
Abstract
Refractoriness to platelet (PLT) transfusion caused by alloimmunization against HLA class I antigens constitutes a significant clinical problem. Thus, it would be desirable to have PLT units lacking HLA antigens on the cell surface. Previously, we showed that the generation of functional HLA class I-silenced (HLA-universal) PLTs from CD34+ cells, using a short hairpin RNA (shRNA) to target β2-microglobulin (β2m) transcripts, is feasible. Here, we assessed the capacity of HLA-silenced PLTs to escape HLA antibody-mediated cytotoxicity in vitro and in vivo. Generation of megakaryocytes (MKs) and PLTs was performed by thrombopoietin-mediated differentiation of HLA-silenced CD34+ cells within 10 days. Lymphocytotoxicity and antibody-dependent cellular cytotoxicity (ADCC) reporter assays using anti-HLA antibodies and a mouse model for PLT refractoriness were used to assess the immune-evasion capability of HLA-universal MKs and PLTs. To mimic PLT refractoriness in vivo, NOD/SCID/IL-2Rγc–/– mice were injected with specific anti-HLA antibodies followed by the infusion of 1×106 HLA-universal MKs. In vivo PLT generation was evaluated by flow cytometry using anti-CD42a and CD61 antibodies. Cells expressing a nonspecific shRNA were used as control. Lymphocytotoxicity and ADCC reporter assays showed that HLA silencing protects MKs against HLA antibody-mediated complement-dependent and cell-mediated cytotoxicity. In lymphocytotoxicity assays, 80–90% of HLA-expressing MKs but only 3% of HLA-silenced MKs were lysed. In the circulation of mice, HLA-expressing and HLA-silenced MKs showed PLT production in the absence of anti-HLA antibodies, with human PLT frequencies of up to 0.5% within the PLT population. However, in presence of anti-HLA antibodies HLA-expressing MKs were rapidly cleared from the circulation of mice, whereas HLA-silenced MKs escaped HLA antibody-mediated cytotoxicity and produced PLTs that were detectable up to 11 days. Our data show that HLA-silenced PLTs are efficiently protected against HLA antibody-mediated cytotoxicity and prevent PLT refractoriness in vivo. Provision of HLA-silenced PLTs may become an important component in the management of patients refractory to PLT transfusion.
Introduction
P
The use of stem cells as a source for in vitro PLT production emerged as a promising strategy to solve other limitations of PLT transfusion such as donor shortage, limited shelf-life, and risk of infection (Reems, 2011). Successful production of functional PLTs has already been achieved from CD34+ hematopoietic progenitor cells (HPCs), human embryonic stem cells, and induced pluripotent stem cells (Matsunaga et al., 2006; Takayama et al., 2010; Lu et al., 2011; Reems, 2011). Large-scale in vitro production of PLTs from genetically modified stem cells or HPCs would allow the generation of genetically engineered designer PLTs for future clinical application. However, the allogeneic origin of the PLT cell source remains an obstacle to the application of such products.
In one study, we showed that the generation of HLA class I-silenced (HLA-universal) PLTs from CD34+ progenitor cells, using an RNA interference (RNAi) approach to target β2-microglobulin (β2m) transcripts, is feasible. Also, HLA-universal PLTs were shown to be functionally similar to blood-derived PLTs (Figueiredo et al., 2010).
In the current study, we evaluated the ability of HLA-universal PLTs to escape HLA alloantibody-mediated cytotoxicity, which is the major cause of immune-mediated PLT refractoriness (Sacher et al., 2003; Hod and Schwartz, 2008). In general, alloantibodies are thought to mediate PLT destruction by two mechanisms: (1) antibody-mediated phagocytosis and/or (2) antibody-mediated complement-dependent cytotoxicity (CDC) (Handin and Stossel, 1974; Klein and Blajchman, 1982; Semple et al., 2007; Hod and Schwartz, 2008). It is assumed that the main mechanism of PLT clearance is the ingestion of antibody-coated PLTs by phagocytic cells through the Fc receptor (Lim et al., 2002). Both mechanisms lead to the rapid destruction of allogeneic PLTs in refractory patients and hence impede or even abolish the therapeutic effect of PLT transfusion (Hod and Schwartz, 2008). In this study we show that HLA-universal megakaryocytes (MKs), the direct precursors of PLTs, and PLTs are protected from antibody-mediated cytotoxicity both in vitro and in vivo.
