
Abstract
Several cancer vaccine efforts have been directed to simultaneously cotarget multiple tumor antigens, with the intent to achieve broader immune responses and more effective control of cancer growth. Genetic cancer vaccines based on in vivo muscle electro-gene-transfer of plasmid DNA (DNA-EGT) and adenoviral vectors represent promising modalities to elicit powerful immune responses against tumor-associated antigens (TAAs) such as carcinoembryonic antigen (CEA) and human epidermal growth factor receptor-2 (HER2)/neu. Combinations of these modalities of immunization (heterologous prime–boost) can induce superior immune reactions as compared with single-modality vaccines. We have generated a dual component–dual target genetic cancer vaccine consisting of a DNA moiety containing equal amounts of two plasmids, one encoding the extracellular and transmembrane domains of HER2 (ECD.TM) and the other encoding CEA fused to the B subunit of Escherichia coli heat-labile toxin (LTB), and of an adenoviral subtype 6 dicistronic vector carrying the same two tumor antigens gene constructs. The CEA/HER2 vaccine was tested in two different CEA/HER2 double-transgenic mouse models and in NOD/scid-DR1 mice engrafted with the human immune system. The immune response was measured by enzyme-linked immunospot assay, flow cytometry, and ELISA. The CEA/HER2 vaccine was able to break immune tolerance against both antigens. Induction of a T cell and antibody immune response was detected in immune-tolerant mice. Most importantly, the vaccine was able to slow the growth of HER2/neu + and CEA+ tumors. A significant T cell response was measured in NOD/scid-DR1 mice engrafted with human cord blood cells. In conclusion, the CEA/HER2 genetic vaccine was immunogenic and able to confer significant therapeutic effects. These data warrant the evaluation of this vaccination strategy in human clinical trials.
Introduction
A
Human epidermal growth factor receptor-2 (HER2)/neu and carcinoembryonic antigen (CEA) represent ideal TAAs, as they have various biological properties associated with the malignant phenotype of cells. HER2/neu is a member of a family of transmembrane receptor tyrosine kinases involved in signal transduction pathways that regulate cell growth and proliferation (Zhou and Hung, 2003). CEA is an immunoglobulin superfamily cell surface glycoprotein that mediates intercellular adhesion through homophilic interactions (Hammarström, 1999). Deregulated overexpression of CEA may contribute to tumorigenesis through the inhibition of cell differentiation and the disruption of tissue architecture (Ilantzis et al., 2002). Both antigens are overexpressed in malignant tissues, as opposed to low-level expression in normal tissues. In particular, HER2/neu and CEA are overexpressed in a significant percentage of epithelial tumors (as shown in Supplementary Table S1; supplementary data are available online at
These antigens are naturally immunogenic in patients with cancer (Disis et al., 2000; Nagorsen et al., 2000; Rentzsch et al., 2003). A spontaneous, albeit weak, T cell response against these antigens in patients with metastatic colorectal cancer and breast cancer has been reported. Thus, these proteins are suitable for therapeutic vaccination to induce long-lasting cytotoxic T and B cell responses that may exert therapeutic effects. Several clinical trials for cancer immunotherapy are being targeted against these proteins, among them studies based on the use of peptides, dendritic cells, and recombinant viral vectors have been reported (Turriziani et al., 2012).
Genetic vaccines are emerging among the most promising methodologies for cancer treatment (Aurisicchio et al., 2011a,b, 2012). Intramuscular delivery of DNA plasmids has been hampered by the poor efficiency of gene transfer as a result of the DNA degradation or lack of inefficient transduction of muscle cells. Combining intramuscular injection of plasmid vaccine with intramuscular electro-gene-transfer (DNA-EGT) increases the efficiency of gene expression and improves immune response in animals relative to simple intramuscular injection (Aurisicchio et al., 2011a,b, 2012; Impellizeri et al., 2012). Adenoviral vectors have been shown to be quite efficient in generating cell-mediated immunity (CMI) against foreign antigens. However, a major liability of the adenoviral vectors is their capacity to rapidly induce a neutralizing antibody response to capsid proteins, which limits their use in revaccination (Dharmapuri et al., 2009).
