
Abstract
Interleukin-12 (IL-12) is an immunostimulatory cytokine that has shown strong antitumor effects in animal models of liver cancer. In order to overcome the severe toxicity associated with its systemic administration, we had previously tested different strategies based on IL-12 gene transfer to tumor cells or to the surrounding liver tissue. We obtained promising results both with a recombinant Semliki Forest virus (SFV) vector expressing high levels of IL-12 (SFV-IL-12) after intratumoral injection and with a plasmid vector [pTonL2(T)-mIL12] that allows liver-specific and inducible IL-12 expression. The aim of the present study was to compare the antitumor responses induced by both systems in a clinically relevant animal model of hepatocellular carcinoma (HCC) developed in L-PK/c-myc transgenic mice. These animals overexpress the c-myc oncogene in their livers, giving rise to spontaneous hepatic tumors with latency, histopathology, and genetic characteristics similar to human HCCs. We observed that intratumoral inoculation of SFV-IL-12 induced growth arrest in most tumors, providing 100% survival rate, in contrast to no survival in control animals. Similar results were obtained with hydrodynamic injection of pTonL2(T)-mIL12 after long-term induction of IL-12 expression in the liver. However, tumor arrest was less evident in plasmid-treated mice and the survival rate was slightly lower, despite higher and more sustained levels of IL-12 and IFN-γ in serum. The fact that SFV-IL-12 was able to induce both apoptosis and a type-I IFN response specifically in the tumor could explain why short-term IL-12 expression from this vector was sufficient to mediate an antitumoral response comparable with long-term IL-12 expression driven by pTonL2(T)-mIL12. Since SFV-IL-12 could reduce the possible toxicity associated with long-term IL-12 expression, we believe that this vector could have a potential application for HCC gene therapy.
Introduction
I
Although IL-12 can be delivered systemically as recombinant protein, this strategy has shown toxicity in clinical trials (Atkins et al., 1997). A safer approach is based on the use of gene therapy vectors that are able to express IL-12 at the local tumor site. Intratumoral administration of first-generation adenoviral vectors expressing IL-12 has shown strong antitumoral effects in preclinical models of primary and metastatic HCCs (Caruso et al., 1996; Andrews et al., 2000; Barajas et al., 2001; Schmitz et al., 2005; Waehler et al., 2005). Moreover, a phase I clinical trial based on this strategy showed low toxicity and tumor infiltration by effector immune cells, resulting in one objective tumor remission in a patient with advanced HCC (Sangro et al., 2004). Despite these encouraging results, the antitumoral efficacy in this setting seemed to be limited by low and transient transgene expression in the adenovirally transduced tumors (Penuelas et al., 2005). Short-term IL-12 expression is inherent to first-generation adenoviral vectors, since they trigger immune-mediated death of infected cells because of expression of viral antigenic proteins. Moreover, short-term transgene expression is also a consequence of the type of cells that are targeted by intratumoral vector administration, because expression from transduced tumor cells will decrease proportionally to tumor remission. A dose-dependent effect of IL-12 has been reported by us and other groups (Zabala et al., 2004; Rodriguez-Madoz et al., 2005); therefore, a therapeutic strategy based on intratumoral IL-12 expression would benefit from vectors that allow high levels of IL-12 before transduced cells die or are eliminated by the immune system.
In this regard, we previously demonstrated that a cytopathic viral vector derived from Semliki Forest virus (SFV-IL-12) is able to express very high levels of IL-12 during a short time interval, inducing potent antitumoral responses (Rodriguez-Madoz et al., 2005). The SFV vector is based on a viral RNA genome in which the region coding for the structural proteins has been replaced by a heterologous gene (Liljestrom and Garoff, 1991). Recombinant SFV RNA can be transcribed in vitro and transfected into cells, leading to its replication and production of a subgenomic RNA from which the heterologous protein will be expressed at high levels. SFV recombinant RNA can be packaged into viral particles (vp) by cotransfecting it into cells together with two helper RNAs that code for the capsid and the envelope proteins (Smerdou and Liljestrom, 1999). SFV vectors expressing IL-12 have shown to be very efficient in inducing antitumoral responses in several transplantable tumor models, such as colon adenocarcinoma, melanoma, or cervical cancer and orthotopic HCC models (Colmenero et al., 2002; Chikkanna-Gowda et al., 2005; Rodriguez-Madoz et al., 2005; Riezebos-Brilman et al., 2009; Quetglas et al., 2012, 2013). Moreover, SFV vectors were able to express more IL-12 and achieve stronger antitumoral activity compared with adenoviral-based vectors, probably because of their capacity to induce apoptosis in tumor cells (Rodriguez-Madoz et al., 2005; Guan et al., 2006).
In conjunction with the former approach, we have also been working on a conceptually different therapeutic strategy based on long-term peritumoral IL-12 expression. Delivering vectors directly to hepatocytes can achieve long-term expression of IL-12 for liver cancer gene therapy, but this strategy requires the use of inducible promoters, since constitutive expression of IL-12 could be highly toxic. We previously constructed a nonviral vector that expresses IL-12 in a liver-specific and doxycycline (Dox)-inducible manner [pTonL2(T)-mIL12] (Zabala et al., 2004). This vector was transferred with high efficiency into mouse livers using hydrodynamic injection and it remained in these organs for several months without apparent side effects (Reboredo et al., 2008a,b). Regular administration of Dox led to high and persistent IL-12 levels in these animals, resulting in complete tumor regression in a mouse model of metastatic HCC (Zabala et al., 2004). In the preclinical L-PK/c-myc HCC transgenic mouse model (Zabala et al., 2007), mice develop spontaneous HCC in a step-wise process as a consequence of c-myc overexpression, an oncogene that is activated in many human HCCs (de La Coste et al., 1999; Lee et al., 2004), resulting in tumors that closely resemble human HCC in terms of biology, histology, and gene expression profile (Cartier et al., 1992; de La Coste et al., 1999; Lee and Thorgeirsson, 2004; Lee et al., 2004). We previously demonstrated antitumoral efficacy in this model using a similar IL-12 hydrodynamic injection strategy.
