
Abstract
Human papillomavirus (HPV) infection is responsible for all cervical cancer cases, other anogenital cancers, and head and neck tumors. The epidemiological relevance of HPV-induced tumors reinforces the need for the development of therapeutic antitumor vaccines. Clinical trials with different vaccine formulations, particularly DNA vaccines, have provided promising results but have still been unable to achieve the immunogenicity required for use in infected patients. In experimental conditions, anticancer HPV-specific vaccines induced E7-specific CD8+ T-cell responses but did not confer full therapeutic antitumor protection in mice with transplanted HPV-expressing TC-1 cells, which are the most frequently used nonclinical protection correlate for antitumor effects. Our group has developed a DNA vaccine strategy based on the fusion of HPV oncoproteins to the herpes virus gD protein. This vaccine promoted the induction of antigen-specific cytotoxic CD8+ T-cell responses and partial antitumor therapeutic effects based on the blockade of coinhibitory signals and the enhancement of coactivation mechanisms. In the present study, we report conditions leading to full therapeutic antitumor effects using the TC-1 cell murine model after a single vaccine dose. The combination of a coadministered plasmid encoding IL-2, optimization of the coding sequence for mammalian cells, and the use of different delivery routes resulted in enhancements of the E7-specific cytotoxic CD8+ T-cell responses and full therapeutic protection under experimental conditions. The combination of these strategies augmented the potency of the DNA vaccine formulation to levels not previously achieved by other therapeutic antitumor vaccines under similar experimental conditions, including some that have been taken to clinical trials.
Introduction
The available prophylactic HPV vaccines are based on the induction of antivirus antibodies that target capsid proteins. These vaccines are effective in preventing HPV infection with two specific oncogenic virus types (16 and 18), but they do not confer any protection to previously infected people and do not have effects on virus-induced cellular lesions. Moreover, the actual contribution of these vaccines in the reduction of cancer rates is expected to be observed only at the long-term because of the slow malignization progress. In addition, an epidemiologically significant decrease in the incidence of the disease will require massive vaccination, which is not presently occurring, particularly at developing countries, because of the high price of the vaccines.
The stimulation of E6- or E7-specific cytotoxic CD8+ T-cell responses is considered the best immunological correlate for antitumor protective immunity (van Duikeren et al., 2012). The efficacy of vaccines targeting HPV-associated tumors can be experimentally assessed in murine models that utilize either transgenic mouse strains that are genetically modified to express the HPV oncoproteins or, more frequently, isogenic mouse strains that are transplanted with malignant cells expressing the HPV oncoproteins. The most widely employed animal model is the C57BL/6 mouse strain and the TC-1 cell lineage, which consists of epithelial lung cells expressing the HPV-16 E6 and E7 oncoproteins and a copy of activated H-ras. This cell can be transplanted into the animals either subcutaneously or intravenously, resulting in a single tumor cell mass at the body flank or pulmonary nodules, respectively (Lin et al., 1996). Thus far, this tumor challenge animal model represents the most frequently used protection correlate for anticancer vaccines before testing potential vaccine formulations under clinical conditions (Hung et al., 2008).
Several therapeutic vaccines targeting HPV-induced tumors have been submitted to clinical trials. However, no formulation has convincingly demonstrated antitumor effects that could lead to the development of safe and effective immunotherapeutic approaches for humans (de Jong et al., 2002; Garcia et al., 2004; Einstein et al., 2007; Kenter et al., 2008; Welters et al., 2008; Van Doorslaer et al., 2009; Ma et al., 2010; Bagarazzi et al., 2012). In most cases, these vaccines were previously tested under preclinical conditions with the murine TC-1 cell challenge model, and the effects were expressed either as preventive tumor protection or, under therapeutic conditions, as tumor growth delay or reduction in the number of pulmonary nodules (Chen et al., 2000; Chu et al., 2000; Cheng et al., 2001; Zwaveling et al., 2002; Karanam et al., 2009; Zong et al., 2009). In all cases, complete therapeutic antitumor protection was not achieved, or it required several vaccines doses, particularly under the more restrictive condition, in which cells are implanted subcutaneously.
During the last few years, our group has been pursuing therapeutic DNA vaccines targeting HPV-induced tumors. The basic strategy has been based on the observation that the fusion of HPV oncoproteins to the herpes virus gD protein promoted a marked enhancement of antigen-specific cytotoxic CD8+ T-cell responses associated with antitumor protective effects (Lasaro et al., 2005; Diniz et al., 2010; Porchia et al., 2011). Through binding to herpes virus entry mediator (HVEM), gD promotes NF-kB activation and the blockade of coinhibitory pathways mediated by B and T lymphocyte attenuator (BTLA) and CD160, leading to enhanced antigen-specific activation of CD8+ T cells (Lasaro et al., 2008; Sciortino et al., 2008; Lasaro and Ertl, 2009; Steinberg et al., 2011). Using the TC-1 cell challenge model, our initial vaccine formulations based on the HPV-16 E7 oncoprotein required at least 4 intramuscular (i.m.) vaccine doses (100 μg of DNA per dose) to achieve a therapeutic protection of 40% in mice challenged subcutaneously with TC-1 cells (Lasaro et al., 2005). Subsequent modifications of the vaccine formulation, based on the incorporation of tandem fusion of additional HPV oncoproteins, coadministration of adjuvant plasmids encoding different cytokines, or the use of alternative administration routes, resulted in increased antitumor therapeutic effects (Diniz et al., 2010; Diniz and Ferreira, 2011). Thus far, full therapeutic antitumor effects have been achieved in mice challenged with the TC-1 cells after three i.m. doses (100 μg/dose) of the DNA vaccine, which encode three HPV-16 oncoproteins (E5–E7) genetically fused to the HSV gD protein, and were coadministered with plasmids encoding IL-12 or GM-CSF (Diniz et al., 2010). The same vaccine formulation conferred 50% therapeutic protection with 50-fold less DNA (2 μg of DNA/dose) after administration with a gene gun (Diniz and Ferreira, 2011).