Materials and Methods
CD34+ progenitor cell isolation and culture
Peripheral blood CD34+ progenitor cells were isolated from apheresis products of eight healthy HLA-typed HPC donors. Informed consent was obtained from all donors as approved by the local ethics committee of Hannover Medical School (Hannover, Germany). The donors received recombinant granulocyte-colony stimulating factor (G-CSF, 10 μg/kg body weight) (lenograstim; Chugai Pharma, London, UK) over 4 days to mobilize HPCs into the peripheral blood. CD34+ cell isolation was performed with a human CD34 microbead kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Isolated CD34+ cells were cultured for 24 hr in TFSI medium, consisting of RPMI 1640 medium (BioWhittaker/Cambrex, Hessisch Oldendorf, Germany) supplemented with 5% human AB serum (C.C. Pro, Neustadt, Germany), thrombopoietin (TPO; 100 ng/ml), Fms-like tyrosine kinase 3-ligand (Flt3-L; 100 ng/ml), stem cell factor (SCF; 100 ng/ml), and interleukin (IL)-6 (50 ng/ml) to keep the cells undifferentiated. All recombinant cytokines were purchased from PeproTech (Rocky Hill, NJ).
Lentiviral constructs and vector production
A lentiviral vector was used to stably express a short-hairpin RNA (shRNA) for β2m silencing as previously described (Figueiredo et al., 2010). Briefly, expression cassettes encoding an shRNA targeting β2m transcripts (shβ2m) (Supplementary Table S1; supplementary data are available online at
Lentiviral vector transduction
CD34+ cells (3×105 per well) in TFSI medium were seeded into RetroNectin (Takara, Otsu, Japan)-coated 24-well plates and infected at a multiplicity of infection of 20 in the presence of protamine sulfate (8 μg/ml; Sigma-Aldrich, Steinheim, Germany). After 16 hr, the CD34+ cells were washed with fresh TFSI medium. Transduction efficiency was calculated by assessing the percentage of GFP-expressing cells.
Generation of HLA-universal MKs and PLTs
After transduction, 5×105 nontransduced, shNS- transduced, and shβ2m-transduced progenitor cells were cultured in RPMI 1640 supplemented with 5% AB serum, TPO (100 ng/ml), and Flt3-L (20 ng/ml) for 48 hr. Subsequently, CD34+ progenitor cells expressing GFP as a marker for successful transduction were isolated by a flow cytometry–based cell-sorting system (MoFlo or XDP sorter; Beckman Coulter). GFP-positive cells were maintained in culture in the presence of TPO (100 ng/ml) for an additional 8–10 days (days 10–12 of differentiation) to allow the differentiation into MKs and the release of PLTs. To assess the effect of irradiation on the capacity of mature MKs to produce PLTs, some MK populations were irradiated with a dose of 30 Gy 24 hr before characterization.
Characterization of HLA-universal megakaryocytes, proplatelets, and platelets
Flow cytometry
The in vitro generation of HLA-universal MKs and PLTs was assessed as previously described (Figueiredo et al., 2010). Briefly, MKs and PLTs were identified by flow cytometry after staining with allophycocyanin (APC)/cyanine 7 (Cy7)-conjugated anti-CD41 (BioLegend, London, UK) and phycoerythrin (PE)-labeled anti-CD42a (BD Biosciences) or anti-CD42a–PE and APC-conjugated anti-CD61 (BioLegend) antibodies, respectively. The PLT gate was adjusted according to forward scatter (FSC) and side scatter (SSC) profiles of blood-derived PLTs from healthy volunteers. HLA class I expression of shNS- and shβ2m-transduced MKs was measured with the APC-labeled w6/32 antibody (AbD Serotec, Düsseldorf, Germany).
In addition, polyploidization of MKs was assessed by propidium iodide (PI; Sigma-Aldrich) staining. Therefore, 1×105 cells were stained with anti-CD41–APC/Cy7, washed twice with phosphate-buffered saline (PBS), and fixed/permeabilized with Cytofix/Cytoperm (BD Biosciences). Cells were then stained with PI solution (10 μg/ml in PBS) containing RNase A (10 U/ml; Sigma-Aldrich). DNA content of CD41+ cells was analyzed by flow cytometry.
Real-time polymerase chain reaction
β2m mRNA levels of shNS- and shβ2m-transduced MKs were analyzed as previously described (Figueiredo et al., 2010). Briefly, total RNA was isolated from cells derived from the CD34+ progenitor cells (RNeasy mini kit; Qiagen, Hilden, Germany) and reverse transcribed to cDNA, using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Darmstadt, Germany). Real-time polymerase chain reaction (PCR) was used to measure β2m transcript levels. The constitutively expressed gene encoding glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as reference standard for normalization of mRNA levels. All real-time PCR analyses were performed in triplicate.
Fluorescence microscopy
The morphology of MKs was assessed by fluorescence microscopy. Imaging of viable GFP-positive proplatelet-bearing MKs was performed with an Olympus IX81 microscope (Olympus, Hamburg, Germany) with a GFP filter set and a ×40 objective. Images were acquired with a digital B/W camera (Olympus) and analyzed with CellM image software (Olympus).