In order to move a step closer to translational cancer gene therapy, we reasoned that a comparative study between our two promising therapeutic approaches in a clinically relevant model of cancer was required. In the present study we compared the antitumoral efficacy of short-term expression of IL-12 in tumor cells using SFV-IL-12 versus sustained peritumoral IL-12 expression using pTonL2(T)-mIL12 in L-PK/c-myc transgenic mice with spontaneous HCC. Our results show that both approaches were able to induce a strong antitumoral effect in this valuable preclinical animal model. However, tumor arrest was more evident and survival rate was slightly higher in the SFV-IL-12-treated mice. Several factors, such as induction of apoptosis and type I IFN responses in tumor cells by the SFV vector, could be responsible for this remarkable antitumoral efficacy.
Materials and Methods
L-PK/c-myc transgenic mice
A transgenic mouse colony was established in the Center for Applied Medical Research (Pamplona, Spain) by crossing L-PK/c-myc (+/−) males (Cartier et al., 1992) with 129S2/SvHsd wt females (Harlam, Barcelona, Spain). Littermates were genotyped as described previously (Cartier et al., 1992). Heterozygous L-PK/c-myc mice (28% of littermates) were fed with a 75% carbohydrate-rich diet (Safe, Augy, France) to enhance c-myc expression in the liver (de La Coste et al., 1999). About 60% of these animals developed single and multifocal hepatic tumors by 7–8 months. We selected animals with comparable tumor burden for antitumoral and mechanistic studies. All animals were kept under standard pathogen-free conditions and received care according to the ethical institution's guidelines. To assess tumor development and progression, L-PK/c-myc mice underwent laparotomy and tumor volume was determined according to the formula: volume=(D×d 2)/2, where D is the longest tumor diameter and d is the shortest tumor diameter. Tumor growth rate was calculated dividing the increment of tumor volume by the number of weeks of treatment.
Production of plasmids and viral vectors
Plasmids pSFV-enhIL-12 and pSFV-Luc, containing genes coding for mouse IL-12 and firefly luciferase, respectively, have been previously described (Rodriguez-Madoz et al., 2005). Vector RNA synthesis and transfection into BHK-21 cells by electroporation was performed as described previously (Liljestrom and Garoff, 1991). Packaging of recombinant RNA into SFV vp was performed as described (Smerdou and Liljestrom, 1999). Briefly, BHK-21 cells were coelectroporated with recombinant SFV vector RNA (SFV-enhIL-12 or SFV-Luc) and both SFV-helper-S2 and SFV-helper-C-S219A RNAs, which provided in trans the capsid and envelope proteins, respectively. Electroporated cells were incubated at 33°C during 48 hr, supernatants were collected, and SFV vp were purified by ultracentrifugation through a 20% sucrose cushion at 100,000×g during 90 min. The titer of SFV-Luc and SFV-enhIL-12 (designated in this article as SFV-IL-12) recombinant virus stocks was determined by indirect immunofluorescence of infected BHK-21 cells using a rabbit polyclonal antiserum specific for the nsp2 subunit of SFV replicase as primary antibody. Plasmid pTonL2(T)-mIL12 carrying the murine IL-12 gene under the control of a liver-specific Dox-inducible system (Zabala et al., 2004) was purified by using EndoFree Maxi Kit (Qiagen, Valencia, CA).
Treatment scheme of HCC-bearing mice
L-PK/c-myc transgenic mice underwent laparotomy, and tumors were counted and measured as described above. Animals were allowed to recover during 17 days before treatment was started. In the case of SFV-treated animals, each tumor nodule was injected at day 0 with 108 vp of SFV-IL-12 diluted in 50 μl of saline buffer. Control animals received the same dose of SFV-Luc, or the same volume of saline buffer, respectively. Fifty days after treatment a second intratumoral dose of 108 vp of SFV-IL-12, SFV-Luc, or an equivalent volume of saline was delivered to each tumor nodule in its respective treatment groups after laparotomy. In the case of plasmid-treated animals, they were injected with 100 μg of pTonL2(T)-mIL12 using the hydrodynamics-based procedure (Zhang et al., 1999) 5 days before giving Dox (day −5). Induction of IL-12 expression was initiated at day 0 by giving 2 mg/ml Dox (Sigma, St. Louis, MO) in drinking water containing 5% sucrose (Panreac, Barcelona, Spain), and was maintained for 10 days (round 1) and 5 days (rounds 2 and 3) of each cycle as shown in Fig. 2A. All animals were euthanized at day 135 and tumors were again measured and counted.
Determination of in vivo luciferase expression
Mice treated with SFV-Luc were anesthetized, injected intraperitoneally with 100 μl
Determination of IL-12 and IFNγ levels
mIL-12 and IFNγ levels in serum were quantified by OptEIA Mouse IL-12 and IFN-γ ELISA Sets (BD Biosciences, San Diego, CA), respectively, according to the manufacturer's instructions.
Processing of tissue samples and immunohistochemistry
Liver and tumor samples were fixed in 10% buffered formaldehyde for 24 hr, washed with PBS, and embedded in paraffin. Four- to six-micrometer sections were gradually hydrated with ethanol at 100%, 96%, 80%, and 70% in distilled water and stained with hematoxylin and eosin according to standard procedures. For immunohistochemistry, samples were embedded and frozen in Tissue-Tek OCT (Sakura, Zoeterwoude, The Netherlands). Cryostat 8-μm-thick sections were cut, air-dried, and fixed with 4% formaldehyde for 2 min. Endogenous peroxidase was quenched using 3% hydrogen peroxide in methanol. Tissue sections were incubated O/N at 4°C with a rat monoclonal antibody specific for murine IL-12 (clone C15.6; BD Pharmingen, San Diego, CA). A rabbit antirat polyclonal antiserum (Dako, Carpinteria, CA) followed by the antirabbit EnVision system (Dako) was used for detection. Peroxidase activity was revealed using DAB+ Sustrate Chromogen System (Dako).