In the present study, we report the optimization of a DNA vaccine encoding the HPV-16 E7 oncoprotein genetically fused to the HSV gD protein aiming to maximize the antitumor therapeutic effects using mouse TC-1 tumor cells as a protection correlate. On the basis of gene codon optimization, the coadministration of a plasmid encoding IL-2 and intradermal administration, we have achieved significant antitumor therapeutic effects compared with other DNA vaccines that target tumors induced by HPVs.
Materials and Methods
Mice
Female C57BL/6 mice aged 6–8 weeks were supplied by the Animal Breeding Center of the Biomedical Sciences Institute of the University of São Paulo and housed at the Parasitology Department of the University of São Paulo. All of the procedures involving handling and euthanasia followed the recommendations for the proper use and care of laboratory animals from the University of São Paulo Animal Ethics Committee (protocol number 136-2012).
The TC-1 cell line
The TC-1 cell line was kindly provided by Dr. T.C. Wu (John Hopkins University, Baltimore, MD). This cell line was transfected with a retroviral vector encoding v-HA-ras and E6 and E7 of HPV-16 (Lin et al., 1996). The TC-1 cells were propagated in Dulbecco's modified Eagle's medium supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 2 mM nonessential amino acids, 10 mM HEPES buffer, 50 units/ml penicillin/streptomycin, and 10% fetal bovine serum (FBS), and were kept at 37°C at 5% CO2. Before inoculation, the TC-1 cells were harvested by trypsinization, washed twice, and suspended in serum-free medium at the proper cell concentration.
DNA vaccines
The DNA vaccine encoding HPV-16 E7 genetically fused after amino acid 244 of the HSV-1 gD protein has been described previously (Lasaro et al., 2005). The gene sequence encoding the gDE7 chimeric protein was removed and cloned into a pVAX1 vector (Invitrogen) after digestion with HindIII (plasmid pgDE7). The DNA vaccine pgDE7h was constructed using a synthetic gene that carries the HPV-16 E7 gene fused at the same permissive site to HSV-1 gD with the gene sequence adapted to human codon usage. The codon optimization was performed following the algorithms of OptimumGene Optimization System (GenScript). Amino acids 24 and 26 from the HPV-16 E7 sequence were changed to glycines to eliminate any residual pRb-binding effect of the E7 protein. The synthetic gene was designed to contain the restriction sites PstI and BglII at the N′ and C′ terminus, respectively, for subsequent cloning into a pUMVC3 vector (Aldevron). The plasmids pVAX1 and pUMVC3 contain a CMV promoter and a kanamycin resistance gene. The plasmid encoding murine IL-2 was kindly provided by Dr. Sergio Costa Oliveira (University of Sao Paulo, Ribeirão Preto, Brazil) and encodes murine IL-2 in a pcDNA3 plasmid background (Invitrogen). The plasmid pcDNA3 contains a CMV promoter and an ampicillin resistance gene.
Immunization regimens and tumor cell challenge
Groups of 4–10 mice were challenged subcutaneously with 7.5×104 TC-1 cells suspended in 100 μl of serum-free medium and injected into the left rear flank of the animals. The mice were vaccinated with the DNA vaccines from 6 hr (same day) to 14 days after implantation of the tumor cells. The mice received one or two vaccine doses in weekly intervals. Tumor growth was monitored by visual inspection and palpation three times a week for a period of 60 days. The animals were scored as tumor-bearing when the tumors reached a size of approximately 2 mm in diameter. The mice were euthanized once the tumors exceeded a diameter of 1 cm or became necrotic. For the i.m. immunizations, each dose contained 10–50 μg of DNA vaccine alone or admixed with 10–50 μg of plasmid encoding IL-2. The DNA was resuspended in phosphate buffered saline (PBS) at a concentration of 1 μg/μl, divided into two 50 μl aliquots, and applied into the tibialis anterior muscle of each mouse hind limb. The intradermal immunization was performed as previously described (Diniz and Ferreira, 2011). Briefly, 2 μg of DNA-coated gold particles (1 μg DNA/bullet) was delivered to the shaved abdominal region of the mice using a helium-driven gene gun device (Biomics) with 400 psi charge pressure. The mice received one bullet of DNA vaccine and one bullet of plasmid encoding IL-2 in each dose in the same inoculation site.
Intracellular cytokine staining
Intracellular IFN-γ staining was performed using blood samples collected 14 days after vaccine administration. The blood samples were treated for lysis of red blood cells, centrifuged at 1,000×g for 5 min, and resuspended in RPMI medium supplemented with 10% FBS and 10−6 M 2-mercaptoethanol. The cells were cultured at a concentration of 106 cells for 6 hr at 37°C in 96-well round-bottom microtiter plates with 10 μg/ml of Brefeldin A (GolgiPlug; BD Biosciences). Some samples received 1.5 μg/ml of the E7-specific RAHYNIVTF peptide (amino acids 49–57) (Feltkamp et al., 1993) as an in vitro stimulus. After washing, the cells were incubated for 30 min at 4°C with 50 μl of a 1:100 dilution of an fluorescein isothiocyanate (FITC)-conjugated anti-CD8a. The cells were fixed and permeabilized with Cytofix/Cytoperm for 20 min at 4°C, washed with Perm/Wash buffer, and incubated in the same buffer for 30 min at 4° C with 50 μl of a 1:100 dilution of a PE-labeled anti-IFN-γ. The buffers and antibodies were purchased from BD Biosciences. After washing, the cells were resuspended in PBS and examined by flow cytometry using a FACS Calibur (BD Biosciences). The data were analyzed using FlowJo software (TreeStar). The percentages of CD8+ cells that stained positive for IFN-γ were determined.