Imaging of MK polyploidy was performed on GFP-positive MKs (2×105 cells per well), which were incubated in poly-
Platelet activation assays
PLTs generated from CD34+ progenitor cells were stimulated with adenosine 5′-diphosphate (ADP, 25 mmol/liter) for 10 min and immediately fixed with Cytofix (BD Biosciences). To assess PLT activation by flow cytometry, PLTs were stained with fluorescein isothiocyanate (FITC)-labeled PAC-1 antibody specific for activated glycoprotein IIb–IIIa (GPIIb–IIIa; BD Biosciences).
Complement-dependent cytotoxicity assay
Lymphocytotoxicity tests were used to assess CDC in vitro. Therefore, 3×103 viable shNS- or shβ2m-expressing MKs were plated in 1 μl of PBS in Terasaki trays (Nunc, Roskilde, Denmark) together with 1 μl of complement-binding donor-specific anti-HLA antibodies (One Lambda, Canoga Park, CA) diluted 1:10 with PBS. One microliter of PBS or nonspecific anti-HLA antibody (One Lambda) was used as negative control and 1 μl of Kontroll-HLA pos. reagent (Bio-Rad, Dreieich, Germany) was used as positive control. Trays were incubated for 30 min at room temperature, followed by the addition of 5 μl of rabbit complement (Bio-Rad). After 60 min of incubation at room temperature, 5 μl of FluoroQuench dye (One Lambda) was added to stain and fix cells. After 15 min cell lysis was analyzed with an Olympus IX81 fluorescence microscope (Olympus) with FITC and Texas Red filters to detect viable cells (green) and lysed cells (orange). For evaluation, samples were blinded and frequencies of lysed cells were estimated by two independent individuals.
Antibody-dependent cellular cytotoxicity reporter assay
An antibody-dependent cellular cytotoxicity (ADCC) reporter bioassay kit (Promega, Madison, WI) was used to assess Fc receptor-mediated ADCC in vitro. The assay was performed according to the manufacturer's instructions. Briefly, 12.5×103 viable shNS- or shβ2m-expressing MKs were incubated with donor-specific anti-HLA antibody (1 μg/ml; One Lambda), anti-HLA-ABC (purified w6/32; AbD Serotec), or nonspecific HLA antibody (One Lambda) and 75×103 ADCC bioassay effector cells (effector-to-target ratio, 6:1). All antibodies used were IgG isotype antibodies. ADCC bioassay effector cells are engineered Jurkat cells that stably express the FcγRIIIa (immunoglobulin-γ Fc region receptor IIIA) as well as an NFAT (nuclear factor of activated T cells) response element driving expression of firefly luciferase for quantification of ADCC activity. After 6 hr of incubation, luciferase assay reagent was added to the cells and luciferase activity in the effector cells was quantified by luminescence readout.
Mouse model for platelet refractoriness
Animal treatment
To assess the functionality of PLTs in vivo we used a mouse model of PLT refractoriness (Supplementary Fig. S1). For this purpose, specific anti-HLA antibodies (3 μg/g of body weight; One Lambda) were injected intravenously into 8- to 10-week-old NOD/SCID/IL-2Rγc–/– mice and allowed to distribute within the circulation of mice for 20 min. Subsequently, 1×106 shNS- or shβ2m-expressing MKs (day 10 of differentiation) were infused into the mice via the tail vein. Mice that received anti-HLA antibody only, MKs only, or MKs in presence of nonspecific anti-HLA antibodies were used as controls. Four animals were treated per condition. All mice were maintained under specific pathogen-free conditions in the animal facility of the Helmholtz Center for Infection Research (Braunschweig, Germany). All animal experiments in this study have been performed in agreement with the local government of Lower Saxony (Germany).
Detection of human MKs and PLTs in mouse peripheral blood
One, 3, 6, and 11 days posttransplantation, 40 μl of peripheral blood (PB) was collected from all mice. Red blood cells (RBCs) in PB were lysed with RBC lysis buffer (BD Biosciences). PB cells were then washed with PBS and treated with an anti-mouse Fc receptor-blocking reagent (Miltenyi Biotec). Human MKs and PLTs in mouse PB were detected by flow cytometry on staining with anti-human CD41–APC/Cy7 and CD42a–PE or CD42a–PE and CD61–APC, respectively. For biodistribution assays, mice that received human cells were sacrificed 13 days after MK transfusion to collect mouse PB, lung, spleen, and bone marrow (BM) harvested from the femur (n=8). Tissues isolated from mice without human MK infusion were used as negative control (n=2). Cell suspensions from all tissues were prepared with cell strainers (BD Biosciences). RBCs in the BM were lysed with RBC lysis buffer and the remaining cells were washed with PBS. The presence of human MKs or PLTs in mouse PB, lung, spleen, or BM was assessed by staining for human CD41, CD42a, and/or CD61 as described previously and flow cytometric analysis. Data were analyzed with FACSDiva software (BD Biosciences).