The majority of cancer vaccine regimens under development consist of repeated injections of the same vaccine. More recently, there has been a growing body of evidence to suggest that heterologous prime–boost immunization regimens are capable of generating higher amplitude and more durable immune responses, and that they exert better prophylactic and therapeutic efficacy in a variety of preclinical disease models (Lu, 2009; Vasconcelos et al., 2012). The heterologous prime–boost approach, particularly when conducted with two different gene-based delivery systems, has been shown to induce a strong CMI response, which is the type of immunity expected to be most involved in the control of cancer cell growth. Data, from both preclinical and clinical vaccination studies against infectious agents, have demonstrated that DNA vectors appear to be the most effective at priming when followed by boosting with a viral vector, being associated with significantly greater immune responses than seen with either agent alone (Woodland, 2004). Hence, combined treatment with DNA-EGT and adenoviral vaccine may give rise to superior immune responses that may result in increased therapeutic efficacy.
In light of these data, we have taken the approach to develop a heterologous cancer vaccine regimen consisting of DNA-EGT injection followed by boosting with an adenoviral vector. An adenovirus subtype 6 (Ad6) vector has been selected because of the substantially lower frequency of preexisting neutralizing antibodies to Ad6 in the human population as compared with Ad5 (Colloca et al., 2012; and our unpublished data). Furthermore, the immunogenicity of Ad6 is similar to that of Ad5, it usually presents an excellent genetic stability profile, and is amenable to large-scale production.
In this study, we show that a heterologous DNA-EGT/adenovirus vaccine regimen targeting CEA and HER2/neu results in an increase in immunogenicity and therapeutic efficacy in two CEA/HER2 double-transgenic mouse models. Moreover, a strong cellular response could be detected in immunocompromised mice engrafted with the human immune system. Our data support the evaluation of a heterologous prime–boost regimen based on DNA-EGT and adenoviral vector targeting CEA and HER2/neu in human clinical trials.
Materials and Methods
Animals and cell lines
Six-week-old BALB/c, C57BL/6, and CB6 mice were purchased from Charles River Breeding Laboratories (Calco, Italy). CEA.Tg mice (H-2b) were provided by J. Primus (Vanderbilt University, Nashville, TN) (Clarke et al., 1998). BALB/NeuT mice (Lucchini et al., 1992) were provided by G. Forni (University of Turin, Turin, Italy) and bred under specific pathogen-free conditions by Charles River Breeding Laboratories. BALB/NeuT and CEA-NeuT (BALB/NeuT×CEA.Tg) were screened for the presence of the transgene by duplex PCR as described (Gallo et al., 2005). B6.HGR mice (Piechocki et al., 2003) carry the full-length wild-type cDNA of human HER2 under the whey acidic protein (WAP) promoter. B6.HGR×CEA.Tg mice (CEA-hHER2) were screened by PCR as previously described (Piechocki et al., 2003; Conforti et al., 2009).
NOD-scid-DR1 mice were bred at Merck Research Laboratories (Whitehouse Station, NJ) by back-crossing the transgenic B10-DR1 mice onto NOD/scid mice from the Jackson Laboratory. Mice were maintained in the barrier facility. All procedures performed on the animals were approved by the institutional animal care and use committee. At the end of the treatment period and before necropsy, mice were killed by administration of compressed CO2 gas in cylinder as indicated by the American Veterinary Medical Association (AVMA) Panel on Euthanasia (2013) and according to the guidelines described by the U.K. Coordinating Committee on Cancer Research (1998). The experiments were conducted according to EU Directive EC86/609 on the protection of animals used for experimental and other scientific purposes, which was ratified by Italian legislation (DL no. 116/92) on February 19, 1992.
The MC38-hCEA/hHER2 cell line was generated by transfecting MC38-hCEA cells (Mennuni et al., 2005) with pcDNA3-hHER2/neu plasmid followed by clone selection with hygromycin. The JY cell line is an Epstein–Barr virus (EBV)-immortalized B cell lymphoblastoid line and was purchased from the American Type Culture Collection (ATCC, Manassas, VA).
Engraftment of NOD/scid-DR1 mice with human cord blood cells
The procedure followed was as described elsewhere (Camacho et al., 2007). Cord blood was purchased from the National Disease Research Interchange (NDRI, Philadelphia, PA), and separated by lymphocyte separation medium (ICN Biomedicals, Aurora, OH) density gradient to obtain mononuclear cells (MNCs). Briefly, NOD/scid-DR1 mice were engrafted with HLA-A2 human cord blood MNCs (CBMNCs) at 5 days of age by intrahepatic inoculation with 2–5 million CBMNCs, after light irradiation (60–70 rad).