RNA isolation and quantitative real-time polymerase chain reaction
Total RNA was extracted from individual tumor samples 48 hr after treatment using TRI Reagent (Sigma). DNase I-treated RNA was retrotranscribed to cDNA with M-MLV in the presence of RNase OUT (Invitrogen, Carlsbad, CA). For real-time polymerase chain reaction, 2 μl of cDNA was incubated with specific oligonuclotides using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). The following oligonucleotides (sense/antisense) were used: 5′-TCTYTCYTGYCTGAAGGAC-3′/5′-CACAGRGGCTGTGTTTCTTG-3′ (IFN-α), 5′-ATGAGTGGTGGTGCAGGC-3′/5′-ACCTTCAAATGCAGTAGATTCA-3′ (IFN-β), and 5′-CTGTTCTCGACGCGTCGTC-3′/5′-GAGGTGTTTCCACGACCC-3′ (SFV genomic RNA). GAPDH was used to normalize gene expression using oligonucleotides 5′-GATGGTGAAGGTCGGTGTG-3′ (sense) and 5′-CTTCCACGATGCCAAAGTTG-3′ (antisense). mRNA expression values were represented as 2ΔCt.
Statistical analysis
All error terms are expressed as the standard deviation of the mean value. Significance levels for comparison of differences between groups were analyzed using the Kruskal–Wallis test followed by the Mann–Whitney U-test. In Fig. 3C, Kaplan–Meyer survival analysis was assessed. In Fig. 6B, data were compared using the Mann–Whitney U-test. All statistical analysis tests were performed using SPSS v.21 software.
Results
Histopathology of HCCs developed in PK/c-myc transgenic mice
In the present study, we used transgenic mice that overexpress the c-myc oncogene controlled by a liver-specific pyruvate kinase (L-PK) promoter, whose activity is enhanced in the presence of insulin and glucose (Cartier et al., 1992). Tumor progression in these animals is a step-wise process that involves different carcinogenic phases, resembling human HCCs (de La Coste et al., 1999). Heterozygous L-PK/c-myc mice were fed with a carbohydrate-rich diet after they reached 30 days of age. About 70% of them developed spontaneous liver tumors in 7–8 months, as it was previously observed (Zabala et al., 2007). At this time, tumor sections from some animals were histologically analyzed. As shown in Fig. 1a, hepatocytes in normal liver are arranged in rows of one cell between sinusoids, whereas in liver tumors, cells are predominantly arranged in rows of 2–5 cells (trabeculas) and in a globular pattern (acini) (Fig. 1c). Tumor cells contained larger nuclei than normal hepatocytes and showed a higher mitotic rate (Fig. 1d). Tumors lacked the normal portal vein distribution and two different areas can be distinguished within them: a zone with high mitotic activity and a less stained zone that is likely associated with rich glycogen accumulation (Fig. 1b). In spite of differences in tissue organization, tumor cells had a morphology that is similar to normal hepatocytes, indicating that these HCCs were well differentiated (Fig. 1c and d). All of these observations confirmed that this model greatly resembles differentiated human HCCs of grade I–II (Edmondson and Steiner, 1954).
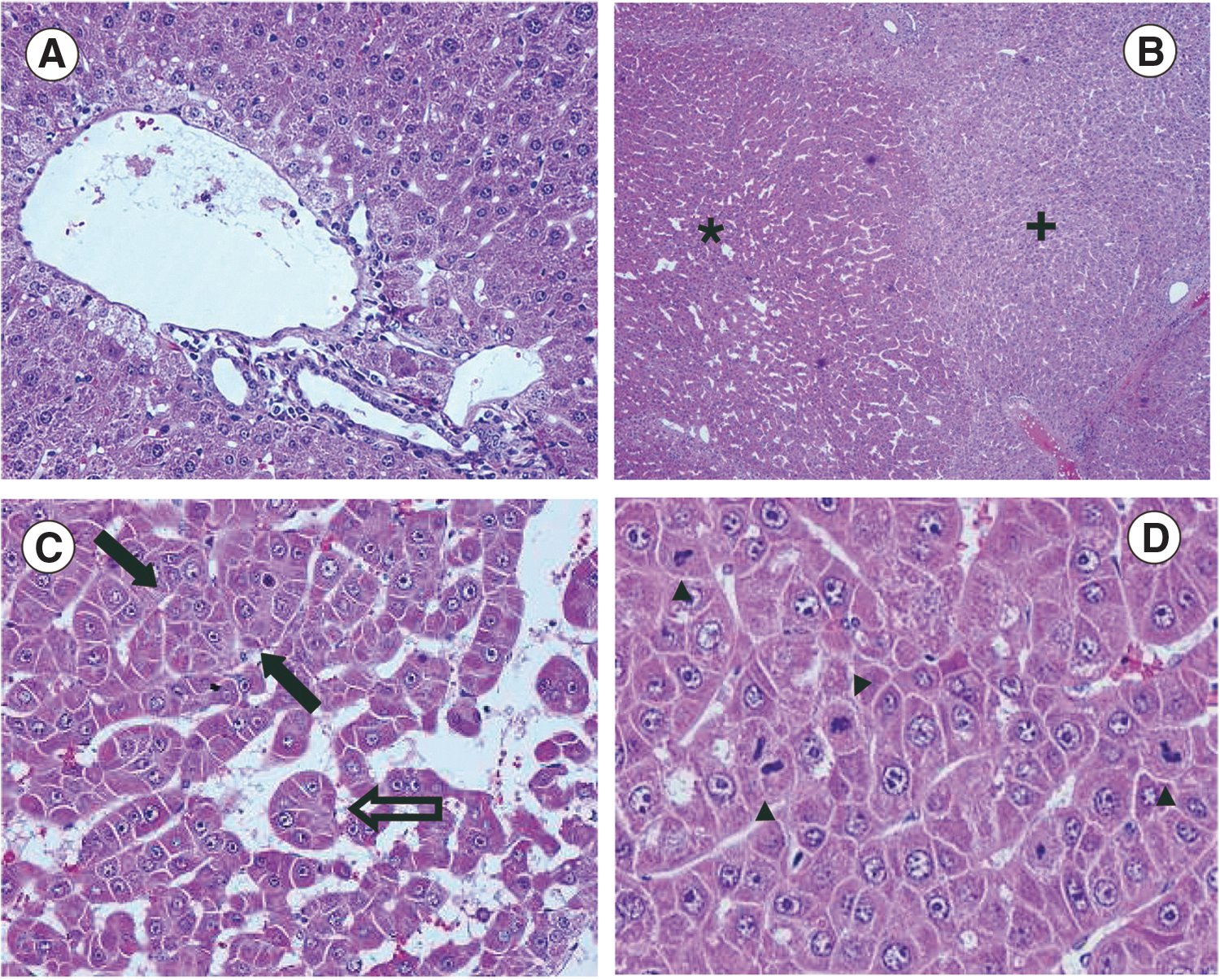
Histological analysis of HCC developed in L-PK/c-myc mice.