Statistical analysis
Statistical analyses were performed using two-tailed t tests run on GraphPad Prism 5 software (GraphPad Software, San Diego, CA). Levels of significance were expressed as p values (*p<0.05, **p<0.01, and ***p<0.001).
Results
Generation of HLA-universal megakaryocytes and platelets
In our previous study we described the successful generation of HLA class I-silenced PLTs from CD34+ progenitor cells, which we termed HLA-universal PLTs (Figueiredo et al., 2010). In this study, we further characterized HLA-universal MKs and PLTs in vitro and in vivo to address their capacity to escape antibody-mediated cytotoxicity. HLA-universal MKs used in this study were characterized on the basis of the expression of CD41 (GPIIb–IIIa) and CD42a (GPIX), as well as morphological features such as polyploidy and pro-PLTs. As previously reported, we observed no significant differences between HLA-silenced and nonsilenced MKs regarding phenotype and morphology. Differentiation of CD34+ cells expressing shNS or shβ2m yielded a mean of 62 and 70% of mature CD41+CD42a+ MKs, respectively, on day 10 of differentiation (Fig. 1B). More than 28% of both HLA-silenced and nonsilenced MKs were shown to be polyploid, with a DNA content higher than 4n (Fig. 1C and D). In addition, shNS- and shβ2m-transduced MKs produced long pro-PLT extensions starting on day 10 of differentiation, similar to nontransduced MKs (Fig. 1E). As expected, we observed significant differences regarding the expression of HLA class I molecules. shβ2m-expressing MKs showed a downregulation of β2m transcripts up to 91% (p<0.001; Fig. 2A) and HLA class I surface expression up to 64%, as compared with shNS-expressing MKs (p<0.001; Fig. 2B). Additional data showed that antigen expression of various HLA loci was similarly downregulated in shβ2m-expressing MKs (Supplementary Fig. S2).

Characterization of CD34+ progenitor cell-derived HLA class I-silenced megakaryocytes (MKs). CD34+ progenitor cells expressing control nonsense short-hairpin RNA (shNS) or shRNA targeting β2-microglobulin transcripts (shβ2m) were cultured for 10 days in the presence of thrombopoietin (TPO, 100 ng/ml).

Silencing of HLA class I expression in CD34+ progenitor cell-derived MKs.
HLA-universal PLTs were characterized by the expression of CD42a and CD61 (GPIIIa) as well as in vitro functional assays. On day 12 of differentiation, a mean of 30% of cells in the PLT population derived from shNS- or shβ2m-expressing MKs were shown to be CD61+CD42a+ (Fig. 3B). Furthermore, the generated PLTs were shown to be functional in vitro, as indicated by upregulation of activated GPIIb–IIIa (PAC-1) on stimulation with ADP, which is a major PLT agonist (Fig. 3C).

Characterization of CD34+ progenitor cell-derived HLA class I-silenced PLTs. PLTs derived from shNS- or shβ2m-expressing CD34+ progenitor cells were identified on the basis of FSC/SSC characteristics of blood-derived PLTs and CD42a and CD61 expression on day 12 of differentiation culture.
HLA-universal megakaryocytes are protected from antibody-mediated complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity in vitro
The susceptibility of HLA-universal MKs to undergo anti-HLA antibody-mediated CDC was analyzed by means of a lymphocytotoxicity assay that is routinely used in clinical transfusion settings (Levin et al., 2003; Buakaew and Promwong, 2011). In this assay, cells are incubated with antibodies (Supplementary Table S2) that mediate CDC on binding to their specific antigen. As expected, incubation of shNS-expressing MKs with specific anti-HLA antibodies and complement resulted in high cell lysis rates, with 87±6% lysed cells, similar to lysis rates observed in positive control samples (97±3% lysed cells) (Fig. 4). Background lysis levels of shNS-expressing MKs were 7±3% and 8±3% in samples with PBS alone or nonspecific anti-HLA antibody controls, respectively. Incubation of HLA-silenced MKs with PBS (negative control, NC), nonspecific anti-HLA antibodies, or positive control (PC) reagent led to lysis rates similar to the corresponding nonsilenced MK samples (NC, 7±3%; isotype antibodies, 8±3%; PC, 92±3%). However, when HLA-silenced MKs were incubated in the presence of specific anti-HLA antibodies and complement, cell lysis was completely abrogated in comparison with nonsilenced MKs (p<0.001) and lysis rates were equivalent to background lysis levels (Fig. 4). These data show that HLA-silenced MKs are efficiently protected from antibody-mediated CDC.