CD8+ T-cell-mediated in vivo cytolytic activity
The in vivo cell lysis assay was carried out using splenocytes from naive mice as target cells, as previously described (Barber et al., 2003). These cells were stained with 0.5 or 5 μM of carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen). The population stained with 5 μM CFSE was pulsed for 40 min at 37°C with 1 μM of HPV-16 E7 peptide, which comprises the immunodominant MHC-I epitope for H-2Db mouse (RAHYNIVTF). The peptide-pulsed and nonpulsed cells were mixed and injected intravenously at 2×107 cells/mouse that had been previously challenged with 7.5×104 TC-1 cells. The mice were then immunized with one dose of the different immunization regimens 14 days after the vaccine administration. One day after the transfer of CFSE-labeled cells, splenocytes were harvested and analyzed for CFSE expression by flow cytometry in a FACSCalibur (BD Biosciences).
Statistical analyses
An ANOVA test was employed, followed by Tukey's posttest when individual data points were compared. Log-rank tests were performed whenever survival curves were compared. All p-values<0.05 were considered significant.
Results
Enhancement of the pgDE7-induced therapeutic antitumor effects after coadministration of an IL-2 encoding plasmid
Our attempts to enhance the therapeutic antitumor effects induced by a DNA vaccine encoding HPV-16 oncoproteins were carried out using a plasmid encoding the E7 oncoprotein that was genetically fused to the HSV-1 gD protein. Our previous results indicated that the coadministration of plasmids encoding IL-12 or GM-CSF could enhance the antitumor therapeutic effects of a gD-based DNA vaccine, as demonstrated with the TC-1 cell challenge model (Diniz et al., 2010). In the present study, we evaluated the adjuvant effect of a plasmid encoding IL-2, a cytokine known to promote proliferation of different T-cell populations, including cytotoxic CD8+ T cells (Waldmann, 2006). The administration of 3 i.m. doses (100 μg) of pgDE7 conferred 70% therapeutic antitumor protection to C57BL/6 mice with previously transplanted TC-1 tumor cells. Furthermore, the coadministration of a plasmid encoding IL-2 (pIL-2) with pgDE7 conferred full therapeutic protection after a single i.m. vaccine dose (100 μg of each plasmid) when administered 6 hr after TC-1 cell transplantation (Fig. 1A). No antitumor protection was detected in mice immunized with pIL-2 (200 