Comparison of the antitumoral efficacy of SFV-IL-12 and pTonL2(T)-mIL12 in L-PK/c-myc mice with HCCs
We previously showed that hydrodynamic administration of pTonL2(T)-mIL12, a plasmid encoding inducible IL-12, to the liver of L-PK/c-myc transgenic mice carrying HCCs inhibited tumor growth after long-term induction of IL-12 expression (Zabala et al., 2007). Although we have not observed significant toxic effects associated with this therapy (Reboredo et al., 2008a), high and persistent levels of IL-12 and its mediator IFNγ could result in latent side effects in humans. Therefore, we decided to compare this therapeutic strategy with one that is based on short-term tumor-directed IL-12 expression. For that purpose, we employed intratumoral administration of the SFV-IL-12 cytopathic viral vector, which was previously shown to have antitumoral potency against liver tumors (Guan et al., 2006; Rodriguez-Madoz et al., 2009; Quetglas et al., 2013). To this end, four groups of L-PK/c-myc mice with tumors of 4.33±1.93 mm in diameter were established with a similar distribution of males and females (Supplementary Table S1; Supplementary Data are available online at
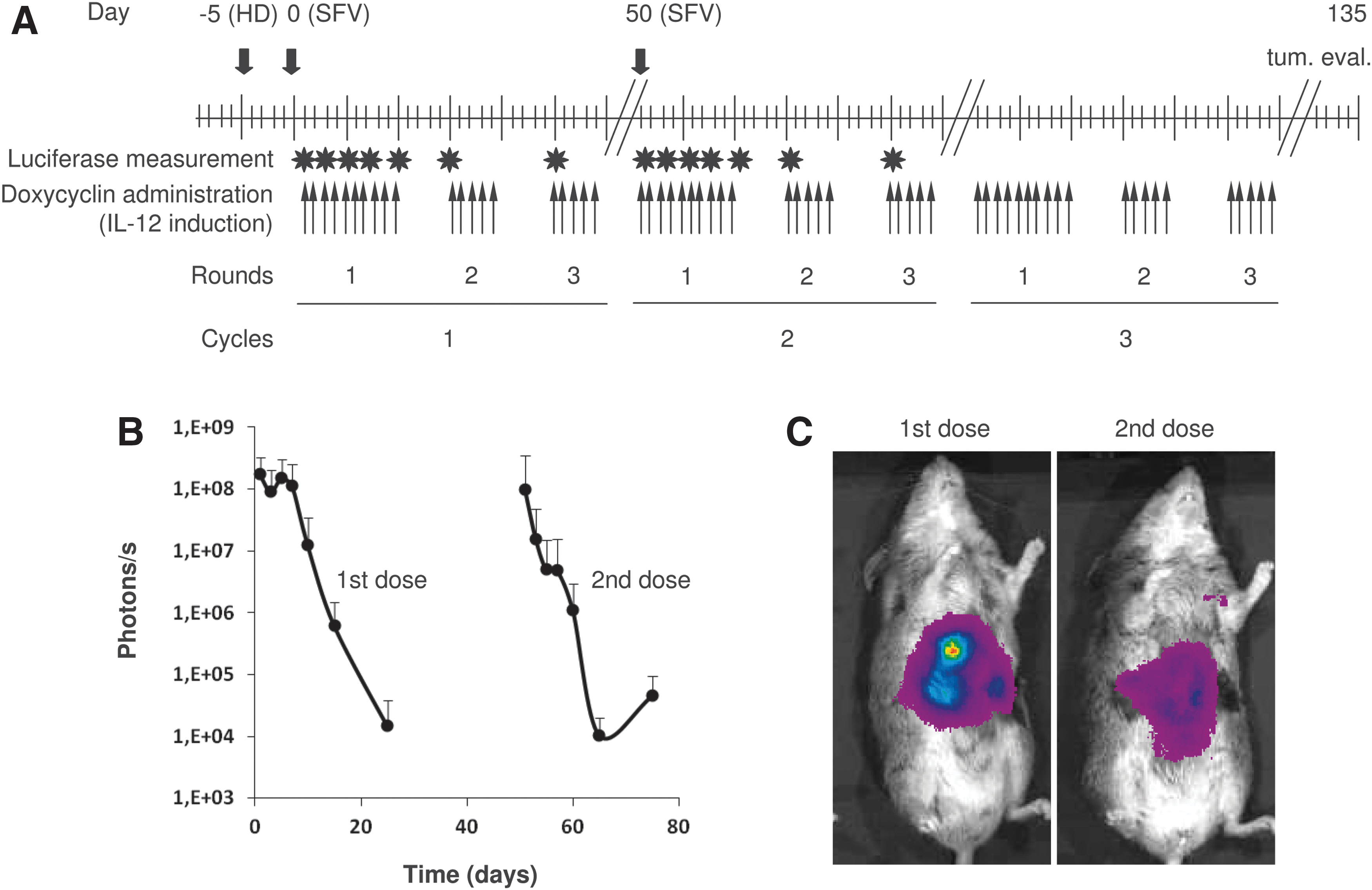
Study design and evaluation of SFV infectivity in HCC.