HLA-silenced MKs are protected from anti-HLA antibody-mediated complement-dependent cytotoxicity. Lymphocytotoxicity assays were performed with shNS- or shβ2m-expressing MKs (day 10 of differentiation), using complement-fixing anti-HLA antibodies according to the HLA type of HPC donors.
In addition, the protective capacity of HLA silencing against ADCC, the second mechanism of antibody-mediated cell destruction, was investigated. Therefore, we used the ADCC reporter bioassay kit from Promega, which was specifically developed for sensitive and robust detection of the ADCC mechanism of action of antibodies toward target cells (Parekh et al., 2012). All antibodies used in this assay, as well as the HLA type of the MKs and PLTs used, are listed in Supplementary Table S2. In this assay, engineered Jurkat cells that stably express FcγRIIIa as well as an NFAT response element driving expression of firefly luciferase were used as effector cells. The ADCC activity of effector cells was monitored by luciferase activity readout. As expected, the combination of shNS-expressing MKs as target cells and specific anti-HLA antibodies resulted in high ADCC activity of the effector cells compared with nonspecific antibody controls (Fig. 5). In contrast, the ADCC activity of effector cells incubated with HLA-universal MK targets plus specific anti-HLA antibodies was significantly reduced compared with nonsilenced target cells (Fig. 5). These results show that HLA silencing also provides protection from ADCC in vitro.
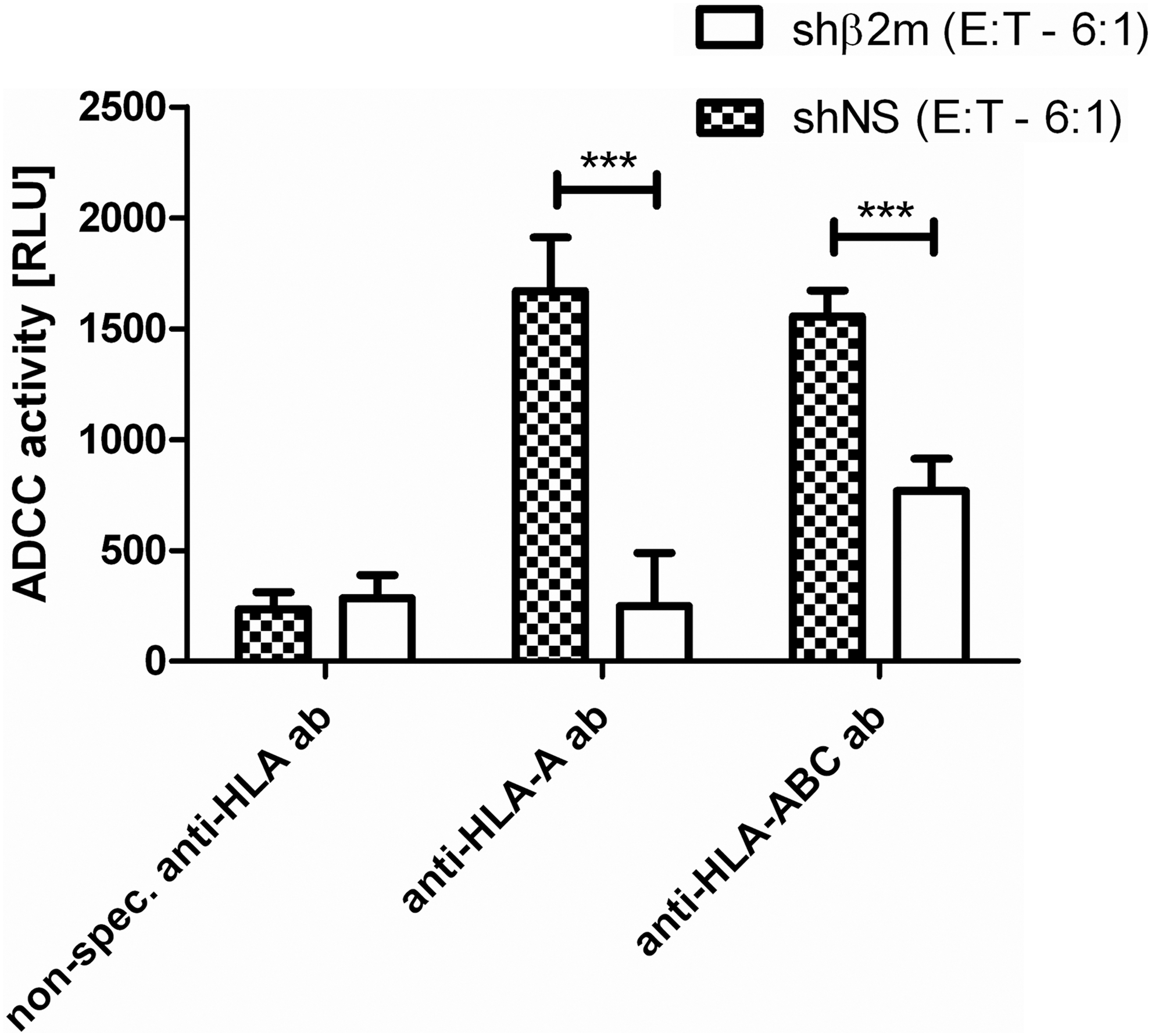
HLA-universal MKs are protected from anti-HLA antibody-dependent cellular cytotoxicity. ADCC reporter assays were performed with shNS- or shβ2m-expressing MKs (day 10 of differentiation) as target cells and anti-HLA IgG antibodies according to the HLA type of HPC donors. Luciferase activity (RLU) was used for quantification of ADCC activity of effector cells. Shown are the means and SD of three independent experiments. ***p<0.001. ADCC, antibody-dependent cellular cytotoxicity; ab, antibody; RLU, relative light units.
