
Abstract
The dendritic cell (DC), a most potent antigen-presenting cell, plays a key role in vaccine therapy against infectious diseases and malignant tumors. Although advantages of viral vectors for vaccine therapy have been reported, potential risks for adverse effects prevent them from being licensed for clinical use. Human parainfluenza virus type 2 (hPIV2), one of the members of the Paramyxoviridae family, is a nonsegmented and negative-stranded RNA virus. We have developed a reverse genetics system for the production of infectious hPIV2 lacking the F gene (hPIV2ΔF), wherein various advantages for vaccine therapy exist, such as cytoplasmic replication/transcription, nontransmissible infectivity, and extremely high transduction efficacy in various types of target cells. Here we demonstrate that hPIV2ΔF shows high transduction efficiency in human DCs, while not so high in mouse DCs. In addition, hPIV2ΔF sufficiently induces maturation of both human and murine DCs, and the maturation state of both human and murine DCs is almost equivalent to that induced by lipopolysaccharide. Moreover, alkylating agent β-propiolactone-inactivated hPIV2ΔF (BPL-hPIV2ΔF) elicits DC maturation without viral replication/transcription. These results suggest that hPIV2ΔF may be a useful tool for vaccine therapy as a novel type of paramyxoviral vector, which is single-round infectious vector and has potential adjuvant activity.
Introduction
Viral vectors are superior to other systems because of their high efficiency of transduction into DCs as a part of the natural infection process and because of their strong DC-stimulatory activity (Jenne et al., 2001; Breckpot et al., 2004). Although several clinical candidates have been reported, their unexpected adverse effects (e.g., severe inflammation and carcinogenesis) or poor immunogenicity has similarly been pointed out (Thomas et al., 2003). Thus, to overcome these problems, it has been desired to develop novel types of viral vectors.
Human parainfluenza virus type 2 (hPIV2), a member of the genus Rubulavirus in the Paramyxoviridae family, is a respiratory pathogen responsible for acute respiratory tract illness, especially among children (Weinberg et al., 2009), but not in adults. The hPIV2 genome is nonsegmented and negative-stranded RNA, which encodes seven proteins. The viral genes line up in the following order: 3′-(leader)-NP-P/V-M-F-HN-L-(trailer)-5′. The P and L proteins bind to nucleocapsid (RNA encompassed by nucleoprotein [NP]) to form the ribonucleoprotein (RNP) complex. The hPIV2 envelope contains two glycoproteins, namely the HN and F proteins, and their cytoplasmic tails bind to the M protein, which lies on the viral envelope (Kawano et al., 2001; Henrickson, 2003). Of the viral envelope proteins, the F protein has fusion activity and it reciprocally works with the HN protein to achieve viral envelope-to-cell or cell-to-cell fusion (Tsurudome et al., 2008). Previous studies demonstrated that Sendai virus (mouse parainfluenza virus type 1) lacking the F gene (SeVΔF) can be self-replicative, but not infect neighboring cells. SeVΔF carrying an exogenous antigen gene efficiently transduces the antigen into DCs and induces antigen-specific immunity in vivo without any side effects (Li et al., 2000; Ferrari et al., 2004; Shibata et al., 2006; Yoneyama et al., 2007; Duan et al., 2009). These data suggest that SeVΔF is safe and effective for vaccine therapy in comparison with conventional viral vectors.
Using reverse genetics technology, we have developed hPIV2 lacking the F gene (hPIV2ΔF) from the recombinant hPIV2 cDNA (Ohtsuka et al., manuscript in preparation), and infectious hPIV2ΔF carrying the jellyfish gene encoding enhanced green fluorescent protein (EGFP) has been successfully recovered from a novel cell line stably expressing hPIV2 F protein. In addition, hPIV2ΔF infects a broad range of cells and shows nontransmissible infectivity, suggesting that hPIV2ΔF is a potentially promising novel paramyxovirus-based vector.
Here we demonstrate the fundamental immunogenic property of hPIV2ΔF, using human and murine DCs in vitro. hPIV2ΔF transduced an exogenous antigen into human DCs more efficiently compared with a conventional retroviral vector in transient assays. Although viral genomic copy numbers of hPIV2ΔF were significantly lower in murine DCs than in human DCs, similar maturation of both DC types was induced after infection. Moreover, DC maturation was found to be induced by preexisting hPIV2ΔF components without viral replication/transcription, implicating adjuvant activity by themselves.
Materials and Methods
Cell lines
The PLAT-gp packaging cell line, kindly provided by T. Kitamura (Division of Cellular Therapy, Institute of Medical Science, University of Tokyo, Tokyo, Japan), was cultured in Dulbecco's modified Eagle's medium (DMEM) (Nacalai Tesque, Kyoto, Japan) containing 10% heat-inactivated fetal bovine serum (GIBCO FBS; Invitrogen, Carlsbad, CA), 1% penicillin–streptomycin, and blasticidin (10 μg/ml). NIH3T3 cells were cultured in DMEM containing 10% heat-inactivated FBS and 1% penicillin–streptomycin. Vero/BC-F cells (a packaging cell line for hPIV2ΔF) (Ohtsuka et al., manuscript in preparation) were cultured in modified Eagle's medium (MEM; Sigma-Aldrich, St. Louis, MO) containing 10% heat-inactivated FBS.
