-4-nitro-5-sulphonyl)-5(phenyl-amino)carbonyl)-2H-tetrazoliumhydroxide)-based colorimetric assay. Briefly, CAR-engineered and mock-engineered T cells were coincubated with tumor cells in triplicates in round bottom microtiter plates for 48 hr. XTT reagent (1 mg/ml) (Cell Proliferation Kit II; Roche Diagnostics, Mannheim, Germany) was added and incubated for 90 min at 37°C. Reduction of XTT to formazan by viable tumor cells was monitored colorimetrically at an absorbance wavelength of 450 nm and a reference wavelength of 650 nm. Specific cytotoxicity of T cells was calculated as follows: cytotoxicity (%)=[1 – OD (experimental wells – corresponding number of effector cells) / OD (tumor cells without effector cells – medium)] × 100. IFN-γ in the culture supernatants of activated T cells was recorded by ELISA utilizing matched pairs of antibodies specific for IFN-γ (clones NIB 42 and B133.5; BD Biosciences). Briefly, IFN-γ was bound to a solid phase anti-human IFN-γ mAb (1 μg/ml) and detected by a biotinylated anti-human IFN-γ mAb (0.5 μg/ml). The reaction product was visualized by a peroxidase-streptavidin-conjugate (1:10,000) and ABTS® (both from Roche Diagnostics) as substrate. The detection limit of the assay was 15 pg/ml for IFN- γ.
Activation-induced cell death
T cells were activated by plate-bound anti-CD3 and anti-CD28 mAbs (each 2μg/ml) or cultivated in medium for 2 to 5 days. Cells were stained for CCR7 by the anti-CCR7 mAb and cell viability was assayed by recording the mitochondrial membrane potential utilizing the JC-1 dye (Invitrogen). Alternatively, cells were stained with the anti-CCR7 mAb and with APC-conjugated Annexin V (Immunotools) and 7-AAD, respectively. T cells were analyzed by flow cytometry and the number of apoptotic CCR7+ and CCR7− was determined. To record CAR-induced activation-induced cell death, CAR T cells (2.5×106 total cells) were cultivated in the presence of 0.5 μg/ml of the anti-idiotypic mAb BW2064/399 and 0.25 μg/ml of a goat anti-mouse IgG antibody (Southern Biotechnology), respectively. After 72 hr, cells were stained for CAR and CCR7 with a FITC-conjugated F(ab’)2 anti-human IgG1 (Southern Biotechnology) and PE-conjugated anti-CCR7 antibody, respectively. Cell viability was recorded by staining with APC-conjugated Annexin V and 7-AAD, respectively. T cells were analyzed by flow cytometry and the number of CAR+/− and CCR7+/− AnnexinV+ cells were determined.
Granzyme/perforin recording
To monitor expression of cytolytic effector molecules, sorted CD4+ and CD8+ T cells were stained with an APC-conjugated anti-CCR7 mAb (clone 150503; R&D, Minneapolis, MN) and subsequently fixed and permeabilized using the “Cytofix/Cytoperm kit” (BD Biosciences). Permeabilized cells were incubated either with PE-conjugated anti-perforin (clone δG9; Ancell, Bayport, MN), granzyme A (clone CB9; BD Biosciences), or granzyme B mAbs (clone GB11; Serotec, Oxford, UK) and analyzed by flow cytometry. Cells were stained with phycoerythrin-conjugated anti-nuclear factor-κB (NF-κB) (Clone K10-895.12.50, BD Biosciences) at room temperature for 30 min. Stained cells were analyzed by flow cytometry using a FACS Aria equipped with DIVA 6.0 software. Dead cells were gated out based on forward/side scatter and positive 7-AAD staining.