HLA-universal megakaryocytes increase human PLT counts in mice refractory to human PLTs
In a next step, we assessed whether HLA-universal PLTs are protected from antibody-mediated cytotoxicity in vivo. For this purpose, we used NOD/SCID/IL-2Rγc–/– (NSG) mice that were injected with specific anti-HLA antibodies to make them “refractory” to human PLTs. Control mice received PBS only or nonspecific anti-HLA antibodies. Specific and nonspecific anti-HLA antibodies as well as the HLA type of MKs and PLTs used in these experiments are listed in Supplementary Table S2. Injection of 1×106 shNS- or shβ2m-expressing MKs into mice, in the absence of anti-HLA antibodies, showed that both HLA-silenced and nonsilenced MKs have the capacity to generate PLTs in vivo (Fig. 6). Twenty-four hours after injection, we observed human PLT (CD42a+CD61+ cell) frequencies of 0.3±0.14% and 0.35±0.13% within the PLT population of mice that received nonsilenced or HLA-silenced MKs, respectively. Similarly, both shNS- and shβ2m-expressing MKs produced PLT frequencies of 0.325±0.095% and 0.375±0.096%, respectively, in the presence of nonspecific anti-HLA antibody controls. However, no human PLTs were detected in the PLT population of mice that were injected with shNS-expressing MKs together with specific anti-HLA antibodies, indicating that human cells were rapidly destroyed in the circulation of these mice. In contrast, injection of HLA-silenced MKs into mice that also received specific anti-HLA antibodies resulted in human PLT frequencies of 0.35±0.129% (p<0.01), similar to PLT frequencies observed in the absence of anti-HLA antibodies (Fig. 6). Human PLTs were detectable in these mice for up to 11 days (data not shown). These results demonstrate that HLA-silenced MKs and PLTs are protected from antibody-mediated cytotoxicity in vivo and increase PLT counts in mice refractory to human PLTs.

HLA-silenced PLTs escape antibody-mediated cytotoxicity in the circulation of living mice. NOD/SCID/IL-2Rγc–/– mice were injected intravenously with specific anti-HLA antibodies (to make them refractory to human PLTs) followed by infusion of 1×106 shNS- or shβ2m-expressing MKs. Mice that received antibody only were used as negative control (NC) and mice that received shNS- or shβ2m-expressing MKs only or in presence of nonspecific anti-HLA antibodies were used as positive controls. Mouse peripheral blood (PB) was harvested 24 hr after injection of MKs. Human PLTs were identified on the basis of the coexpression of human (h)CD42a and hCD61. As an additional control, the PB of mice that received MKs only was stained with isotype controls for anti-hCD42a and anti-hCD61 antibody.
It is important to note that in all animal experiments, mice behaved normally and did not show any adverse effects up to 2 month after injection of genetically engineered MKs. Furthermore, on day 13 after injection no human cells (CD41+, CD42a+, or CD61+) could be detected in PB, BM, lungs, or spleen of mice (Supplementary Figs. S3 and S4).