Generation of CD14-positive monocyte-derived DCs
Monocyte-derived dendritic cells (MoDCs) were generated as described previously with minor modifications (Elkord et al., 2005). Peripheral blood was obtained from healthy adult volunteers with informed consent after approval by the Human Research Ethics Committee of the Mie University had been obtained (No. 2402). Peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation with Ficoll-Paque PLUS (GE Healthcare Life Sciences, Piscataway, NJ). CD14-positive monocytes were purified from PBMCs by positive selection with CD14 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). To generate immature MoDCs, CD14-positive cells were cultured in RPMI 1640 medium (Nacalai Tesque) containing 10% heat-inactivated FBS, 1% penicillin–streptomycin, 50 μM 2-mercaptoethanol, and human granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4 (each 50 ng/ml; both from Miltenyi Biotec) for 7 days at 37°C. Half the medium was replaced with fresh medium every 3 days. After 7 days, the purity of immature MoDCs (CD11c- and HLA-DR-positive cells) was >90% on flow cytometric analysis (data not shown).
Generation of bone marrow-derived DCs
Six- to 8-week-old female C57BL/6 and BALB/c mice were purchased from CLEA Japan (Tokyo, Japan). The animals were housed in a specific-pathogen-free condition facility in accordance with the guidelines of Mie University (Tsu, Japan). Bone marrow-derived dendritic cells (BMDCs) were generated as described previously with minor modifications (Inaba et al., 1992). BM cells were isolated from the femurs and tibias of mice, and to eliminate red blood cells (RBCs) BM cells were incubated in RBC lysis buffer (phosphate-buffered saline [PBS] containing 150 mM NH4Cl, 10 mM KHCO3, and 100 μM EDTA). After washing, BM cells were then cultured in RPMI 1640 medium containing 10% heat-inactivated FBS, 1% penicillin–streptomycin, 50 μM 2-mercaptoethnol, and mouse GM-CSF and IL-4 (each 20 ng/ml; both from Miltenyi Biotec) for 7 days at 37°C. Half the medium was replaced with fresh medium every 2 days. After 7 days, the purity of immature BMDCs (CD11c-positive cells) was >80% on flow cytometric analysis (data not shown).
Preparation of hPIV2ΔF and retrovirus carrying the EGFP gene
Generation of the plasmid pEGFP-hPIV2ΔF and recovery of the hPIV2ΔF/EGFP viruses after transfection into Vero/BC-F cells were performed as described elsewhere (Ohtsuka et al., manuscript in preparation). After 7 days, supernatant containing hPIV2ΔF/EGFP virus was recovered by centrifugation at 2000 rpm for 10 min, concentrated by ultracentrifugation at 28,000 rpm for 30 min at 4°C in a Beckman SW28 swinging bucket rotor with sterilized Beckman Ultra-Clear centrifuge tube (Beckman Coulter, Brea, CA), and then resuspended in PBS.
For inactivation of hPIV2ΔF/EGFP, β-propiolactone (BPL; Wako Pure Chemical Industries, Kyoto, Japan) was added to the purified supernatant at a final concentration of 0.05% (v/v) and incubated for 16 hr at 4°C. After incubation for 2 hr at 37°C, ultracentrifugation was then performed. Inactivation of hPIV2ΔF/EGFP was examined as follows: 1 ml of BPL-inactivated hPIV2ΔF/EGFP (BPL-hPIV2ΔF/EGFP) (1×106 plaque-forming units [PFU]) was added to 1 ml of medium of cultured Vero/BC-F cells, followed by incubation for 5 days, and 1 ml of culture supernatant was added to new Vero/BC-F cells to cultivate for an additional 5 days. EGFP expression in the cells was analyzed by fluorescence microscopy. A hemagglutination (HA) assay was performed as previously described (Tsurudome et al., 2008). Briefly, each 100 μl of hPIV2ΔF/EGFP and BPL-hPIV2ΔF/EGFP stock was serially 2-fold diluted in PBS and added into a U-type 96-well plate. After 100 μl of 1% guinea pig erythrocytes was added into each well, the plate was incubated for 2 hr at 4°C.
For hPIV2ΔF/EGFP titration, hPIV2ΔF/EGFP genomic RNA was isolated from hPIV2ΔF/EGFP viral stock, using a High Pure viral RNA kit (Roche Applied Science, Indianapolis, IN), according to the manufacturer's instructions. One microgram of the isolated viral RNA was reverse transcribed, using an hPIV2 NP gene-specific primer (5′-CAACATTCAATGAATCAGT-3′) and SuperScript II reverse transcriptase (Invitrogen). Subsequently, quantitative real-time PCR was performed as described later.