Plasmid constructs and adenoviral vectors
Plasmids pV1J/CEA-LTBopt and Ad5/CEA-LTBopt carry the codon usage-optimized cDNA of CEA fused to the B subunit of Escherichia coli heat-labile toxin (LTB) and has been previously described (Facciabene et al, 2007). pV1J/ratHER2-ECD.TM and Ad5/ratHER2-ECD.TM carry codon usage-optimized cDNA encoding the rat extracellular and transmembrane domains of HER2 (rat ECD.TM) and has been described elsewhere (Gallo et al., 2007; Cipriani et al., 2008). For the construction of pV1J/HER2-ECD.TM, the cDNA corresponding to the human HER2-ECD.TM region was amplified by PCR, using sequence-specific primers from plasmid pV1J/HER2 that contains the entire codon-optimized cDNA of HER2. The forward primer carried an EcoRV restriction site at its 5′ end, whereas the reverse primer carried an SalI restriction site. The amplified DNA fragment was restricted with EcoRV and SalI and was directionally cloned into the linearized pV1J vector. Key steps involved in the construction of V932 are described in the text that follows.
Construction of adenoviral shuttle vectors
Shuttle plasmid pNEBAd6-CEA-LTB/HER2.ECD.TM was constructed by removing the dicistronic expression cassette from polyMRK-CEA-LTB/HER2ECD.TM SV40 by SpeI and AflII and inserting it in the same restriction sites of pNEBAd6-2HCMVnefMCMVgagpol. The monocistronic shuttle vectors pNEBAd6-CEA-LTB and pNEBAd6-HER2ECD.TM were constructed by removing the expression cassettes by SpeI and AflII and inserting them in the ClaI site of the pNEBAd6 vector. All the ends were modified by fill-in with T4 DNA polymerase. The genetic structure of pNEBAd6-CEA-LTB/HER2ECD.TM was verified by restriction enzyme analysis.
Construction of preadenoviral plasmid
To construct preadenoviral vector pV932, the transgene-containing fragment was liberated from shuttle plasmid pNEBAd6-CEA-LTB/HER2ECD.TM by digestion with restriction enzymes PacI and PmeI and gel purified. The purified transgene fragment was then cotransformed into E. coli strain BJ5183 with linearized (ClaI-digested) adenoviral backbone plasmid pAd6MRKDE1DE3. Plasmid DNA isolated from BJ5183 transformants was then transformed into competent E. coli DH5α for screening by restriction analysis. The desired plasmid pV932 was verified by restriction enzyme digestion and DNA sequence analysis.
Immunization
Mice were injected with 5–50 μg of plasmid DNA in a 50-μl volume in mouse quadriceps muscle followed by electroporation as previously described (Facciabene et al., 2007). Similarly, adenovirus injections (108–1010 viral particles [VP]/mouse, in a 50-μl volume) were carried out in quadriceps.
Peptides
Lyophilized CEA and HER2/neu peptides were purchased from JPT Peptide Technologies (Berlin, Germany) and resuspended in dimethyl sulfoxide (DMSO) at 40 mg/ml. Pools of peptides consisting of 15 amino acids overlapping by 11 residues were assembled as previously described (Mennuni et al., 2005). Peptides and pools were stored at −80°C.
Antibody detection and titration
Sera for antibody titration were obtained by retro-orbital bleeding. ELISA was performed with human CEA protein as previously described (Mennuni et al., 2005; Cipriani et al., 2008). Anti-TAA serum titers were calculated as the reciprocal limiting dilution of serum producing an absorbance at least 3-fold greater than the absorbance of autologous preimmune serum at the same dilution.