Discussion
Allogeneic PLT transfusions are used as standard treatment for patients with severe thrombocytopenia or PLT dysfunction syndromes (Sharma et al., 2011). In particular, patients with major bleeding episodes due to trauma or surgery or patients suffering from bone marrow suppression highly depend on a constant supply of allogeneic PLTs to prevent fatal hemorrhage (Stephan et al., 1999; Blajchman et al., 2008; Zaffar et al., 2013). A promising strategy for constant and safe supply of blood products independent of blood donation is the in vitro production of blood cells including erythrocytes and PLTs from stem and progenitor cells (Giarratana et al., 2011; Reems, 2011). Hence, several research groups have been dedicating their work to the in vitro manufacturing of PLTs and have successfully derived functional PLTs from HPCs, embryonic stem cells, and induced pluripotent stem cells (Matsunaga et al., 2006; Takayama et al., 2010; Lu et al., 2011). Furthermore, with the development of bioreactors, significant success has been made regarding large-scale production of PLTs for future clinical application (Sullenbarger et al., 2009). However, as in the case of blood-derived PLTs, the use of in vitro-generated allogeneic PLTs would face the problem of PLT refractoriness in alloimmunized patients. Here, the use of genetically engineered progenitor cells as the source for PLT production offers the possibility to generate designer PLTs with low immunogenicity, especially for patients requiring multiple transfusions. The high level of polymorphism within the HLA locus is the major reason for alloimmunization on transplantation/transfusion of allogeneic products or after pregnancy (Ayala García et al., 2012). Accordingly, in the case of PLT transfusion, HLA class I molecules are the major target of antibody-mediated alloimmune responses leading to PLT transfusion refractoriness (Sacher et al., 2003; Hod and Schwartz, 2008). Therefore, HLA molecules are an attractive target to reduce the immunogenicity of in vitro-generated PLTs. Previously, we demonstrated the successful in vitro production of HPC-derived HLA class I-silenced PLTs, which were shown to be functionally similar to blood-derived PLTs (Figueiredo et al., 2010). To efficiently reduce HLA class I surface expression, we used a combination of lentiviral gene transfer and RNAi technology to target the conserved β2m molecule, which is an essential part of dimeric HLA class I molecules (Figueiredo et al., 2010). In this study, we assessed the capacity of HLA-silenced MKs and PLTs to escape anti-HLA antibody-mediated cytotoxicity, which is the main cause of immune-mediated PLT refractoriness (Sacher et al., 2003). Therefore, we produced HLA-universal MKs and PLTs as previously described (Figueiredo et al., 2010). Morphologic and phenotypic analysis confirmed the successful production of mature MKs and PLTs from HLA-silenced CD34+ progenitor cells. Using platelet activation assays we demonstrated that HLA-universal MKs generate functional PLTs in vitro. In a next step we showed that HLA class I silencing efficiently prevents antibody-mediated PLT destruction in vitro and in vivo. Alloantibodies are generally thought to cause PLT destruction by either CDC or Fc-receptor-mediated phagocytosis, with the latter mechanism considered to be more relevant (Lim et al., 2002). Using in vitro lymphocytotoxicity assays, we showed that HLA-silenced MKs are resistant to anti-HLA antibody-mediated CDC, whereas most nonsilenced MKs were lysed. These data strongly suggest that HLA-silenced PLTs derived from HLA-silenced MKs are equally protected from antibody-mediated CDC. In addition, we used an in vitro ADCC reporter assay to assess the effect of HLA silencing on Fc receptor-mediated PLT destruction. Our results show that whereas nonsilenced MKs elicit high ADCC activity of effector cells in the presence of specific anti-HLA antibodies, ADCC activity toward HLA-universal MKs was strongly reduced. These data suggest that HLA silencing also provides efficient protection against Fc receptor-mediated ADCC.
Furthermore, a mouse model for PLT refractoriness was used to assess the immune evasion capacity of HLA-silenced MKs and PLTs in vivo (Supplementary Fig. S1). To mimic PLT refractoriness, NOD/SCID/IL-2Rγc–/– mice were injected intravenously with specific anti-HLA antibodies. Transfusion of MKs is an alternative strategy to increase PLT counts in thrombocytopenia situations. Previously, it has been shown that mature human MKs infused into the circulation of living mice generate PLTs in vivo (Chen et al., 2009). Because in vitro PLT production is still less efficient than in vivo PLT generation (Reems, 2011), we decided to infuse mature MKs into the circulation of mice to allow in vivo PLT production. As a positive control for in vivo PLT generation, nonsilenced and HLA-silenced MKs were infused into mice in the absence of anti-HLA antibodies. Our data show that within 24 hr of MK infusion both nonsilenced and HLA-silenced MKs produced similar frequencies of human PLTs in the circulation of mice. However, when nonsilenced MKs were infused into mice that also received specific anti-HLA antibodies, no human PLTs (Fig. 6) or MKs (data not shown) were detected in mouse PB 24 hr postinfusion. These data strongly suggest that nonsilenced human MKs and PLTs were rapidly destroyed in the circulation of these mice by an anti-HLA antibody-mediated mechanism mimicking PLT refractoriness. In contrast, HLA-silenced MKs produced human PLTs in the circulation of mice despite the presence of specific anti-HLA antibodies (Fig. 6). After 24 hr, HLA-silenced PLTs were detectable in these mice at frequencies similar to control mice. This difference in HLA-universal and HLA-expressing PLT frequencies could not be observed in the presence of nonspecific anti-HLA antibody controls, which confirms the specificity of the antibody mechanism of action. These results demonstrate that HLA-silenced MKs and PLTs are resistant to PLT refractoriness in vivo. Because NOD/SCID/IL-2Rγc–/– mice are deficient in the C5 component of the complement system (Foreman et al., 2011), the rapid antibody-dependent destruction of MKs and PLTs in these mice is likely mediated by Fc receptor-mediated phagocytosis, which is thought to be the major mechanism of PLT destruction in PLT-refractory patients (Lim et al., 2002). We have covered this lack of the mouse model by carefully addressing the CDC mechanism of action with lymphocytotoxicity tests, as mentioned previously.