A replication-incompetent vesicular stomatitis virus G glycoprotein (VSV-G)-pseudotyped retrovirus (ReV) was generated as described previously (Suzuki et al., 2012) with minor modifications. Briefly, PLAT-gp cells (Kitamura et al., 2003), which stably express gag-pol of Moloney murine leukemia virus, were grown to about 50% confluency and the medium was replaced with DMEM containing 10% heat-inactivated FBS and no blasticidin. PLAT-gp cells were transfected with plasmid encoding the VSV-G envelope protein driven by the cytomegalovirus (CMV) promoter and a retroviral vector pMX-EGFP, which lacks gag-pol and env (Onishi et al., 1996), using FuGENE 6 (Roche Applied Science) according to the manufacturer's instructions. The medium was replaced every 24 hr and the supernatant was harvested 48, 72, 96, 120, and 144 hr after transfection. The supernatant harvested at each time point was filtered and concentrated by ultracentrifugation at 16,500 rpm for 90 min at 4°C. The virus was then resuspended in PBS and stored at 4°C. Viral titer was determined by flow cytometry as described below. NIH3T3 cells were plated in a 12-well plate in 2 ml of DMEM with 10% heat-inactivated FBS. After the viral stock was serially 10-fold diluted, NIH3T3 cells were infected with the virus in the presence of Polybrene (8 μg/ml; Sigma-Aldrich) for 6 hr at 37°C, followed by replacement with fresh medium. At 48 hr postinfection, the viral titer was calculated on the basis of the number of EGFP-positive cells as determined by flow cytometry.
Viral transduction into DCs
MoDCs and BMDCs (2×105 cells per well) were plated in a 24-well plate and infected either with hPIV2ΔF/EGFP or ReV/EGFP at various multiplicities of infection (MOIs) for 48 hr. DCs were harvested and washed with FACS (fluorescence-activated cell-sorting) buffer (PBS containing 2% FBS). EGFP expression in DCs was analyzed by flow cytometry.
Flow cytometry
MoDCs and BMDCs unstimulated or stimulated either with lipopolysaccharide (LPS; Sigma-Aldrich), hPIV2ΔF/EGFP, or BPL-hPIV2ΔF/EGFP for 48 hr were harvested, and Fc receptors were blocked with TruStain FcX (BioLegend, San Diego, CA) in FACS buffer for 5 min. DCs were then stained with respective antibodies and washed with FACS buffer. The antibodies used were as follows: phycoerythrin (PE)-conjugated anti-CD40, CD80, CD86, HLA-A, and H-2Kb antibodies, peridinin chlorophyll protein (PerCP)/Cy5.5-conjugated anti-HLA-DR and I-A/I-E antibodies, and allophycocyanin (APC)-conjugated anti-CD11c antibody. PE-, PerCP/Cy5.5-, and APC-conjugated subclass-matched antibodies were used as the isotype controls, respectively. All antibodies used for flow cytometry were purchased from BioLegend. Data were acquired on a FACSCalibur (BD Biosciences, San Jose, CA) and analyzed with CellQuest software (BD Biosciences).
ELISA
Culture supernatant was harvested from MoDCs and BMDCs unstimulated or stimulated either with LPS, hPIV2ΔF/EGFP, or BPL-hPIV2ΔF/EGFP for 48 hr and stored at −80°C until use. Human and mouse IL-6 and IL-12p40 were measured according to the manufacturer's instructions (eBioscience, San Diego, CA).
Quantitative real-time PCR
To determine hPIV2ΔF/EGFP titer and viral genomic RNA copy numbers, real-time PCR was performed to amplify hPIV2 NP-intergenic-P sequences. Briefly, one-tenth of the reaction solution containing the viral cDNA product was added to TaqMan gene expression master mix (Applied Biosystems/Invitrogen) containing the probe and primers, giving a final reaction volume of 20 μl. PCR assay was performed in triplicate on a StepOnePlus real-time PCR system (Applied Biosystems/Invitrogen), according to the manufacturer's instructions. StepOne software v2.1 (Applied Biosystems/Invitrogen) was used to analyze the real-time PCR data. Cycle conditions were set as follows: initial template denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 sec, and annealing/extension at 60°C for 1 min. Data were expressed as the numbers of hPIV2ΔF/EGFP NP gene copies. Primers used to measure hPIV2 genomic copy numbers were 5′-ACACACTCATCCAGACAAATCAAAC-3′ and 5′-TGTGGAGGTTATCTGATCACGAA-3′. The probe used was 5′-AAGCACCGGATTTCTAACCCGTCCG-3′.
To detect viral transcripts, total RNA was extracted from MoDCs or BMDCs infected with hPIV2ΔF/EGFP for 48 hr, using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNAs were synthesized from the same amounts of total RNA, using oligo(dT)20 primer and SuperScript II reverse transcriptase. Subsequently, real-time PCR was performed with specific primers for the hPIV2 NP gene and for the human or murine GAPDH gene. Each reaction contained 10 μl of 2× Power SYBR green master mix (Applied Biosystems/Invitrogen), forward and reverse primers, and 2 μl of the cDNA product. Real-time PCR was performed in triplicate as described previously. Primers used to measure the transcripts of hPIV2 NP, human GAPDH, and murine GAPDH were 5′-ACCAGTATCAGTAGCAAAGC-3′ and 5′-TAGCGGTTTGCTAGCAAAGATC-3′, 5′-GTGAAGGTCGGAGTCAACGGA-3′ and 5′-GGTGAAGACGCCAGTGGACTC-3′, and 5′-CCCTTATTGACCTCAACTACATGGT-3′ and 5′-GAGGGGCCATCCACAGTCTTCTG-3′, respectively. Cycle conditions were described previously. The levels of hPIV2ΔF/EGFP NP transcripts in each species with increased MOIs were measured by the ΔΔC T method and normalized by the expression of each GAPDH reference gene. Data are shown as the relative levels of NP gene transcripts of hPIV2ΔF/EGFP.
Statistical analyses
All data are shown as means±SD. Significant differences among groups were evaluated by one-way analysis of variance (ANOVA), followed by the Bonferroni test with R software for Windows. p<0.05 was considered statistically significant.