The kinetics of infectivity of SFV in HCC nodules was tested with SFV-Luc and visualized with a CCD camera at different time points posttreatment (Fig. 2B and C). All animals showed high luciferase activity during 5 days after the first dose of SFV-Luc, which temporally decreased rapidly. Mice were also able to express luciferase after the second dose of SFV-Luc, although at lower levels than after the first dose. This could be because of the development of an immune response against the SFV vector (Rodriguez-Madoz et al., 2007).
In order to evaluate the therapeutic efficacy of each treatment, we measured the tumor sizes at the end of the experiment (day 135 for surviving mice, or time of death for the rest of the mice; Supplementary Table S1). Tumors that were treated with saline or SFV-Luc grew as expected, with both groups showing a similar average tumor burden (p=0.815; Fig. 3A and Supplementary Table S1). In stark contrast, treatment with SFV-IL-12 induced a significant reduction in the average tumor volume compared with saline (p=0.006) or SFV-Luc (p=0.047) groups. A similar antitumoral effect was observed in mice that were treated with pTonL2(T)-mIL12, although in this case the overall reduction in tumor size was only significant versus the saline control group (p=0.049) but did not reach significance versus the SFV-Luc group (p=0.27). In fact, the tumor burden in the pTonL2(T)-mIL12 group seemed to be slightly higher than that in SFV-IL-12-treated mice (Supplementary Table S1 and Fig. 3A). Since some of the control animals did not survive until the end of the experiment (3 and 1 mice treated with saline and SFV-Luc, respectively; Supplementary Table S1), a better comparison of the effect of each treatment can be obtained by tracking the rate of tumor growth/week after treatment. As shown in Fig. 3B, mice treated with SFV-IL-12 showed a growth rate that was significantly lower than that of control animals (p=0.001 vs. saline and p=0.036 vs. SFV-Luc). Although the lowest tumor growth rate was observed in the SFV-IL-12 group, it was not significantly different from the pTonL2(T)-mIL12 group (p=0.122). All mice receiving SFV-IL-12 or pTonL2(T)-mIL12 survived until the end of the experiment, while 88% and 62% of mice receiving SFV-Luc and saline, respectively, were still alive at day 135 (Supplementary Table S1). However, at this time point, most of these animals had reached a tumor burden that was higher than 2,000 mm3, a tumor size that is considered an ethical criterion for euthanasia in our study (Zabala et al., 2007). We calculated the estimated time at which each mouse should have been euthanized assuming that tumor growth was constant along the study (the Estimated end point column in Supplementary Table S1). We then determined the “estimated survival” rate for each group (Fig. 3C) by taking into account the “estimated end point” for each animal, or the time of death if the mouse died before day 135. Mice that had received IL-12 therapy showed a significantly higher “estimated survival” rate, which reached 100% (p=0.001 vs. saline) and 88% (p=0.004 vs. saline) for SFV-IL-12- and pTonL2(T)-mIL12-treated animals, respectively (Fig. 3C). Interestingly, SFV-Luc was also able to improve the estimated survival of mice to 55%, but this was significantly lower than that observed in the SFV-IL-12 (p=0.021) or pTonL2(T)-mIL12 (p=0.032) groups, respectively, and did not show significance versus the saline group (p=0.983). As previously observed, animals that were treated with saline showed a very low “estimated survival” rate, with no animals expected to remain alive at the end of the experiment (Fig. 3C).

Antitumoral efficacy of IL-12 treatments in HCC.
IL-12 and IFNγ expression in SFV-IL-12- and pTonL2(T)-mIL12-treated mice
It has been previously shown by us that the antitumor effect mediated by IL-12 expression was dose dependent (Barajas et al., 2001; Wang et al., 2004; Zabala et al., 2004; Rodriguez-Madoz et al., 2005). In order to determine how IL-12 levels provided by SFV-IL-12 and pTonL2(T)-mIL12 correlated with the antitumoral effects observed in L-PK/c-myc mice bearing HCC, we determined the level of this cytokine and its mediator IFN-γ, in serum at different times after treatment. pTonL2(T)-mIL12-treated mice showed relatively high levels of IL-12 during most of the induction regime (Fig. 4A). However, these levels decreased during the course of each induction cycle and were about 100–1,000-fold lower during the third induction cycle. In contrast, the levels of IL-12 in animals treated with SFV-IL-12 were initially high, and comparable with those observed in pTonL2(T)-mIL12-treated mice, but decreased rapidly after 3 days, becoming undetectable by day 10 (Fig. 4A). The second dose of SFV-IL-12 resulted in a much lower expression of IL-12 that could be detected only for 1 day. Interestingly, similar levels of IFN-γ could be detected during the first 10 days of treatment in both pTonL2(T)-mIL12 (+Dox) and SFV-IL-12-treated mice (Fig. 4B). However, IFN-γ levels decreased more rapidly after the second dose of SFV-IL-12. In accordance with our previous reports, we could not detect IFN-γ during the third induction cycle in pTonL2(T)-mIL12-treated mice (Zabala et al., 2007). Neither IL-12 nor IFN-γ was detected in saline and SFV-Luc-treated mice (data not shown). IL-12 expression was also analyzed by immunohistochemistry in liver sections of mice euthanized 48 hr after treatment with pTonL2(T)-mIL12 (+Dox), SFV-IL-12, or saline (Supplementary Fig. S1). As expected, the first group showed a pattern of scattered positive cells throughout the liver. In the case of SFV-IL-12-treated animals, expression was observed in groups of cells showing a morphology compatible with tumor area. Many of these cells seemed to be associated to necrotic areas. No staining was observed in liver sections of control mice. Overall, these data suggest that short-term IL-12 expression driven by SFV-IL-12 could be sufficient to trigger an efficient antitumor response, which was comparable with that obtained with long-term IL-12 expression.