Cytokine intracellular staining and cytotoxic T lymphocyte assay
The peripheral immune response was measured as previously described (Giannetti et al., 2006). Briefly, peripheral blood mononuclear cells (PBMCs) or scratched splenocytes were treated with ACK lysing buffer (Life Technologies, Carlsbad, CA) for red blood cell lysis and resuspended in 0.6 ml of RPMI–10% fetal calf serum (FCS) and incubated with the indicated pool of peptides (5-μg/ml final concentration of each peptide) and brefeldin A (1 μg/ml; BD Biosciences, San Jose, CA) at 37°C for 12–16 hr. Cells were then washed and stained with surface antibodies. After washing, cells were fixed, permeabilized, and incubated with fluorescein isothiocyanate (FITC)-conjugated interferon (IFN)-γ antibodies (BD Biosciences), fixed with formaldehyde (1% in phosphate-buffered saline [PBS]), and analyzed on a FACSCalibur flow cytometer, using CellQuest software (BD Biosciences). DMSO and staphylococcal enterotoxin B (SEB, cat. no. S-4881; Sigma-Aldrich, St. Louis, MO) at 10 μg/ml were used as internal negative and positive controls of the assay, respectively.
Cytolytic activity was assessed by culturing spleen cells isolated from NOD/scid-DR1 mice with the peptides in microtiter wells for 7 days. After washing the cells once, they were incubated with the chromium-51-labeled EBV-transformed cell line JY (HLA-A2+), with or without peptides in a 4-hr lytic assay. Cell-free supernatant was collected and the release of chromium by lysed cells was measured with a scintillation counter. Percent lysis was calculated as follows: percent lysis=100×(experimental – spontaneous lysis)/(maximum lysis – spontaneous lysis).
Statistical analysis
Log-rank test and a two-tailed Student t test were used where indicated. All analyses were performed in JMP version 5.0.1 (SAS Institute, Cary, NC).
Results
Immunogenicity of CEA/rat HER2 vaccine
To verify whether it is possible to simultaneously break tolerance to HER2/neu and CEA in an immunologically tolerant mouse model, BALB/NeuT mice (BALB/c background, H-2d) and CEA.Tg mice (transgenic for human CEA, C57BL/6 background, H-2b) were crossed and double-transgenic mice from the F1 progeny (CEA-NeuT) were subjected to repeated immunizations with a mixture of plasmids pV1J/ratHER2-ECD.TM and pV1J/CEA-LTB. CB6 mice (BALB/c×C57BL/6) with the same genetic background were used as the nontolerant control group. Mice were subjected to five weekly injections of 50 μg of each construct; all vaccinations were accompanied by EGT (Fig. 1A). Two weeks after the last DNA-EGT, mice were boosted with the Ad5-ratHER2-ECD.TM and Ad5-CEA-LTB combination, 109 VP each. The amplitude of the CEA and rat HER2/neu-specific immune responses was analyzed by intracellular staining for IFN-γ. The immune response to CEA was determined with pools of peptides covering amino acids 137–327 and 497–703. The response to rat HER2/neu was measured with pool of peptides covering amino acids 1–651. The results in Fig. 1B and C show that the vaccination could induce a strong CMI response to both TAAs in CB6 mice and that immune tolerance significantly affected its amplitude, because the response against CEA and rat HER2/neu in CEA-NeuT mice was severely blunted. Similarly, the antibody titer against CEA was significantly lower in CEA-NeuT mice. CEA/– littermates were used as control, showing tolerance only to CEA antigen. As shown in other models, the adenovirus boost induced a steady increase in the immune response (p=0.002; Fig. 1C).