Results
Transduction of hPIV2ΔF and ReV into MoDCs
To assess the transduction efficiency of hPIV2ΔF/EGFP, we infected MoDCs with hPIV2ΔF/EGFP or ReV/EGFP at various MOIs for 48 hr. Much stronger EGFP expression in hPIV2ΔF/EGFP-infected MoDCs was observed by fluorescence microscopy, compared with that in ReV/EGFP-infected MoDCs in similar replication-incompetent single-round infectious systems (Fig. 1A). The percentages of EGFP-positive MoDCs infected with hPIV2ΔF/EGFP were 59.9, 73.1, and 77.8% at MOIs of 25, 50, and 100, respectively, whereas those infected with ReV/EGFP were 4.4, 11.2, and 25.2% at MOIs of 25, 50, and 100, respectively (Fig. 1B). Moreover, the mean fluorescence intensity (MFI) of EGFP, reflecting the amount of EGFP protein, in MoDCs infected with hPIV2ΔF/EGFP was much higher than that with ReV/EGFP (Fig. 1C). These data indicate that hPIV2ΔF transduced an exogenous gene into MoDCs more efficiently than ReV at 48 hr after infection, unveiling the great potential of hPIV2ΔF for transient gene delivery.

Comparison of hPIV2ΔF and a retroviral (ReV) vector in terms of EGFP transduction efficiency in MoDCs at various MOIs 48 hr after infection.
Comparison of replication efficiency of hPIV2ΔF in MoDCs and BMDCs
Because previous studies demonstrated that replication/transcription of some hPIVs was suppressed in L929 murine fibrosarcoma cells (Ito et al., 1989; Komada et al., 2000), we used primary murine BMDCs to investigate whether the replication efficiency of hPIV2ΔF/EGFP and the expression level of EGFP were reduced. As shown in Fig. 2A, hPIV2ΔF/EGFP genomic RNA copy numbers were 3.5-, 5.9-, and 4.7-fold lower in murine BMDCs (C57BL/6) than in human MoDCs at MOIs of 25, 50, and 100, respectively, and no significant difference was observed between the mouse strains (C57BL/6 and BALB/c). This finding suggests that viral uptake and/or genomic RNA replication of hPIV2 in murine cells is less efficient than in human cells. We next investigated NP transcripts in BMDCs and MoDCs at each MOI, using the ΔΔC T method of real-time PCR and using each species of GAPDH as a reference gene (Fig. 2B). The NP transcripts increased MOI dependently in both species of cells. Although we could not directly compare the quantity of NP transcripts between murine and human cells, the real-time PCR analysis using the same amount of the cDNA without reference genes showed similar reduction in NP transcripts in murine cells as in NP genomic copy numbers (data not