Induction of cytokines in treated mice. IL-12
Antitumoral effects of SFV-IL-12 could be favored by induction of type I IFN responses and apoptosis of tumor cells
SFV vectors are able to elicit a type I interferon response in infected cells, which contributes to the immune responses induced by these vectors (Hidmark et al., 2006). In order to determine if this type of response was taking place in L-PK/c-myc mice bearing HCC, we measured the mRNA levels of IFN-α and -β in the liver and tumor tissues of SFV-IL-12, pTonL2(T)-mIL12 (+Dox), or saline cohorts after 2 days of treatment. SFV-IL-12 was able to induce IFN-β specifically in tumors (Fig. 5A), which correlated with SFV RNA being mainly detected in this tissue (Fig. 5C), which indicated low vector dissemination to the liver. Interestingly, IFN-α was not induced by SFV-IL-12 but was detected in the livers of pTonL2(T)-mIL12-treated mice (Fig. 5B). This result suggested that this second kind of treatment could be more toxic, since IFN-α can cause myelosuppression, leading to leukopenia and thrombocytopenia (Kirkwood et al., 2002). Another important feature of alphaviral vectors is their capability to induce apoptosis in tumor-infected cells, which can lead to the release of tumor antigens that can be taken up by antigen presenting cells, favoring antitumoral immune responses (Ying et al., 1999). TUNEL analysis showed a significantly higher number of apoptotic cells in tumors from SFV-IL-12-treated mice compared with tumors from saline- or pTonL2(T)-mIL12-treated animals (Fig. 6A and B).
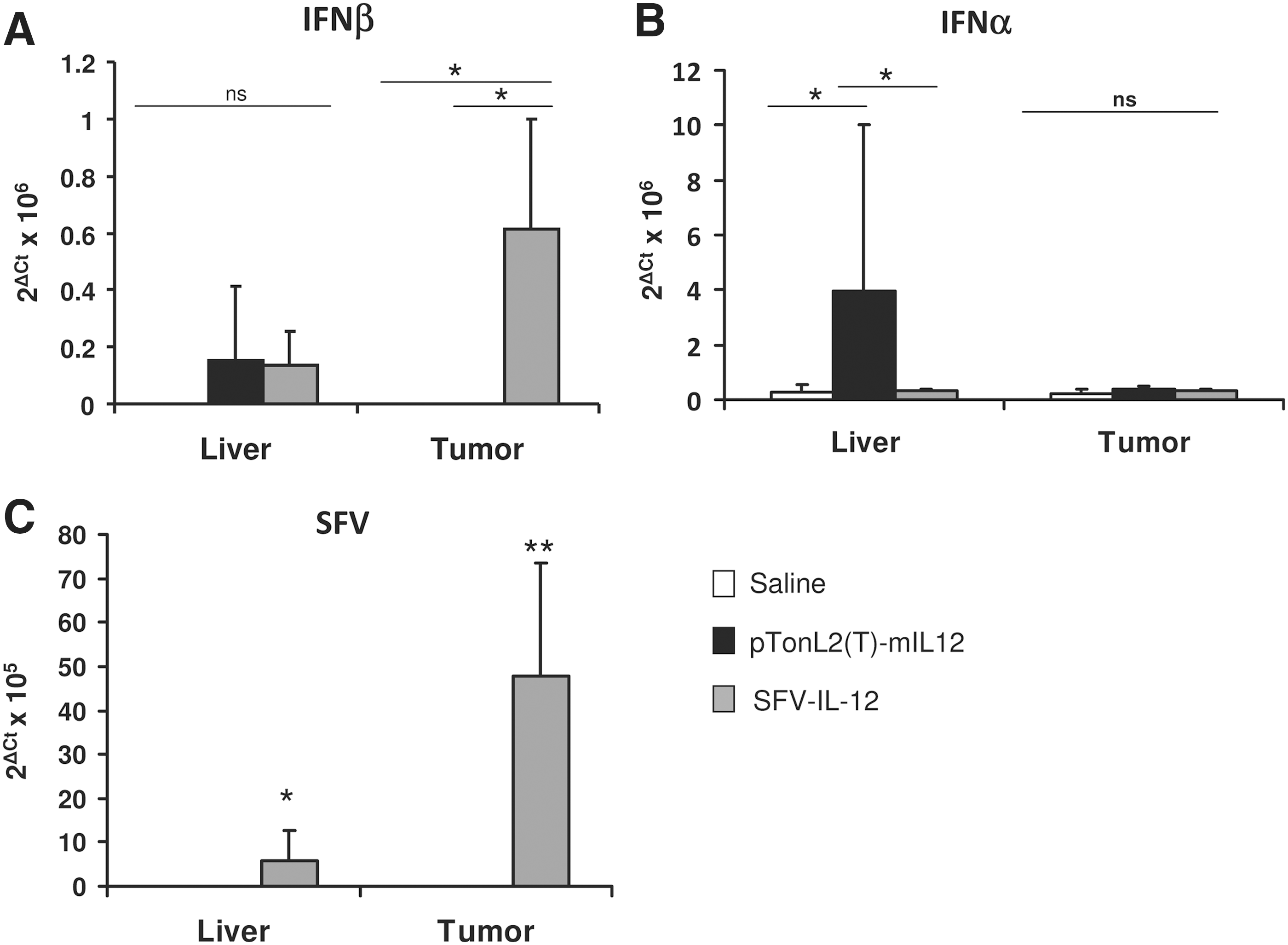
Induction of type I IFN responses. mRNA levels of the indicated IFNs
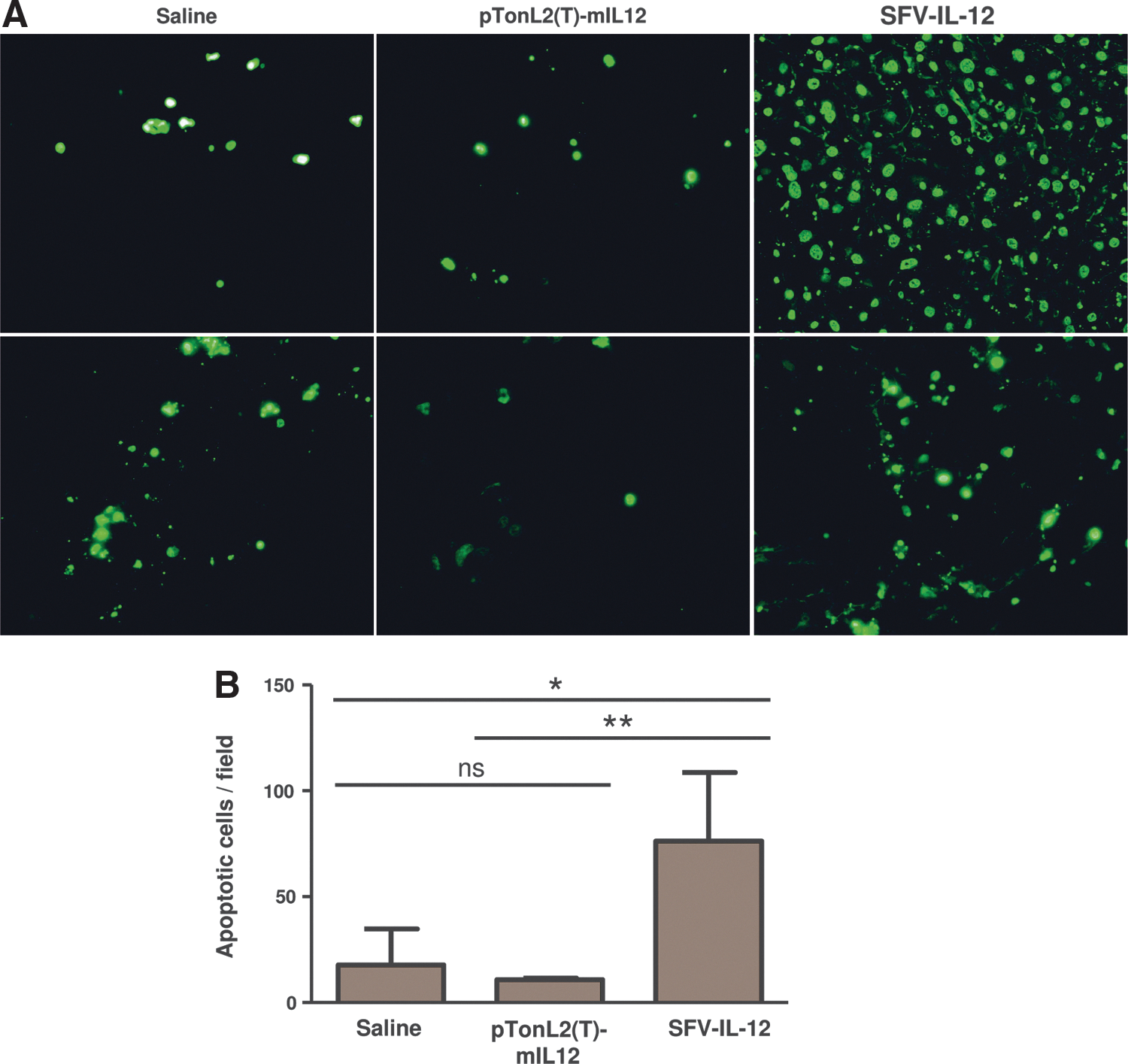
Induction of apoptosis in tumor cells.
Discussion
Immunotherapy constitutes a promising approach for cancer treatment, since the activation of a specific immune response against tumor cells could reach, in theory, all tumor cells present in the body. However, systemic delivery of cytokines with strong antitumoral activity, such as IL-12, can result in acute toxicity (Berraondo et al., 2009). This problem can be avoided by using expression vectors that are able to deliver cytokines locally at the tumor site, or in the adjacent tissue. Toxicity can also be greatly reduced by regulating the expression of cytokines using inducible systems.