μg) alone or with pVAX1 coadministered with pgDE7 (Fig. 1 and data not shown). We also tested the minimal vaccine dose that could confer full therapeutic protection to mice challenged with TC-1 cells. The results shown in Fig. 1B reveal that a single vaccine dose containing 50 μg of each plasmid DNA conferred 100% therapeutic antitumor protection. The antitumor protection dropped to 80% when the DNA was reduced to 50 μg (25 μg of each plasmid vector), while no protection was detected in mice immunized with 10 μg of each plasmid (Fig. 1B).
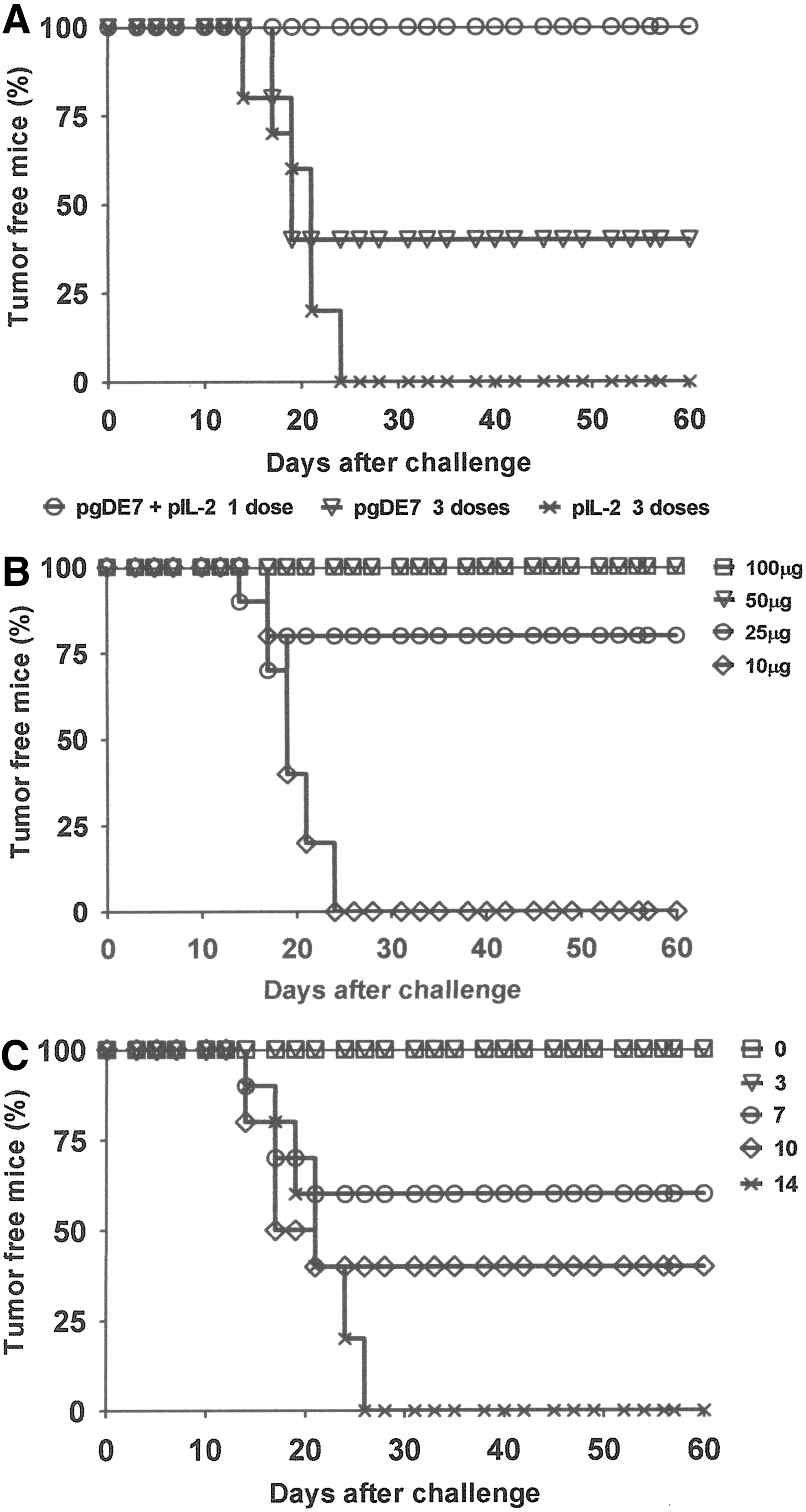
Coadministration of pgDE7 and pIL-2 confers full therapeutic protection to TC-1 cells.
We also tested whether the antitumor protection conferred by the vaccine would be maintained if the immunization was delayed with regard to the transplantation of the TC-1 cells. As indicated in Fig. 1B, full antitumor protection was maintained in the vaccinated mice (single dose containing 50 μg of each DNA vaccine) up to 3 days after the TC-1 cell challenge. Further delay in the administration of the vaccine caused a reduction in the antitumor protection, with 60% therapeutic protection in mice immunized 1 week after the challenge and 40% protection in mice vaccinated 10 days after the TC-1 challenge (Fig. 1C).
Coadministration of pgDE7 and pIL-2 increased activation of E7-specific CD8+ T-cell responses
The therapeutic protection to TC-1 tumor cells correlates to the induction of E7-specific CD8+ T-cell responses (Liu et al., 2000; Lamikanra et al., 2001; Cheng et al., 2002). Thus, we measured the induction of antigen-specific CD8+ T-cell activation in mice challenged with TC-1 and immunized with the different vaccine formulations. As demonstrated in Fig. 2A and B, the coadministration of pIL-2 and pgDE7 increased the activation of E7-specific CD8+ T cells in mice immunized intramuscularly with a single vaccine dose (50 μg of each plasmid) 3 days after the TC-1 cell challenge The adjuvant role of pIL-2 on the antitumor effects conferred by pgDE7 could also be demonstrated by in vivo antigen-specific cytotoxic effects mediated by the E7-specific CD8+ T cells (Fig. 2C). In this assay, splenocytes labeled with CFSE and a synthetic peptide, representing the immunodominant CD8+ T-cell epitope for the H-2Db haplotype, were more efficiently lysed after inoculation into mice immunized with pIL-2 and pgDE7 compared with mice immunized only with pgDE7 (Fig. 2C). These results confirmed that the enhanced therapeutic antitumor protection observed in the mice immunized with pIL-2 and pgDE7 correlated with a more efficient activation of cytotoxic E7-specific CD8+ T cells.