shown). Next, we investigated the transduction efficiencies of hPIV2ΔF/EGFP into BMDCs (C57BL/6). The percentages of EGFP-positive BMDCs were increased in an MOI-dependent manner and about 70% of BMDCs expressed EGFP at an MOI of 100 (Fig. 2C), but a significantly lower amount of EGFP was detected in hPIV2ΔF/EGFP-infected BMDCs by fluorescence microscopy (Fig. 2D) and by flow cytometry (data not shown) compared with those in human MoDCs. Consistent with the previously reported findings (Ito et al., 1989; Komada et al., 2000), the high MOI of hPIV2ΔF/EGFP was required to obtain appropriate transduction efficiency in the study using murine BMDCs to overcome the markedly reduced replication/transcription of hPIV2.
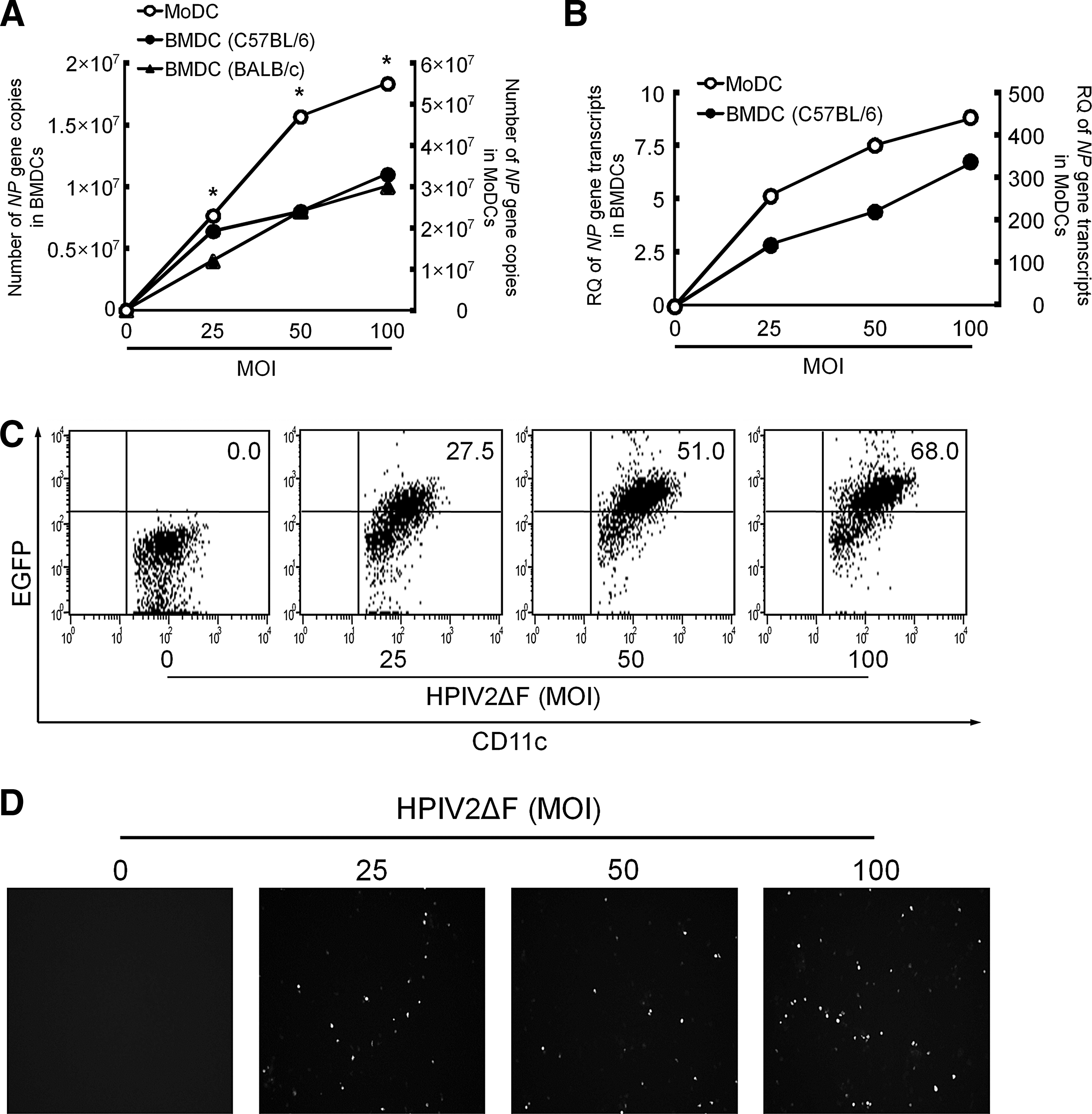
HPIV2ΔF/EGFP shows low replication/transcription capacity in BMDCs.
Maturation states of MoDCs and BMDCs infected with hPIV2ΔF/EGFP
In in vivo DC-targeted vaccines and ex vivo DC therapies, not only transduction of vaccine antigens into DCs, but also DC maturation is pivotal to prime T cells. Thus, we investigated the effects of hPIV2ΔF/EGFP on DC maturation. Figure 3A shows surface expression of CD40, CD86, HLA-A, and HLA-DR on MoDCs stimulated with either hPIV2ΔF/EGFP or LPS, a well-known DC stimulator. In MoDCs, hPIV2ΔF/EGFP stimulation significantly increased the expression levels of their maturation markers, which were nearly comparable to those induced by LPS, compared with unstimulated MoDCs. High amounts of IL-6 and IL-12p40 were secreted from hPIV2ΔF/EGFP-stimulated MoDCs (Fig. 3B). These experiments were also performed in BMDCs (hereafter only C57BL/6 mice were used). Similar to MoDCs, hPIV2ΔF/EGFP-stimulated BMDCs significantly increased the expression of maturation markers and the amounts of cytokines (Fig. 3C and D). These results indicate that hPIV2ΔF/EGFP has strong stimulatory activity for both types of DCs. Interestingly, although the genomic copy numbers of hPIV2ΔF/EGFP in BMDCs were about 3.5 times lower than in MoDCs at an MOI of 25 (Fig. 2A), the expression levels of maturation markers on BMDCs and MoDCs after viral infection, relative to those after LPS stimulation, were nearly equal. These results suggest that DC maturation induced by hPIV2ΔF/EGFP depended not only on replication of the viral genome or de novo synthesis of viral proteins but also on preexisting hPIV2ΔF components (e.g., by binding of the envelope proteins to host receptors or by recognizing the original viral genome).