Mice
All animal experiments were performed according to the Animal Experiments Committee regulations and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz, Recklinghausen, Germany (K17/35-05). CD1−/− nu/nu and NIH-III (Crl:NIH-LystbgFoxn1nu Btk xid ) mice were purchased from Charles River Laboratories (Sulzfeld, Germany). CEA+ C15A3 tumor cells (1×106 cells/mouse) were subcutaneously (s.c.) coinjected with engineered T cells (2.5×105 CAR-modified T cells) into CD1−/− nu/nu mice. Tumor volume was recorded and area under curve determined (Duan et al., 2012). CEA-transgenic mice (kindly provided by D. Gilham, Paterson Institute, Manchester, UK) were used to record T-cell migration. CEA-transgenic mice are immune competent and express CEA under control of the human promoter orthotopically in the gastrointestinal tract and the lung (Eades-Perner et al., 1994). CEA+ Panc02 carcinoma cells marked with click beetle luciferase (CBLuc) were transplanted into the pancreas to induce pancreatic carcinoma. Mouse T cells with anti-CEA CAR were marked with a membrane-anchored variant of Gaussia luciferase (GLuc), separated into CCR7+ and CCR7− subsets and were intravenously applied by a single injection into the tail vein (1×106 CAR-expressing cells/mouse). In vivo imaging was performed upon intra-peritoneal injection of D-luciferin (1.5 mg/mouse) or benzyl-coelenterazine (100 μg/mouse) (PJK GmbH) as substrate for the CBLuc and GLuc, respectively, and visualized using the Photon Imager (Biospace Lab). The exposure time was 300 sec for all recordings. The threshold of bioluminescence signals was automatically determined using the Photo Vision software. Bioluminescence signals were accordingly filtered against background noise. Regions of interest (ROIs) were defined as regions above threshold and automatically gated by predefined program tools. There was no manual gating of ROIs in order to avoid any incoherence. Photon emission intensity (photon/s/sr) was calculated from data of emitted photons from the respective ROI using the Photo Vision software (Biospace Lab).
Immunohistological analyses
Cryostat sections from tumor tissues and isolated spleen cells placed on cover slips were fixed with acetone for 10 min at −20°C. Slides were first incubated with “Fc receptor block” reagent for 15 min, followed by “Background Buster” for 30 min (both Innovex Biosciences, Richmond, CA) to block nonspecific binding. Slides were simultaneously stained with an Alexa-488-conjugated anti-CD3 (17A12), a Brilliant Violett 421-conjugated anti-CCR7 (4B12) (both Biozol, Eching, Germany), and a Dye-Light 549-conjugated F(ab′)2 anti-mouse IgG (gamma) antibody (KPL, Gaithersburg, MD), which detects the CAR extracellular IgG Fc spacer. Specificity of staining was assayed by incubation with the respective isotype-matched control antibodies. Cell nuclei were stained with RedDot2 (Biozol). Slides were mounted with “ImmunoSelect” anti-fading mounting medium (Dianova, Hamburg, Germany). One slide out of a series of tissue sections was routinely stained with haematoxylin–eosin (Carl Roth, Karlsruhe, Germany) to confirm histology. Slides were analyzed using the Confocal Laser Scanning Microscope LSM 510 equipped with ZEN 2009 software (Karl Zeiss, Oberkochen, Germany). A minimum of 100 cells per slide was counted.
Statistical procedures
Statistical analyses were performed using Student's t-test.
Results
CCR7+ memory T cells converted to CCR7− cells upon prolonged stimulation
When engaging, their cognate antigen T cells extensively amplify, which may produce altered phenotype and altered functional properties, both having substantial consequences for the efficacy of adoptive cell therapy in the long-term. To address this issue, CCR7+ T cells were isolated from peripheral blood by magnetic activated cell sorting (MACS), labeled with CSFE, and repetitively stimulated by CD3/CD28 or CD3/IL-2, respectively. As shown in Figure 1a, T cells lost CCR7 expression after several rounds of amplification in vitro. At day 11, the majority of CD3/IL-2 and CD3/CD28 stimulated T cells ceased CCR7 expression. Change in phenotype depends on T-cell amplification since a block in proliferation by mitomycin C prevented the generation of CCR7low cells.