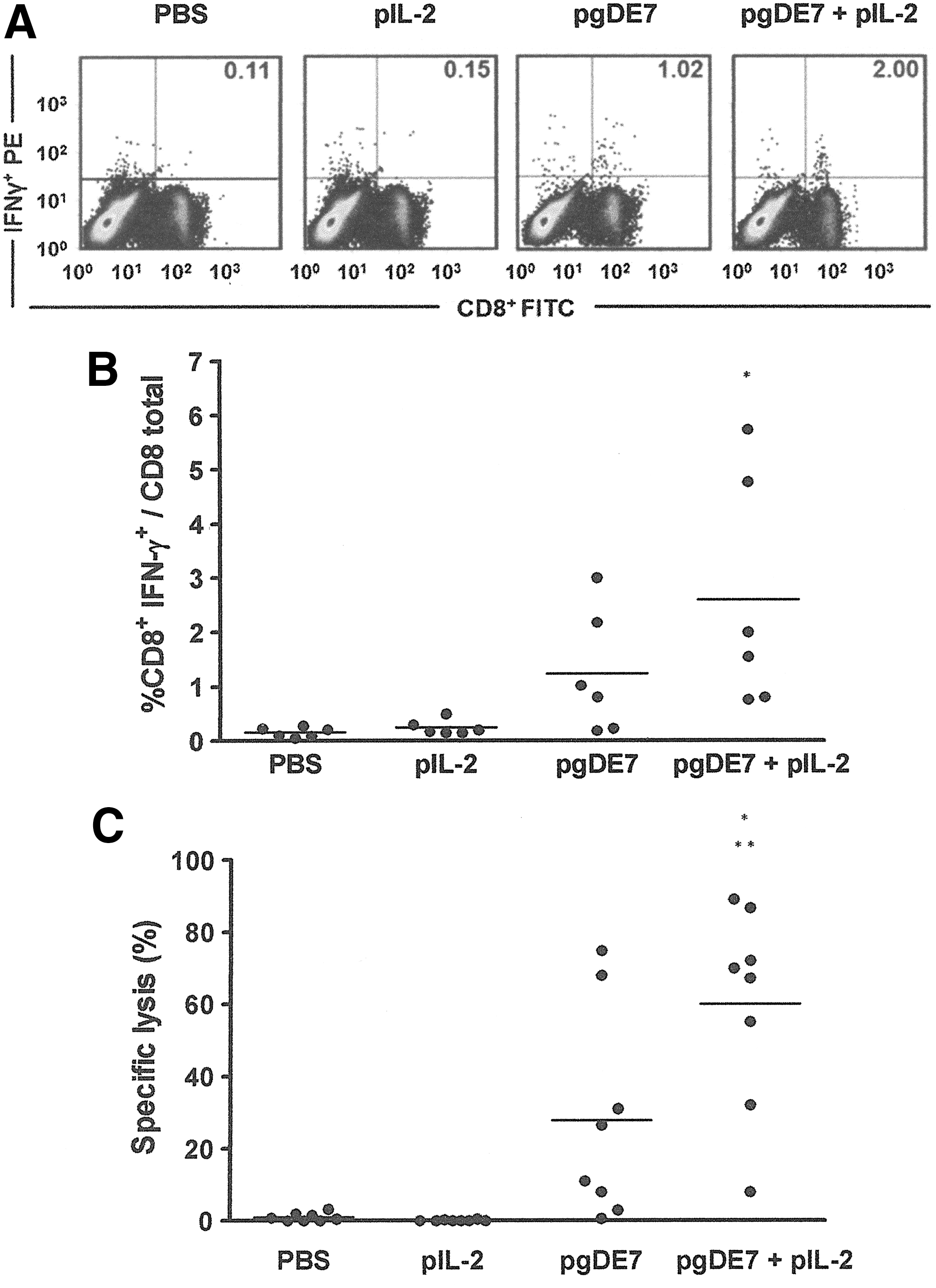
Activation of E7-specific CD8+ T cells in mice immunized with the DNA vaccines. Mice were challenged with TC-1 tumor cells (subcutaneous [s.c.] inoculation of 7.5×104 cells) and 3 days later were immunized intramuscularly with 50 μg of pIL-2 or pgDE7 or with 50 μg of pgDE7 admixed with 50 μg of pIL-2 or PBS as a control. Splenocytes and blood samples were collected 14 days after vaccination.
Codon optimization of the gD-E7 sequence enhances the antitumor effects and E7-specific CD8+ T-cell responses
Further enhancement of the therapeutic antitumor effects induced by the administration of the pgDE7 vaccine was obtained after modification of the gD-E7 nucleotide sequence to optimize codon utilization by mammalian cells and enhance the expression of the encoded antigens by transfected cells. Two-point mutations were also inserted into the E7 sequence (replacement of C24 and E26 by glycines) to avoid residual binding activity of E7 to the target cell-cycle controlling protein (pRb). The synthetic gene was cloned into a plasmid carrying a gene encoding resistance to kanamycin and was compatible with future clinical testing (pgDE7h). When tested in mice previously transplanted with TC-1 cells, pgDE7h induced higher protective immune responses than the original vector. One dose (50 μg) of pgDE7h administered intramuscularly 6 hr after the TC-1 challenge conferred 100% antitumor protection even in the absence of the pIL-2 vector (Fig. 3A). Nonetheless, the antitumor protection conferred by pgDE7h dropped to 70% if the vaccine was delivered 3 days after the tumor challenge (data not shown). The coadministration of the pIL-2 vector further enhanced the pgDE7h antitumor effects and conferred complete therapeutic antitumor protection up to 3 days after the TC-1 cell challenge in mice immunized with 25 μg or 50 μg of each plasmid (Fig. 3B and C, respectively).
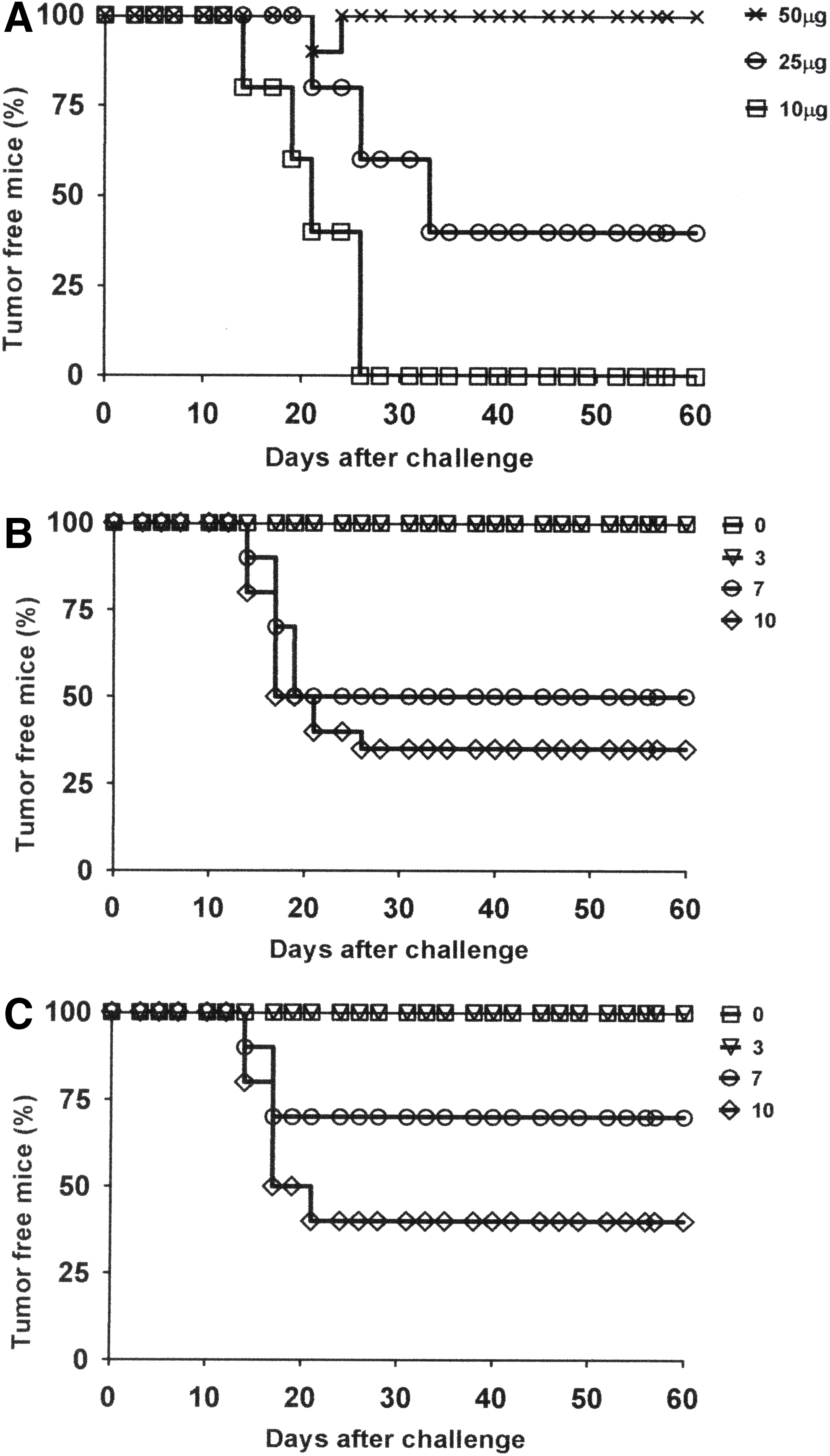
Codon use optimization enhanced the therapeutic antitumor effects of the vaccine.