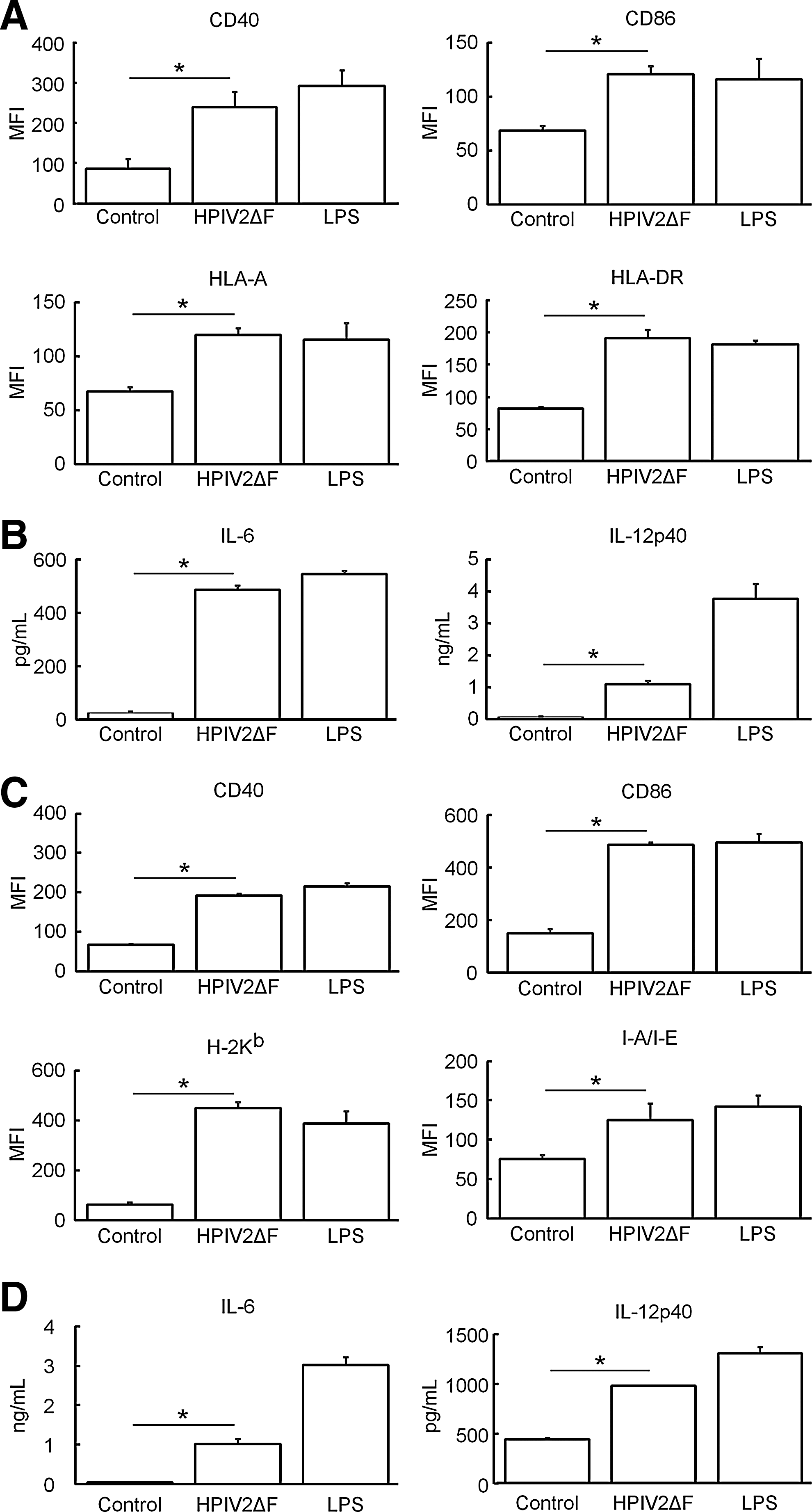
HPIV2ΔF/EGFP-induced DC maturation and maturation state of DCs were comparable to those induced by LPS. MoDCs
Effects of replication-defective hPIV2ΔF/EGFP on BMDC maturation
On the basis of the data described previously, we next investigated whether DC maturation was induced by preexisting viral components without replication/transcription. First, to generate replication-defective hPIV2ΔF/EGFP, we chemically inactivated hPIV2ΔF/EGFP. Because β-propiolactone (BPL) is a chemical reagent that has alkylating activity against adenosine and guanosine of viral genomic RNA, and BPL treatment does not affect viral infectivity (Desbat et al., 2011; Budimir et al., 2012), we chose this inactivation method. Inactivation of hPIV2ΔF/EGFP was confirmed by fluorescence microscopy and no cytopathic effects were observed (Fig. 4A and data not shown), Furthermore, no functional loss of envelope proteins was observed by HA assay (Fig. 4B). Figure 4C shows a comparison of DC maturation states induced by live hPIV2ΔF/EGFP and BPL-inactivated hPIV2ΔF/EGFP (BPL-hPIV2ΔF/EGFP). Although BPL-hPIV2ΔF/EGFP exhibited significantly reduced DC-stimulatory activity compared with live hPIV2ΔF/EGFP, the expression levels of maturation markers on BMDCs stimulated with BPL-hPIV2ΔF/EGFP were significantly enhanced, compared with those on unstimulated BMDCs. Similar findings were also observed in cytokine release (Fig. 4D). These results suggest that preexisting hPIV2ΔF components per se have DC-stimulatory activity leading to DC maturation, albeit viral RNA synthesis may be required to induce DC maturation effectively.