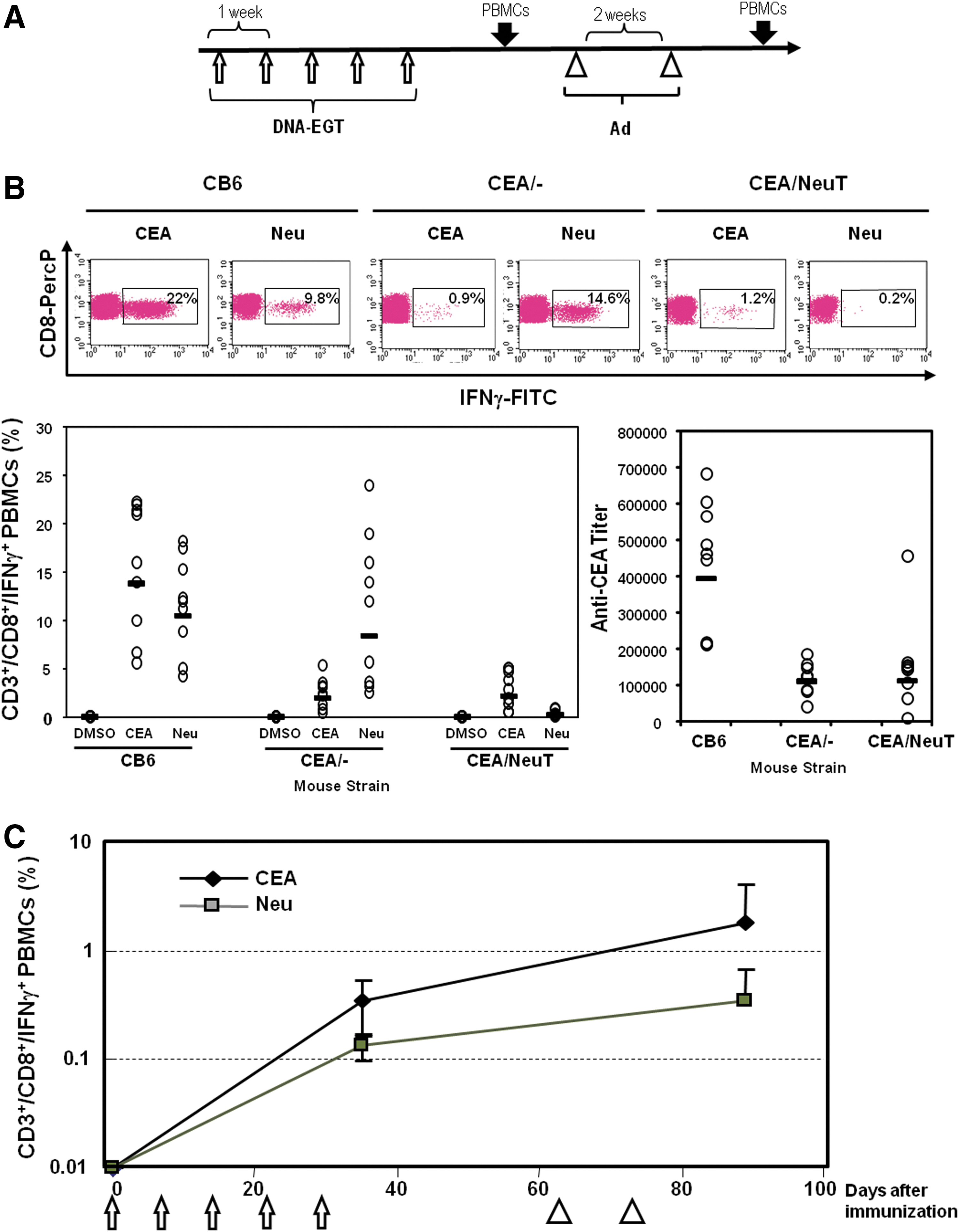
Electro-gene-transfer of plasmid DNA (DNA-EGT) and adenovirus are immunogenic and can break tolerance simultaneously to human epidermal growth factor receptor-2 (HER2)/neu and carcinoembryonic antigen (CEA).
To verify whether the concurrent immunization with two plasmids leads to impaired immune responses because of antigen competition, CEA-NeuT mice were vaccinated with pV1J/ratHER2-ECD.TM, pV1J/CEA-LTB, or a mixture of the two plasmids. CMI responses against rat HER2 and CEA were not significantly affected by coimmunization (p=0.29 and p=0.073, respectively; Fig. 2A). In contrast, the anti-CEA antibody response in covaccinated mice, although still remaining high (antibody titer, approximately 1:40,000), was about 2-fold lower than on vaccination with pV1J/CEA-LTB only (p=0.008; Fig. 2B). These data show that the combined CEA/rat HER2 vaccine is immunogenic and capable of breaking immune tolerance.