Immunization with pgDE7h, alone or admixed with the plasmid encoding IL-2, enhanced the E7-specific cytolytic CD8+ T-cell responses. The mice immunized with pgDE7h or pgDE7h plus pIL-2 showed a dose-dependent enhancement of the frequencies of E7-specific IFN-γ producing CD8+ T cells and the in vivo cytotoxic E7-specific CD8+ T-cell-dependent responses (Fig. 4A–C). Although the coadministration of pIL-2 significantly increased the therapeutic antitumor effects of pgDE7h, the differences observed in the frequencies of CD8+ T-cell-dependent responses with regard to the mice immunized with only pgDE7h, although higher, did not reach a statistical significance (Fig. 4B and C). Collectively, these data demonstrated that pgDE7h codon optimization augmented both the immunogenicity and the therapeutic antitumor effects induced by the vaccine alone or coadministered with pIL-2.

Induction of E7-specific CD8+ T-cell responses in mice immunized with pgDE7h. Mice were challenged with TC-1 tumor cells (s.c. inoculation of 7.5×104 cells) and 3 days later were immunized intramuscularly with 25 or 50 μg of pgDE7h alone or admixed to the same amount of pIL-2. Alternatively, mice were inoculated with PBS as a control. Splenocytes and blood samples were collected 14 days after vaccination.
Administration of pgDE7h and pIL-2 by gene gun further enhanced the therapeutic antitumor effects
As a last step in the search for improvements in the experimental antitumor effects of the DNA vaccine, we tested the impact of the administration method. Mice challenged 3 days earlier with a gene gun device with 1 or 2 doses containing 1 μg of pgDE7h alone or in combination with 1 μg of pIL-2 maintained the antitumor protection. The administration of two doses of pgDE7h conferred 50% antitumor protection to the vaccinated mice (Fig. 5A). The mice immunized with one dose of pgDE7h plus pIL-2 developed 40% therapeutic tumor protection, but 2 doses conferred full therapeutic antitumor protection to the vaccinated animals (Fig. 5A). In addition, the administration of pgDE7h combined with pIL-2 induced a statistically significant increase in the number of E7-specific CD8+ T cells in comparison with the mice immunized with 2 doses (Fig. 5B and C). These results indicated that the therapeutic antitumor protection of pgDE7h was further enhanced after inoculation by the gene gun. Collectively, the present results suggest that pgDE7h is endowed with higher therapeutic antitumor effects under experimental conditions when compared with previously reported DNA vaccines developed for the same purpose.