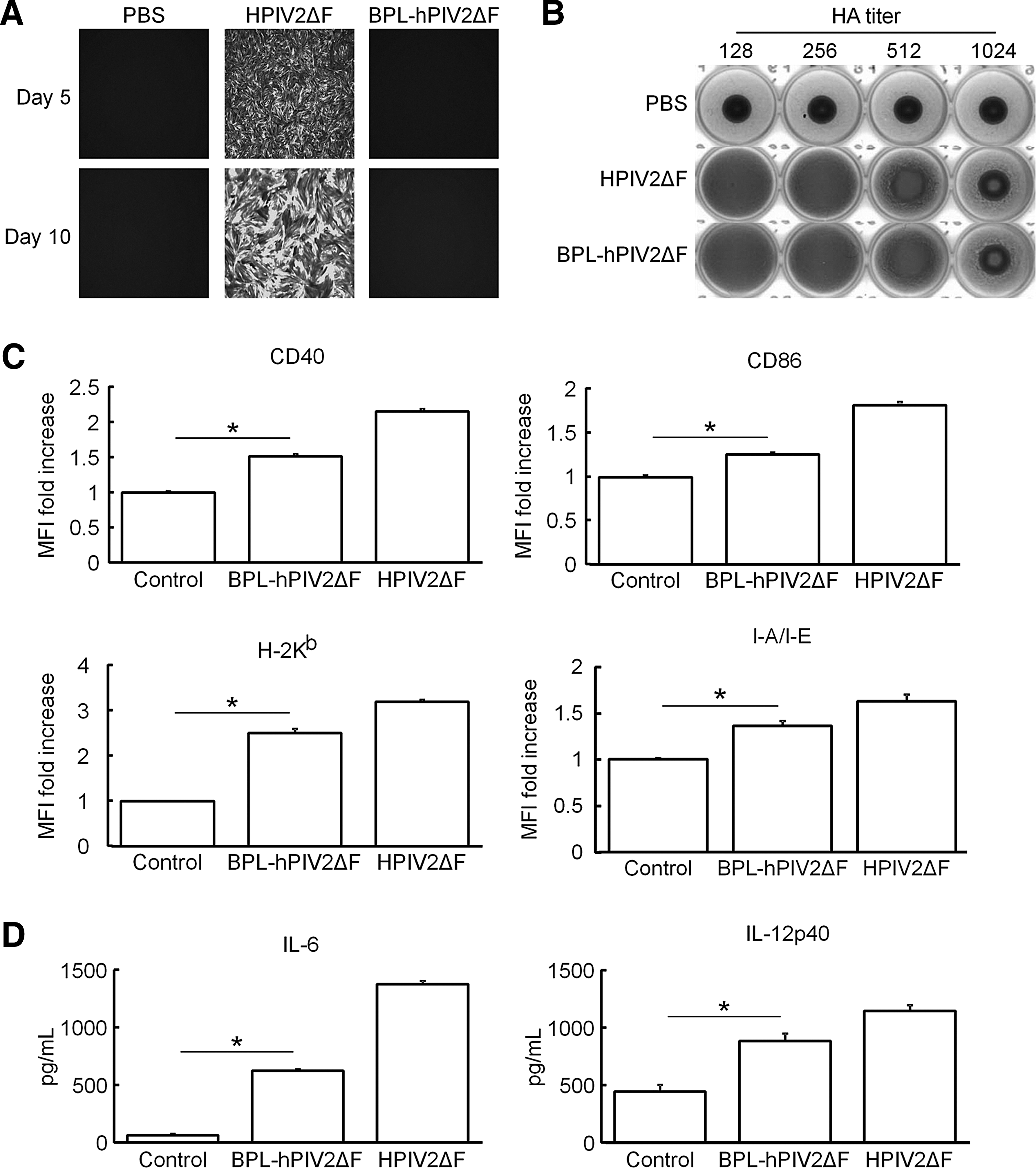
Effects of β-propiolactone (BPL)-inactivated hPIV2ΔF/EGFP on DC maturation. BMDCs were unstimulated or stimulated with either hPIV2ΔF/EGFP or BPL-hPIV2ΔF/EGFP at an MOI of 25 for 48 hr.
Discussion
Previous epidemic data disclosed that about 60% of all hPIV2 infections occurred in children younger than 5 years old, and that the infection rate of hPIV2 was less frequent than that of other hPIVs (Henrickson, 2003). In addition, hPIV2 naturally reinfects healthy adults throughout life (Hall, 2001), which makes it likely that hPIV2-based vectors could be safely administered and escape from responses to the viral backbone. In the present study, we first investigated the transduction efficiency of hPIV2ΔF into human MoDCs. The results showed that hPIV2ΔF at an MOI of 25 more efficiently transduced the EGFP gene into MoDCs in comparison with a conventional retroviral (ReV) vector at 48 hr postinfection. Whereas the transduction efficiency of the ReV vector into nondividing cells, such as DCs, is generally low (Szabolcs et al., 1997; Jenne et al., 2001; Tan et al., 2005), other commonly used vectors, such as an adenoviral vector or an adeno-associated viral vector, require an MOI greater than 100 to achieve sufficient transduction into DCs (Ponnazhagan et al., 2001; Mercier et al., 2003). Furthermore, the entire life cycle of hPIV2 takes place completely out of the nucleus, which excludes the risk of host genome alterations (Henrickson, 2003). Altogether, hPIV2ΔF could be useful as a novel cytoplasmic RNA viral vector to develop recombinant vaccines against various diseases.
Next, we compared the replication/transcription states of hPIV2ΔF in murine BMDCs and human MoDCs. Consistent with the previous reports (Ito et al., 1989; Komada et al., 2000), viral genomic copy numbers and viral transcripts of hPIV2ΔF in BMDCs were significantly lower than those in MoDCs. However, hPIV2ΔF/EGFP transduction of BMDCs showed MOI-dependent propagation, and about 70% of BMDCs expressed EGFP at an MOI of 100. Studies have revealed that the V protein of hPIV2 inhibited interferon (IFN) production and signaling by degrading human STAT2, but not murine STAT2 (Nishio et al., 2001, 2005; Parisien et al., 2001). Furthermore, V-knockout hPIV2 induced higher IFN production and viral replication was restricted in the human cell line (Schaap-Nutt et al., 2010). Although IFN signaling was not investigated in our study, similar mechanisms may be involved in less propagation of hPIV2ΔF in primary murine cells, compared with human cells.