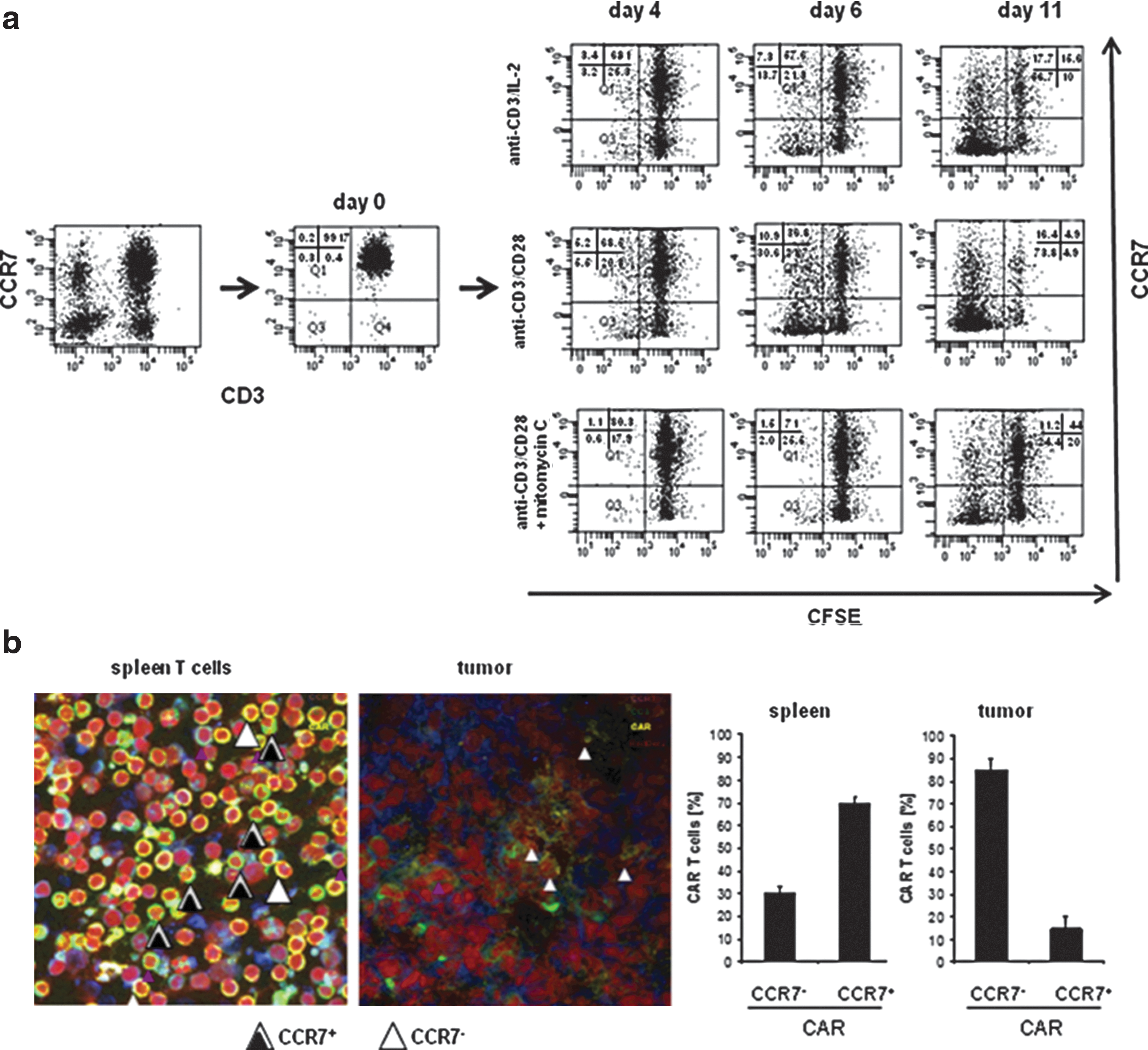
CCR7+ T cells acquire the CCR7− phenotype upon prolonged stimulation.
We asked whether adoptively transferred T cells convert to CCR7− cells in vivo when accumulating at the targeted tumor site. Spleen T cells were engineered with a chimeric antigen receptor (CAR) specific for carcinoembryonic antigen (CEA), which is expressed on a variety of gastrointestinal carcinoma cells, and adoptively transferred into mice with transplanted CEA+ pancreatic carcinoma. Whereas adoptively transferred T cells were predominantly of CCR7+ phenotype, 80% of tumor infiltrating T cells lacked CCR7 (Fig. 1b).
The CCR7− subset of T cells generated by extensive amplification in vitro was predominantly composed of effector memory T cells as indicated by the CD45RAlow CD45ROhigh phenotype and by decreased CD27, CD28, and CD62L compared to CCR7+ central memory T cells (Fig. 2a). Essentially the same data were obtained for CD4+ and CD8+ T cells. CCR7− T cells moreover expressed CD57 in higher frequencies than CCR7+ cells. CCR7− T cells harbored higher amounts of the cytotoxic effector molecules perforin (prf) and granzyme (grz) A and B compared to the corresponding CCR7+ T cells (Fig. 2b), which is in accordance to a previous report on CD57+ T cells (Chattopadhyay et al., 2009). Data imply that CCR7− T cells generated during extensive T-cell expansion may be potent effectors in adoptive cell therapy. CCR7− T cells, however, were prone to massive spontaneous apoptosis compared to CCR7+ cells (Fig. 2c). Apoptosis of CCR7− T cells could not be prevented by CD3/CD28 stimulation; in particular, more CCR7− cells showed breakdown in mitochondrial membrane potential than CCR7+ cells after 2 days, as indicated by loss of JC1 staining, and up to 80% of CCR7− T cells underwent apoptosis after 5 days of stimulation. Substantial cell death, however, limits the therapeutic efficacy of CCR7− T cells asking for strategies to counteract.