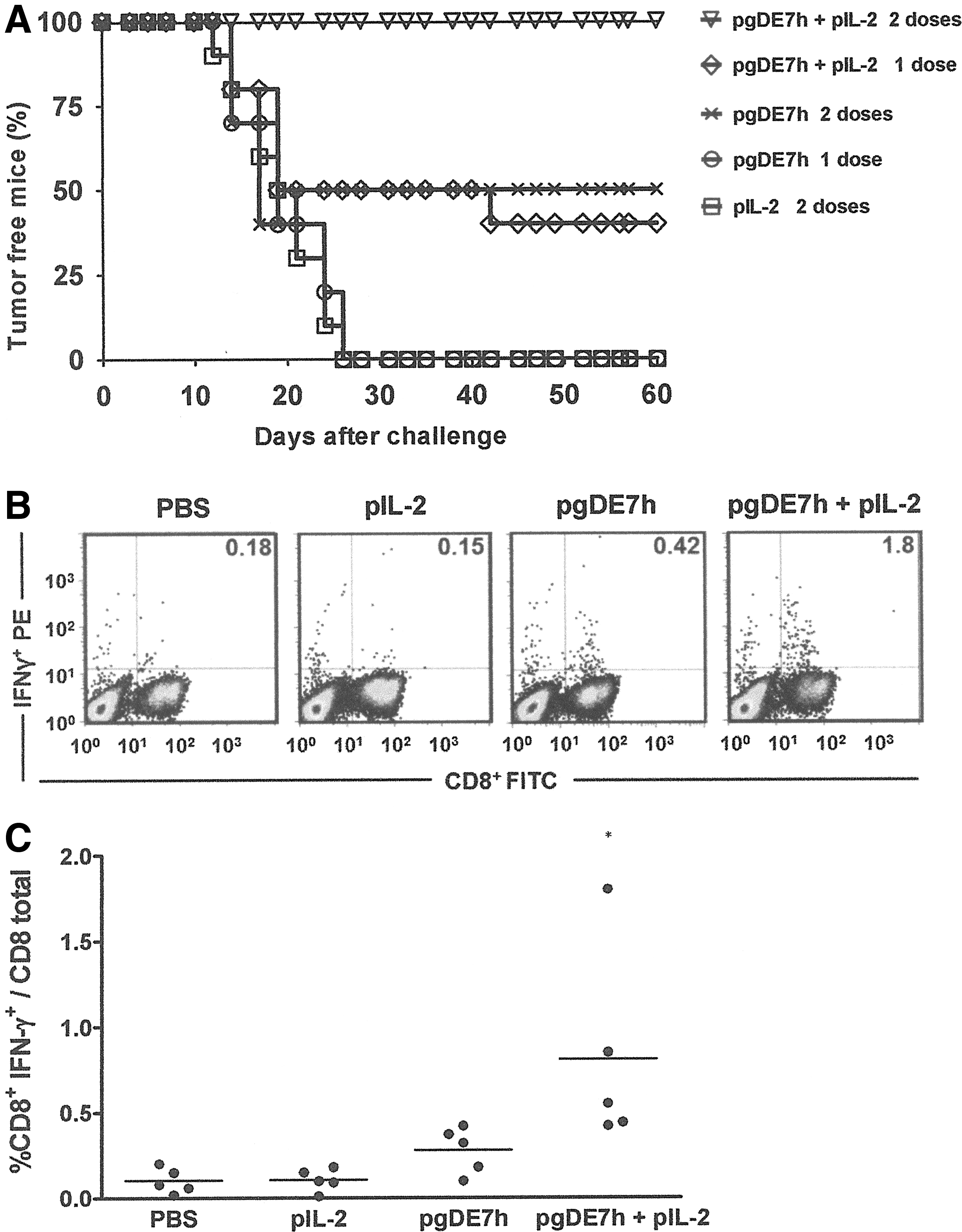
Therapeutic antitumor protection and induction of E7-specific CD8+ T-cell responses in mice immunized with pgDE7h admixed with pIL-2 delivered with a gene gun device. Mice were challenged with TC-1 tumor cells (s.c. inoculation of 7.5×104 cells) and vaccinated 3 days later using a gene gun device.
Discussion
DNA vaccines represent one of the most promising therapeutic anticancer approaches against HPV-induced tumors. Several clinical trials with different vaccine formulations have shown promising results, but none have achieved results that would warrant future licensing for human use. In fact, there is an urgent need for new vaccine formulations that can convincingly and reproducibly show immunogenicity and antitumor protection effects under both experimental and clinical conditions. Therefore, we performed several experiments to enhance the antitumor therapeutic effects of a DNA vaccine that was previously designed and tested by our group. We used the TC-1 cell transplantation model, the monitoring of E7-specific CD8+ T-cell responses, and in vivo measurement of E7-specific cytotoxic activity conferred by the vaccines as protection correlates. Our results demonstrated that the coadministration of a plasmid encoding IL-2, optimization of the codon usage for mammalian cells of the gDE7 gene and, the intradermal delivery by a gene gun device enhanced the potency of the DNA vaccine formulation to levels not previously recorded by other therapeutic antitumor vaccines previously tested under similar experimental conditions, including some that have been taken to clinical trials.
Cytokines have strong immunomodulatory adjuvant properties capable of stimulating both humoral and cellular responses (Dong et al., 1995; Heath, 1995; Iwasaki et al., 1997; Weiss et al., 1998). Although cytokines have great potential and plasticity to augment vaccine-induced immune responses, systemic cytokine administration is costly and can induce adverse effects (Rosenberg et al., 1989; Chow et al., 1997; Kang et al., 2012). The administration of a plasmid encoding cytokines can promote the expression of low but sustained levels of different cytokines, which provide adjuvant effects to vaccines with reduced toxicity. The in vivo expression of IL-15, GM-CSF, IL-18, IL-12, IL-2, and other cytokines augment the humoral and cellular immune responses to different antigens (Geissler et al., 1997; Chow et al., 1998; Song et al., 2000; Toubaji et al., 2007; Zhang et al., 2007; Li et al., 2008). Indeed, previous studies from our group showed that the coadministration of vector encoding IL-12 or GM-CSF enhanced the antitumor effects of a DNA vaccine encoding HPV-16 oncoproteins (Diniz et al., 2010; Diniz and Ferreira, 2011).