Different viral and nonviral vectors expressing IL-12 have shown high antitumoral activity in a variety of tumor models (Tahara et al., 1995; Andrews et al., 2000; Zabala et al., 2007; Quetglas et al., 2010; Vanrell et al., 2011). However, the modest antitumoral responses mediated by vectors tested in clinical trials questions the translation of preclinical results (Sangro et al., 2004; Triozzi et al., 2005; Mahvi et al., 2007; Daud et al., 2008). We think that direct comparisons between available vectors in clinically relevant tumor models may enhance the success of gene therapy vectors in the clinic. For this reason, we directly compared two different IL-12-expressing vectors with different properties in a spontaneous HCC preclinical model developed in L-PK/c-myc transgenic mice (de La Coste et al., 1999; Zabala et al., 2007). For this comparison, we chose two vectors that had previously shown good antitumoral efficacy against liver tumor models, despite having very different features (Zabala et al., 2004, 2007; Guan et al., 2006; Rodriguez-Madoz et al., 2009; Quetglas et al., 2013). The first vector is based on the cytopathic SFV-IL-12 replicon that can provide high locally and transient levels of IL-12 (Rodriguez-Madoz et al., 2005). The second vector is based on a plasmid that can express sustained IL-12 levels in the liver in a regulated fashion [pTonL2(T)-mIL12] (Zabala et al., 2004). Despite their different patterns of cytokine expression (Fig. 4), both vectors were able to induce similar antitumoral responses, resulting in a significant reduction of HCC growth. In the case of SFV-IL-12, tumors received two doses of vector separated by 50 days (Fig. 2A). Although most viral vectors induce an immune response against viral proteins that prevents infection after a second administration, SFV vectors have been shown to have very low immunogenicity (Berglund et al., 1999; Rodriguez-Madoz et al., 2007). In fact, we showed that most animals that received a first dose of SFV-Luc could be re-infected with the same vector, leading to luciferase expression in tumors, although at lower levels than those detected after the first administration (Fig. 2B and C). Moreover, mice that received two doses of SFV-IL-12 were also able to express IL-12 after the second administration, although at lower levels, which also resulted in the detection of IFNγ in the sera of these animals. However, the length of expression of both cytokines in the SFV-IL-12 group was much lower than that observed in pTonL2(T)-mIL12-treated mice (Fig. 4).