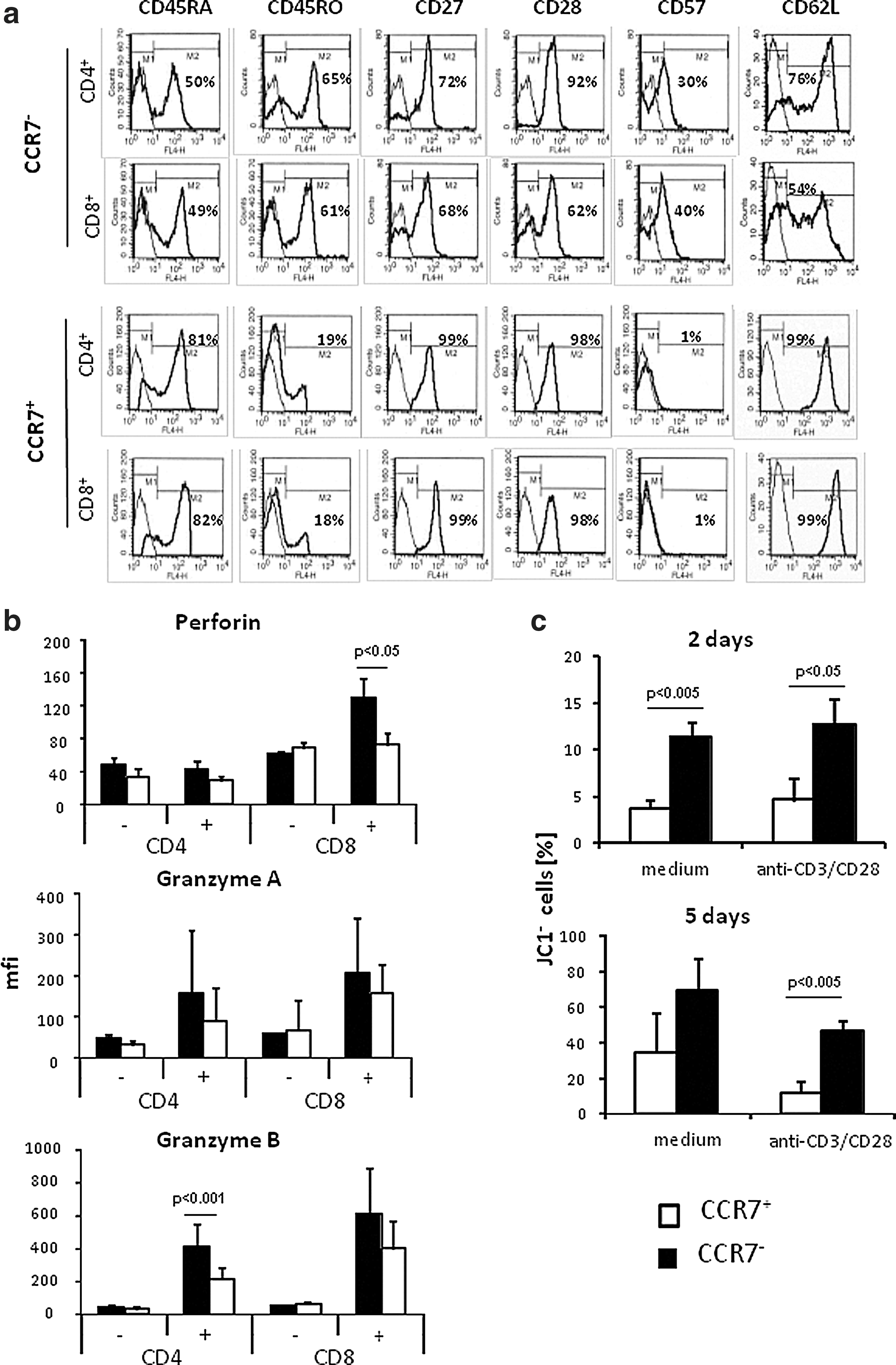
CCR7− T cells represent effector memory cells in terminal differentiation and are prone to apoptosis.