DC maturation is an essential event to trigger immunity. In presenting antigen to naive T cells, costimulatory molecule-related signals from DCs are important for the fate of naive T cells in addition to the signals through the MHC–T cell receptor (TCR) complex (Guermonprez et al., 2002; de Jong et al., 2005). Moreover, T cell polarization is controlled by a number of cytokines from DCs, and they reciprocally regulate the differentiation of naive T cells into each T cell subset (Watford et al., 2003; Feili-Hariri et al., 2005; Zhou et al., 2009). This study demonstrated that hPIV2ΔF stimulation sufficiently increased the expression of MHCs and costimulatory molecules on MoDCs and BMDCs to a level comparable with LPS stimulation. IL-6 and IL-12p40 are proinflammatory cytokines that regulate T cell polarization (Egwuagu, 2009; Kimura and Kishimoto, 2010; Prochazkova et al., 2012). Secretion of both IL-6 and IL-12p40 from MoDCs and BMDCs was significantly increased by hPIV2ΔF stimulation, suggesting functional maturation of DCs. Interestingly, despite the low replication/transcription of hPIV2ΔF/EGFP in BMDCs (Fig. 2A and B), the maturation state of BMDCs was nearly equal to that of MoDCs, in each comparison with LPS stimulation of each cell type, implying that DC maturation was triggered by preexisting hPIV2ΔF components without viral replication/transcription events.
Finally, we genomically inactivated hPIV2ΔF by BPL treatment (BPL-hPIV2ΔF) to investigate whether DC maturation was induced by preexisting hPIV2ΔF components alone. BPL completely inactivated hPIV2ΔF replication/transcription without affecting viral envelope function, suggesting that BPL is a useful viral inactivator in developing safer vaccines. Although the DC-stimulatory activity of the BPL-hPIV2ΔF vehicle itself was attenuated in comparison with live hPIV2ΔF, BPL-hPIV2ΔF still possessed DC-stimulatory activity without viral replication/transcription. In fact, hPIV2ΔF inactivated with BPL at a lower concentration (0.012%, v/v), despite the lack of viral replication/transcription, markedly enhanced its DC-stimulatory activity compared with virus inactivated with 0.05% BPL (data not shown). These findings suggest that hPIV2ΔF, the adjuvanticity of which is still maintained after genomic inactivation, would be useful as a safe vector for recombinant vaccines, and the strength of BPL inactivation might influence viral adjuvanticity, probably by affecting intraviral structure.
Viral inactivation with chemical reagents (e.g., formalin) or physical treatments (e.g., heat and ultraviolet [UV]) is likely desirable for developing safe vaccines, and some inactivated vaccines are currently used. However, vaccine efficacy is still controversial from immunological points of view (Delgado et al., 2009).
In DC monitoring of viral infection, two major sensors exist. One is the family of Toll-like receptors (TLRs), and the other is the family of cytosolic RNA helicases such as retinoic acid-inducible gene I (RIG-I) or melanoma differentiation-associated gene 5 (MDA5) (Meylan et al., 2006; Pichlmair et al., 2006; Barral et al., 2009). In these sensors, murine myeloid DCs detect viral infection by recognizing uncapped 5′-triphosphates of viral single-stranded RNA (ssRNA) or double-stranded RNA (dsRNA) through cytosolic RNA helicases (Kato et al., 2005). Our finding predicts that hPIV2ΔF-induced DC maturation occurs in the absence of viral replication/transcription, suggesting that alternative viral sensors, for example, a TLR- or autophagy-mediated signaling pathway (Bieback et al., 2002; Shirey et al., 2010; Morris et al., 2011), may be involved in DC maturation subsequent to hPIV2ΔF infection. A previous report using Sendai virus (SeV) showed that UV-inactivated SeV (UV-SeV) induced no DC maturation (Okano et al., 2011), suggesting that viral replication/transcription is essential for DC maturation. On the other hand, other reports demonstrated that SeV replication/transcription was not required for the expression of DC maturation markers, whereas it was required for cytokine production (López et al., 2003). Also, UV-SeV induced DC maturation at a level similar to that induced by live SeV (Kurooka and Kaneda, 2007). The discrepancy between these results has not been defined.
In summary, we demonstrated that nontransmissible hPIV2ΔF is a promising viral vector for delivering antigen into DCs in vitro. In addition, their DC-stimulatory activity was not abrogated in the absence of viral replication/transcription. These data indicate that hPIV2ΔF carrying the exogenous gene and BPL-hPIV2ΔF carrying the exogenous protein on the viral surface may be new options for viral vector-based vaccine therapy for human diseases.
Footnotes
Acknowledgments
The authors thank Dr Toshio Kitamura for PLAT-gp cells and Dr. Hiroyuki Miyoshi for pCMV-VSV-G. The authors are grateful to Dr. Yasuhiro Yasutomi for valuable discussion. This work was supported by a Grant-in-Aid for the Regional Innovation R&D Program by the Ministry of Economy, Trade, and Industry of Japan.
Author Disclosure Statement
Some of the authors are patent applicants for BPL-hPIV2ΔF (K.H., M.F., J.O., M.K., and T.N.) and Vero cells stably expressing hPIV2 F protein (M.F., J.O., M.K., and T.N.). M.F. is a founder of Biocomo. M.F., M.K., and T.N. have shares of stock in Biocomo. J.O. is an employee of Biocomo.