Taken together, our data demonstrate that HLA-universal MKs and PLTs are efficiently protected from anti-HLA antibody-mediated cytotoxicity (CDC and Fc receptor-mediated phagocytosis) and prevent PLT refractoriness in vivo. Hence, our approach offers a promising strategy for future clinical PLT supply for patients with immune PLT refractoriness, obviating the need for time-consuming and cost-intensive HLA typing and matching.
In our in vivo experiments, mature MKs were infused into mice to circumvent the relatively low rates of in vitro PLT production. This strategy may also be useful for future clinical application. Importantly, mice did not show any adverse effects after injection of genetically engineered human cells. Furthermore, biodistribution analysis revealed that on day 13 postinfusion no residual human cells were present in the PB, BM, lungs, or spleen of mice (Supplementary Figs. S3 and S4). These data indicate that our MK differentiation cultures probably did not contain potentially cancerogenic precursor cells, which could have engrafted and survived for prolonged periods of time. However, to further eliminate safety concerns about infusing genetically engineered nucleated cells, we assessed the feasibility of irradiating mature pro-PLT-bearing MKs. Irradiation can be used to eliminate any residual precursor or transformed cells that are potentially cancerogenic (Gekas and Graf, 2010). Also, platelet γ or UVA irradiation is a common technique used to increase the clinical safety of transfusion practice (Blajchman et al., 2004). We observed that mature MKs irradiated with a dose of 30 Gy generated functional PLTs in vitro, similarly to nonirradiated controls (data not shown). These results indicate that mature MKs could be irradiated before infusion for clinical purposes.
In addition to the treatment of alloimmunized patients, genetically engineered PLTs could be used to avoid alloimmunization of patients receiving PLT transfusions. This would be of particular interest for patients requiring multiple transfusions and/or patients in need for transplantation of allogeneic cells or tissues. Previous studies have shown that allogeneic platelets per se do not stimulate direct T cell allocytotoxicity (Gouttefangeas et al., 2000); however, it is thought that indirect T cell activation may occur on antigen presentation of peptides derived from allo-HLA molecules (Semple et al., 1995). Reduction in immunogenic molecules (i.e., allogeneic HLA molecules) is likely to reduce the probability of alloimmunization through indirect T cell activation. In the current study, we provide the proof-of-concept for the successful generation of genetically engineered PLTs with reduced immunogenicity. Targeting the conserved β2m molecule via RNAi technology resulted in the efficient downregulation of HLA class I surface expression by MKs and PLTs and conferred protection against anti-HLA antibody-mediated cytotoxicity. Previously we showed that another feasible approach to reduce the expression of HLA class I molecules is direct targeting of HLA class I heavy chains (Figueiredo et al., 2007). This approach would allow the reduction of alloantigen expression by MKs and PLTs and would likely also reduce the risk of alloimmunization by indirect T cell activation.
In conclusion, this study demonstrates that in vitro-generated HLA-universal PLTs are efficiently protected from anti-HLA antibody-mediated cytotoxicity and prevent PLT refractoriness in vivo. Manufactured HLA-universal MKs and PLTs may become an important tool for the treatment of PLT-refractory patients in the near future. Moreover, they may be life-saving in the case of highly alloimmunized patients, in whom even HLA-matched or cross-matched PLT units fail to increase PLT blood counts.
Footnotes
Acknowledgments
The authors are grateful to Stefanie Vahlsing, Angelika Höhne, Ulrike Bröder, and Mathias Neumeyer for technical assistance. The authors thank Radiana Antarianto, who contributed with helpful discussion. This work was supported in part by funding from the German Research Foundation (DFG) for the Cluster of Excellence REBIRTH (From Regenerative Biology to Reconstructive Therapy) (EXC 62) (C.F., C.A.G., C.G.).
Author Disclosure Statement
C. Gras, no competing financial interests exist; K. Schulze, no competing financial interests exist; L. Goudeva, no competing financial interests exist; C.A. Guzman, no competing financial interests exist; R. Blasczyk, no competing financial interests exist; C. Figueiredo, no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.