Combined CD28-ζ-OX40 stimulation counteracted activation-induced cell death and sustained antitumor activity of redirected CCR7− T cells
T cells of different subsets and in different stages of maturation require different costimuli for activation and prevention from apoptosis. While CD28 costimulation in addition to TCR/CD3ζ signaling is sufficient to promote activation of “young” CCR7+ T cells, we hypothesized that CCR7− T cells, which are more progressed in maturation, may require additional “late” costimulation as provided by OX40 to counteract AICD. To combine “early” CD28 costimulation with “late” OX40 costimulation, we generated a CAR with a CD28-CD3ζ-OX40 endodomain (Fig. 3a). The rationale is that an OX40 CAR proved to be more efficient than a 4-1BB CAR to modulate the primary CD3ζ signal upon antigen encounter (Hombach et al., 2011). Adding OX40 at the CAR terminal position leaves the CD28-ζ alignment unaltered and generates basically the same effector functions as the CD28-OX40-ζ alignment (Wilkie et al., 2008). CARs with different signaling domains were expressed with similar efficacies in peripheral T cells (Hombach et al., 2011). CAR signaling as indicated by IFN-γ secretion was substantially enhanced by CD28 and OX40 costimulation; simultaneous CD28 and OX40 costimulation acts synergistically when enhancing signaling of the CAR basic CD3ζ domain (Fig. 3a).
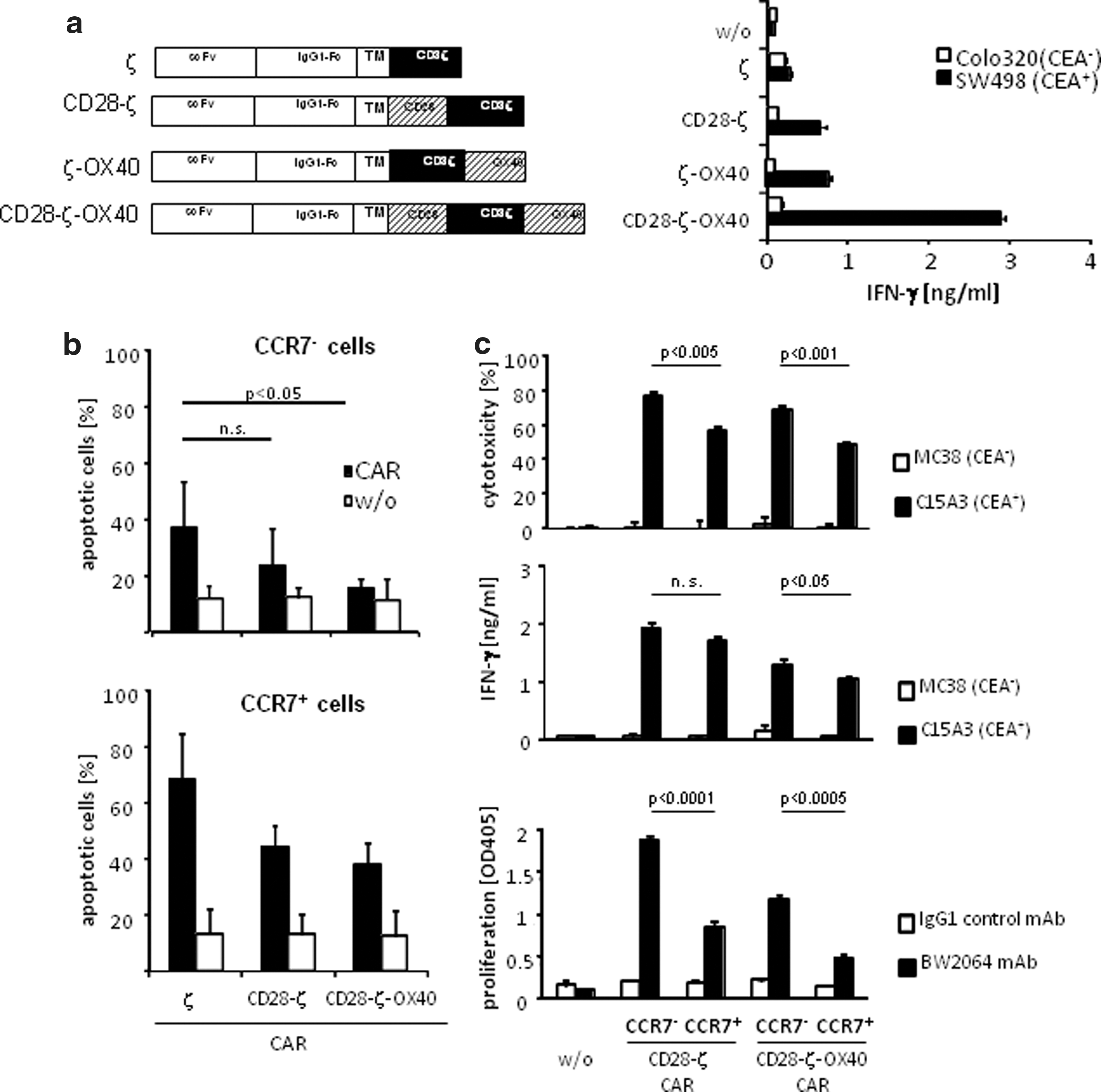
The CD28-ζ-OX40 CAR protects CCR7− T cells from apoptosis and improves redirected activation.