A remarkable result of our study was that transient expression of IL-12 delivered from SFV was able to provide similar antitumoral effects compared with sustained expression of the same cytokine delivered by the pTonL2(T)-mIL12 plasmid. These data clearly indicate that short-term IL-12 expression from SFV-IL-12-transduced tumor cells could be sufficient to trigger an antitumoral response as potent as that provided by sustained IL-12 expression derived from a nonviral vector (Fig. 3). This conclusion is of paramount importance to choose the more suitable vector for clinical application.
A clue for the high antitumoral activity of SFV-IL-12 could be obtained from results observed with control SFV-Luc vector. This vector was able to increase the survival of mice compared with saline-treated mice, although we did not observe significant differences in tumor growth rate between these two groups (Fig. 3). This suggests that the antitumoral effects induced by SFV-IL-12 are not only because of IL-12 and IFN-γ expression and that vector RNA amplification by the viral SFV replicase in tumor cells could also be contributing to the therapeutic effects.
Production of double-stranded RNA in cells infected with SFV vectors functions as a “danger signal” that provides an adjuvant effect to vector-encoded antigens (Ying et al., 1999; Leitner et al., 2000). This danger signal leads to the induction of type I IFNs, which are the main cytokines that are involved in innate immune responses against viral infections. This type of response is known to enhance CTL responses while attracting effector lymphocytes toward infected tissue. In the absence of a vector-encoded antigen, this response could be directed against tumor antigens that are released from infected tumor cells. Indeed, ongoing studies from our group show the absolute need of type I IFN responses for the antitumoral effects of SFV-IL-12 (Hervas-Stubbs S. et al., manuscript in preparation). In the present study, we were able to detect a significant increase of IFN-β specifically in tumors treated with SFV-IL-12 at early times posttreatment (Fig. 5A). We also observed a type I IFN response in mice treated with pTonL2(T)-mIL12 (Fig. 5B), which could be the result of Toll-like receptor 9 activation by plasmid DNA (Akira and Hemmi, 2003). Although this response could also favor the antitumoral efficacy of this strategy, IFN-α production was observed mainly in the liver and could result in toxic effects. A second property of alphaviral vectors that can contribute to their antitumoral efficacy is the induction of apoptosis in tumor cells (Leitner et al., 2004). As shown in Fig. 6, SFV-IL-12 was able to induce apoptosis in a significantly higher number of tumor cells compared with mice treated with pTonL2(T)-mIL12 or saline. Cell death could facilitate the uptake of apoptotic bodies by antigen presenting cells, leading to an enhanced immune response against tumor antigens by a cross-presentation mechanism. In summary, the high antitumoral effects of SFV-IL-12 could be caused by a combination of factors, including high local cytokine expression, type I IFN secretion, and the induction of apoptosis.
Although both types of vectors that were tested in this study provided similar antitumoral effects, neither strategy was able to completely eradicate tumors, suggesting that treatment with a single agent, such as IL-12, is not enough and that more potent strategies are needed. In fact, a previous study by our group showed that the antitumoral activity of pTonL2(T)-mIL12 was limited by the activation of immunosuppressive mechanisms. One possibility to enhance the antitumoral effect of IL-12 could be to combine this cytokine with additional cytokines/factors that are able to enhance effector or memory immune responses or to inhibit suppressor responses. As an example, we have recently shown that a combination of SFV-IL-12 and a CD137 agonistic antibody can enhance antitumoral immune responses against subcutaneous melanoma tumors in mice (Quetglas et al., 2012).
Our use of a clinically relevant animal model that closely resembles human HCCs warrants interest from a translational research perspective. Although both the long-term and short-term expression vectors that we used in this study showed similar effects, the latter has some intrinsic advantages that could favor its clinical use: (i) alphaviral vectors can be produced at high titers and have been tested in clinical trials, showing a high degree of safety (Bernstein et al., 2009; Morse et al., 2010); (ii) phase I clinical trials have proven the feasibility and low toxicity of intratumoral injection of viral vectors into HCC nodules (Sangro et al., 2004, 2010; Penuelas et al., 2005); (iii) short-term IL-12 and IFN-γ expression was sufficient to achieve potent therapeutic effects, which could reduce the toxicity associated with the long-term expression of these cytokines; and finally, (iv) although improbable, a long-term expressing DNA vector such as pTonL2(T)-mIL12 could integrate into the genome of healthy liver cells, while the RNA SFV-IL-12 vector would quickly disappear because of the apoptosis of infected cells concomitant with tumor regression. All of these properties, together with the high antitumoral efficacy already observed in other clinically relevant models of spontaneous HCC (Rodriguez-Madoz et al., 2009), indicate that SFV-IL-12 could be clinically useful. In addition, it has recently been shown that advanced cancer patients could be treated with repeated immunizations of a recombinant alphaviral vector, despite the induction of neutralizing antibodies against the vector (Morse et al., 2010). In our study, we have also observed that it is possible to re-administer our vector, which can probably increase the potency of this type of therapy. Production of clinical-grade SFV-IL-12 will probably constitute one of the main challenges when taking this vector from bench to bedside. However, this challenge is technically feasible, as demonstrated by the production of a similar alphaviral vector for clinical use (Morse et al., 2010). In conclusion, we believe that SFV-IL-12 may have potential application for HCC gene therapy.
Footnotes
Acknowledgments
We would like to thank Christine Perret (Institut Cochin, Paris, France) for kindly providing the L-PK-cmyc transgenic mice strain. We also thank Erkuden Casales for excellent technical assistance, and Laura Guembe, Elena Remirez, Matilde Bustos, and Josu Sola (Clinica Universitaria, University of Navarra, Pamplona) for help with immunohistochemistry and histopathological analysis. We would also like to thank Neethan A. Lobo for kindly revising and editing the article. This work was supported by the Spanish Health Ministry (PI11/02190), Departamento de Educación del Gobierno de Navarra (GNE-VECTORES ALFAVIRUS), and the Center for Applied Medical Research-UTE Project.
Author Disclosure Statement
No competing financial interests exist for any of the authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.