IL-2 is largely used in cancer clinical trials and is approved by the FDA for the treatment of metastatic renal cell carcinoma and malignant melanoma (Aoyama et al., 2012). IL-2 induces T-cell growth factor and promotes CD8+ T-cell killer activity (Farrar et al., 1981; Waldmann, 2006; Tian et al., 2012). In the present work, the coadministration of a plasmid encoding IL-2 enhanced E7-specific CD8+ T-cell activation and in vivo cytotoxic effects and, more relevantly, the therapeutic antitumor effect of the vaccine formulation. Other studies have demonstrated the adjuvant effects of IL-2 when combined with DNA vaccines targeting HPV-induced tumors, but the beneficial effects were observed only after the administration of bicistronic vectors or when the expression of IL-2 was genetically fused to the target antigen (Chow et al., 1998; Inoue et al., 2002; Lin et al., 2007). In our hands, the coadministration of the IL-2 encoding plasmid enhanced both the induction of E7-specific CD8+ T-cell responses and, most relevantly, the therapeutic antitumor effects of the vaccines based on the expression of the gDE7 protein. On the basis of this vaccine formulation, full therapeutic antitumor protection to the TC-1 cell challenge was achieved after a single i.m. vaccine dose containing 50 μg of each plasmid vector. Although the precise anticancer effects endowed by vaccines utilizing fusion of the HPV oncoproteins with the HSV-1 gD protein remain to be fully evaluated, previous experiments have demonstrated that the expression of recombinant gD attached to the outer face of the cytoplasmic membrane of transfected cells blocks a coinhibitory signaling pathway mediated by HVEM and BTLA, contributing to the more efficient activation of CD8+T cells (Lasaro et al., 2008). Under such conditions, the coexpression of IL-2 promotes the expansion of activated E7-specific CD8+ T cells and enhances the antitumor effects, irrespective of expression, in the same transfected cell and fusion with the target antigen.
Among the strategies employed to augment the potency of DNA vaccines is codon optimization of the target gene nucleotide sequence. Codon adaptation to mammalian cells enhances antigen expression, leading to higher immune responses. In our work, the codon optimization of the gDE7 chimeric gene caused an important stimulatory effect on the antitumor protective effects conferred by the DNA vaccine. The mice immunized with pgDE7h showed full therapeutic protection against the TC-1 cell challenge after a single vaccine dose without the coadministration of pIL-2. Nonetheless, the combination of the pgDE7 and pIL-2 further enhanced the antitumor protection effect, as demonstrated by the induction of full therapeutic antitumor protection with lower amounts of DNA and the option to delay the vaccine regimen until after transplantation of the TC-1 cells. The adaptation of codon usage, the modification of the E7 protein sequence to abolish residual oncogenic effects, and the replacement of the plasmid backbone are additional steps toward future testing under clinical conditions to improve both safety and immunogenicity.
More efficient delivery methods can be a pivotal element for a successful clinical trial with DNA vaccines. Intradermal gene gun immunization increases DNA vaccine potency through targeting the plasmid DNA to the skin, where a resident population of antigen-presenting cells (Langerhans cells) is present (Condon et al., 1996). In our hands, the gene gun immunization allowed a drastic reduction in the amount of DNA required to induce full therapeutic protection to the TC-1 cell challenge. Presently, gene gun immunization has been scaled for larger animals, including humans, and is being tested under clinical conditions with a DNA vaccine targeting the induction of immune responses capable of controlling HPV-induced tumors (Ma et al., 2010).
Several clinical studies have been conducted with therapeutic vaccines targeting the HPV E6 and E7 proteins, but none of them have reached licensing (Ma et al., 2010). Recent clinical studies were performed with vaccines based on proteins, peptides, or DNA that could activate immune responses, such as antibodies, CD4+ or CD8+ T cells, but the demonstration of full tumor regression remains to be achieved (de Jong et al., 2002; Goldstone et al., 2002; Sheets et al., 2003; Kenter et al., 2008; Welters et al., 2008; Bagarazzi et al., 2012). Four different DNA vaccines against HPV-induced tumors have been tested in clinical settings. The ZYC-101a encodes HPV-16 and 18 E6 and E7 peptides encapsulated into microparticles, and VGX-3100 encodes consensus HPV-16 and 18 E6 and E7 proteins delivered through the i.m. route followed by in vivo electroporation. Both vaccines have induced cytotoxic T-cell responses to at least one target antigen in approximately 70% of the vaccinated subjects (Sheets et al., 2003; Bagarazzi et al., 2012). Two additional DNA vaccines encoding codon-optimized HPV-16 E7 genetically fused to Mycobacterium tuberculosis Hsp70 and calreticulin, proteins with DC-stimulating effects, have also been submitted to clinical trials, but the immunogenicity results have not yet been published. Both of the vaccines were previously tested under experimental conditions with the TC-1 cell model and could not induce full therapeutic protection in mice, even after repeated immunizations (Chen et al., 2000; Cheng et al., 2001). The present description of an optimized DNA vaccine conferring strong therapeutic antitumor effects under experimental conditions opens perspectives for future clinical trials. The availability of this reagent for trials in humans can improve the chances for the discovery of a safe and efficacious immunotherapy against tumors induced by papillomaviruses.
Footnotes
Acknowledgments
This work was supported by CNPq Instituto Nacional de Ciência e Tecnologia de Vacinas, Fundação de Amparo à Pesquisa do Estado de São Paulo, and Universidade de São Paulo (Vaccine Research Core) Grants. We thankfully acknowledge the helpful technical assistance of E. Gimenes, Institute of Biomedical Sciences, University of São Paulo (USP), Brazil.
Author Disclosure Statement
No competing financial interests exist.