To redirect T cells in an antigen-specific fashion toward CEA+ tumor cells, CCR7− T cells were engineered with a CAR with combined CD28-ζ-OX40 or CD28-ζ signaling domain, respectively. Both CARs recognized CEA+ tumor cells by the same binding domain. CAR-engineered T cells were subsequently separated into the CCR7+ and CCR7− cell subsets. CARs were nearly equally expressed on engineered T cells of both subsets (data not shown). The procedure of modifying with a CAR did not change CCR7 expression of the respective subset of cells. Activation-induced cell death was efficiently reduced when CCR7− cells were stimulated by the CD28-ζ-OX40 CAR compared to the CD28-ζ CAR (Fig. 3b). CD28 costimulation in addition to ζ signaling had no significant effect in reducing the number of apoptotic CCR7− cells. This is in contrast to CCR7+ cells, which gained benefit from CAR-mediated CD28 costimulation but did not from combined CD28-ζ-OX40 stimulation. CD28-ζ-OX40 CAR redirected effector functions of CCR7− T cells were improved upon CAR signaling compared to CCR7+ cells, including IFN-γ secretion, proliferation, and the cytolytic activity (Fig. 3c). Anti-CEA CAR T cells were activated upon coincubation with CEA+ but not with CEA− cells; unmodified T cells were not activated by CEA+ or CEA− target cells. In short term assays, CCR7− T cells showed substantially improved activation, cytotoxicity, and proliferation compared to CCR7+ T cells; CCR7− T cells, however, benefit more than CCR7+ T cells from OX40 cosignaling in repressing apoptosis.
Redirected CCR7− T cells accumulated more at the tumor site than CCR7+ cells and showed improved antitumor activity when redirected by the CD28-ζ-OX40 signaling CAR
CCR7− T cells are less able to reenter the lymph and thereby inefficiently recirculate (Sallusto et al., 1999). Adoptive cell therapy may take advantage of this property of CCR7− T cells since most solid cancer lesions occur in the periphery. To explore their tumor targeting capacity, CCR7− T cells with the CEA-specific CD28-ζ CAR were marked with a membrane-anchored variant of the G-luc to allow in vivo tracking of labeled cells. Solid tumors were established in mice by subcutaneous transplantation of CB-luc-marked CEA+ C15A3 tumor cells. When tumors were established, CAR-engineered CCR7− and CCR7+ T cells were applied by a single i.v. injection into the tail vein. Both T cell subsets were detected at the tumor site as early as at day 2 after injection; CCR7− CAR T cells, however, more efficiently persisted and amplified at least until day 17, while CCR7+ cells were no longer detected at the tumor site (Fig. 4a and b). We simultaneously analyzed the cytolytic capacity of CAR-engineered CCR7− and CCR7+ cells, respectively, in vivo by recording bioluminescence of tumor cells. In contrast to the high accumulation of CAR+CCR7− T cells at the tumor site, the antitumor reactivity of these cells was substantially lower than those of CAR+CCR7+ T cells (Fig. 4a and b), indicating their high tumor accumulating but low antitumor reactivity in vivo when redirected by a CD28-ζ CAR.
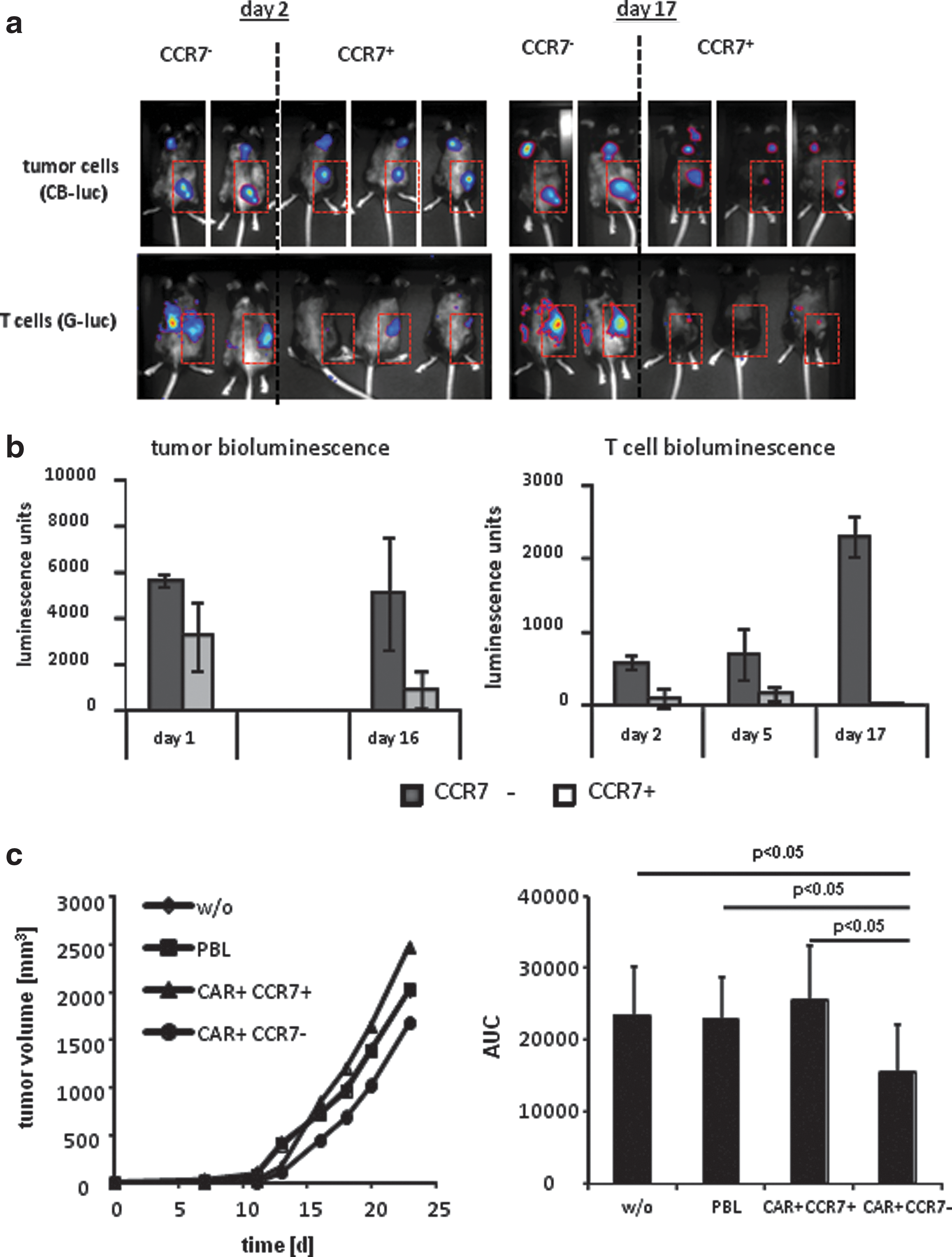
CD28-ζ-OX40 CAR-redirected CCR7− T cells exhibit improved antitumor activities.
To compare the in vivo cytolytic activity of CCR7− vs. CCR7+ T cells that were redirected by the same CD28-ζ-OX40 CAR, both modified T-cell subsets were co-inoculated together with CEA+ C15A3 tumor cells in immune deficient mice. This setting avoids the different homing properties of the T-cell subsets impacting the antitumor response. Mice that received engineered CCR7+ T cells more rapidly developed tumors compared to mice receiving the same number of engineered CCR7− T cells (Fig. 4c), indicating superior tumor cell lysis by redirected CCR7− compared to CCR7+ T cells. The redirected cytolytic activity was specific since nonmodified T cells had no impact on co-inoculated CEA+ tumor cells. Taken together, CCR7− T cells, which are generated by repetitive stimulation of CCR7+ cells, can significantly contribute to the antitumor efficacy of adoptive cell therapy when redirected by a CD28-ζ-OX40 CAR.
Discussion
Prolonged expansion upon repetitive antigen engagement convert naïve and central memory T cells to cells with a CCR7− CD62Llow CD57+ phenotype, which accumulate at the targeted tumor site. This process is of particular relevance when CAR-redirected T cells encounter targeted tumors during adoptive cell therapy. While CCR7− T cells secrete more IFN-γ and harbor more cytolytic effector molecules, they exhibit an extremely high propensity to undergo spontaneous and activation-induced cell death, which substantially limits their therapeutic efficacy. In this work, we revealed that apoptosis of CCR7− T cells can be counteracted by combined CD28-OX40 costimulation as provided by a so-called third-generation CAR. Since T cells of any subset will convert to CCR7− cells upon extensive amplification and antigen engagement, which then need additional OX40 costimulation, harnessing “young” T cells with a CD28-ζ-OX40 CAR may be beneficial for a redirected antitumor attack in the long-term since those cells are rescued from apoptosis after expansion and conversion to a CCR7− phenotype. While CCR7− T cells benefit from a CD28-ζ-OX40 CAR, OX40 cosignaling showed less effect for CCR7+ T cells, whereas CD28 cosignaling, which is efficient for CCR7+ T cells, does not reduce the number of apoptotic CCR7− cells. As a result, CD28-ζ-OX40 CAR redirected CCR7− cells perform superior over CCR7+ T cells in counteracting tumor progression.
There are certainly additional factors that impact the antitumor efficacy; firstly, the improved cytokine secretion and cytotoxicity of engineered CCR7− T cells as recorded by two assays, i.e., the in vitro short-term and the in vivo co-inoculation kill assay. The latter assay records tumor cell elimination while avoiding impact of migration and amplification, which substantially differ for both T-cell subtypes. Secondly, CCR7− T cells more efficiently accumulate and persist at the tumor lesion compared to CCR7+ cells after systemic application. Prolonged persistence in the targeted tumor tissue is likely due to the deficiency of CCR7− T cells to migrate through lymph endothelial cell layers and to re-enter the lymph in contrast to CCR7+ T cells, which repetitively recirculate through lymphoid organs (Sallusto et al., 1999; Müller and Lipp, 2003; Bromley et al., 2005; Klebanoff et al., 2005). Less recirculation may result in prolonged T-cell persistence in the tumor tissue, thereby increasing the probability for tumor cell contact and successful killing. These properties together support that the CD28-ζ-OX40 CAR-redirected CCR7− T cells repress tumor progression with higher efficacy than CCR7+ T cells redirected by the same CAR.
While preclinical models provide strong evidence for using central memory T cells redirected by a CD28-ζ CAR (Klebanoff et al., 2005), those cells convert to CCR7− effector memory T cells upon repetitive antigen engagement and then require the OX40 signal in addition to CD28 to counteract AICD. Since CD28-OX40 CAR-redirected CCR7− T cells produce increased cytolytic capacities, secrete increased levels of proinflammatory cytokines, and accumulate in the periphery without recirculating to the lymph, we assume that adoptive therapy with any T-cell subset, which finally produces CCR7− T cells, will benefit from combined CD28-OX40 cosignaling in the long-term.
Each costimulatory domain, CD28, OX40, and 4-1BB, in series with CD3ζ, initiates distinct but different functions dependent on the antigenic history of the T cell on which the CAR is engineered (Finney et al., 2004; Pulè et al., 2005; Carpenito et al., 2009). OX40 promotes the expression of Bcl-xL and Bcl-2, enhances the survival of antigen-experienced effector T cells, and improves the generation of antigen-specific T-cell memory (Rogers et al., 2001). Studies which compared CARs with different costimulatory domains of the CD28 family revealed substantial differences of the costimulatory domains in preventing from AICD in vitro (Hombach et al., 2001; Abken et al., 2002; Finney et al., 2004; Hombach and Abken, 2011). 4-1BB costimulation, however, improved T-cell persistence, but not antitumor activity, in vivo (Song et al., 2011). Physiologically, the individual costimulatory signals occur in a defined temporal and spatial order; the CAR with two costimulatory domains, in contrast, simultaneously delivers the signals to the engineered T cell, almost independently whether the costimulatory receptor is expressed and costimulatory ligands are present on the antigen-presenting cells.
In addition, to provide appropriate costimulation, several other attempts are currently undertaken to improve trafficking and persistence of redirected T cells at the targeted tumor. CAR T cells were additionally engineered with the CCR2 receptor, which produced enhanced tumor localization and eradication (Moon et al., 2011). Nonmyeloablative lymphodepletion is performed to eradicate repressor T cells and to provide space for homeostatic expansion of transferred T cells, low or medium dose of IL-2 is routinely administered to sustain T cell expansion (Weber et al., 2011). The latter is mandatory to establish adoptively transferred T cells in the long-term, however, accelerates conversion of central memory T cells to CCR7− effector memory T cells, which require the additional OX40 signal to be efficiently protected from activation-induced cell death.
Footnotes
Acknowledgments
The work was supported by the European Commission through the ATTACK project; the Deutsche Forschungsgemeinschaft, Bonn; and the Fortune Program of the Medical Faculty of the University of Cologne. We thank Dr. Martin Hellmich (Institut für Medizinische Statistik, Informatik und Epidemiologie, Uniklinik Köln) for statistical analyses and Danuta Chrobok, Nicole Hoffmann, Frank Steiger, and Samir Tawardos for technical assistance.
Author Disclosure Statement
No competing financial interests exist.