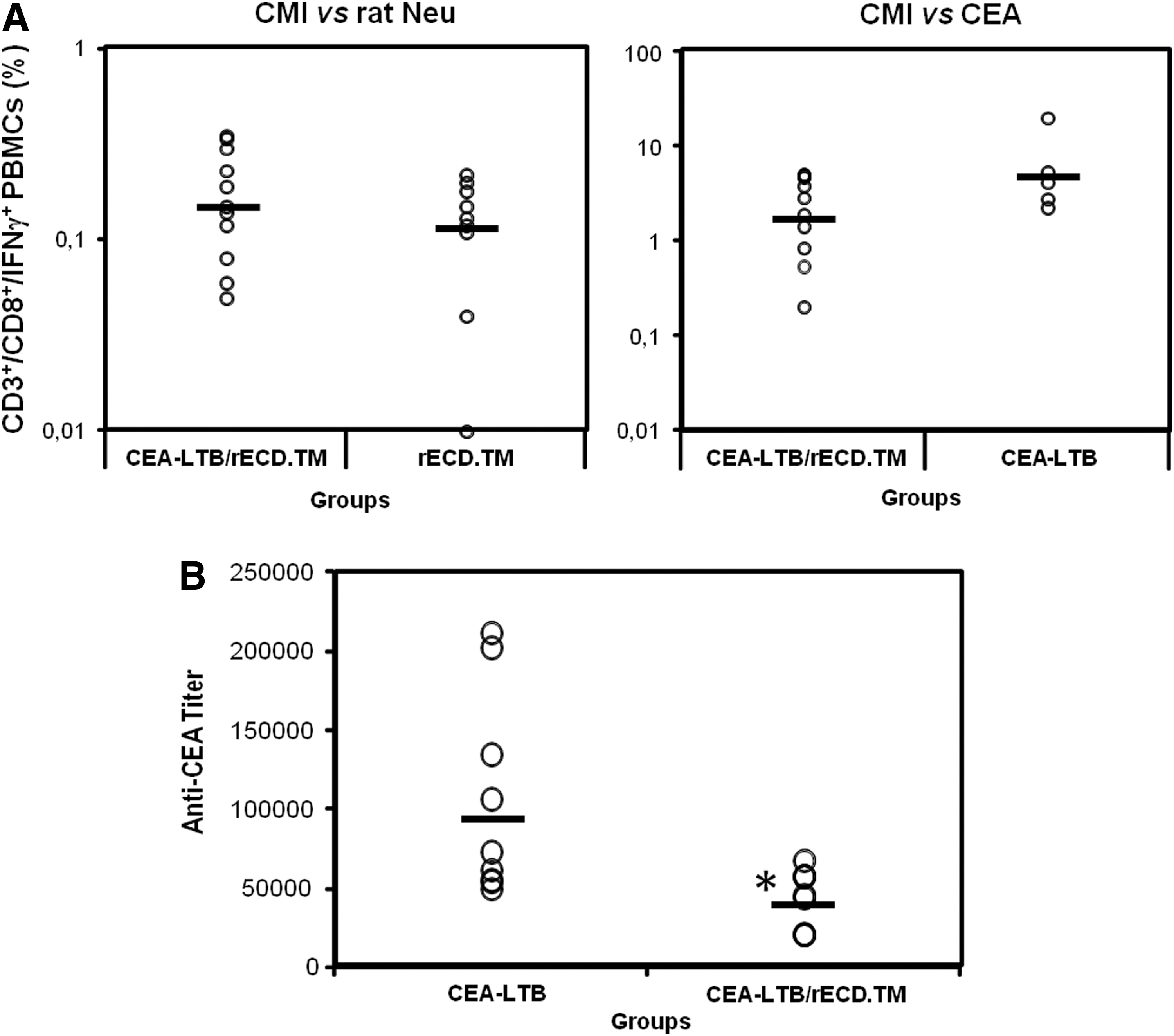
Coadministration of CEA-LTB and rat HER2/neu ECD.TM partially impairs the immune response. Groups of CEA-NeuT mice were vaccinated as indicated in text. After five DNA-EGT, PBMCs and serum were analyzed by intracellular staining for IFN-γ and ELISA, respectively.
CEA/rat HER2 vaccine protects mice from tumor development
BALB/NeuT mice spontaneously develop mammary tumor in all 10 mammary glands (Lucchini et al., 1992). To study the effect of the combination of CEA/rat HER2 vaccine in an immunologically tolerant environment on spontaneous tumor development, groups of CEA-NeuT mice were treated starting at 10 weeks of age. Plasmid DNA vaccine was administered at weeks 10, 11, 12, and 13 by EGT and adenovirus was injected at weeks 15 and 17. By week 22, tumors became palpable in the control group and the development of mammary gland lesions was monitored. The vaccinated group remained tumor free until week 31 (Fig. 3A). The control group developed tumors in at least six mammary glands by week 34. The vaccine conferred a significant antitumor effect, with mice having an average of two palpable lesions at week 38 (Fig. 3B).
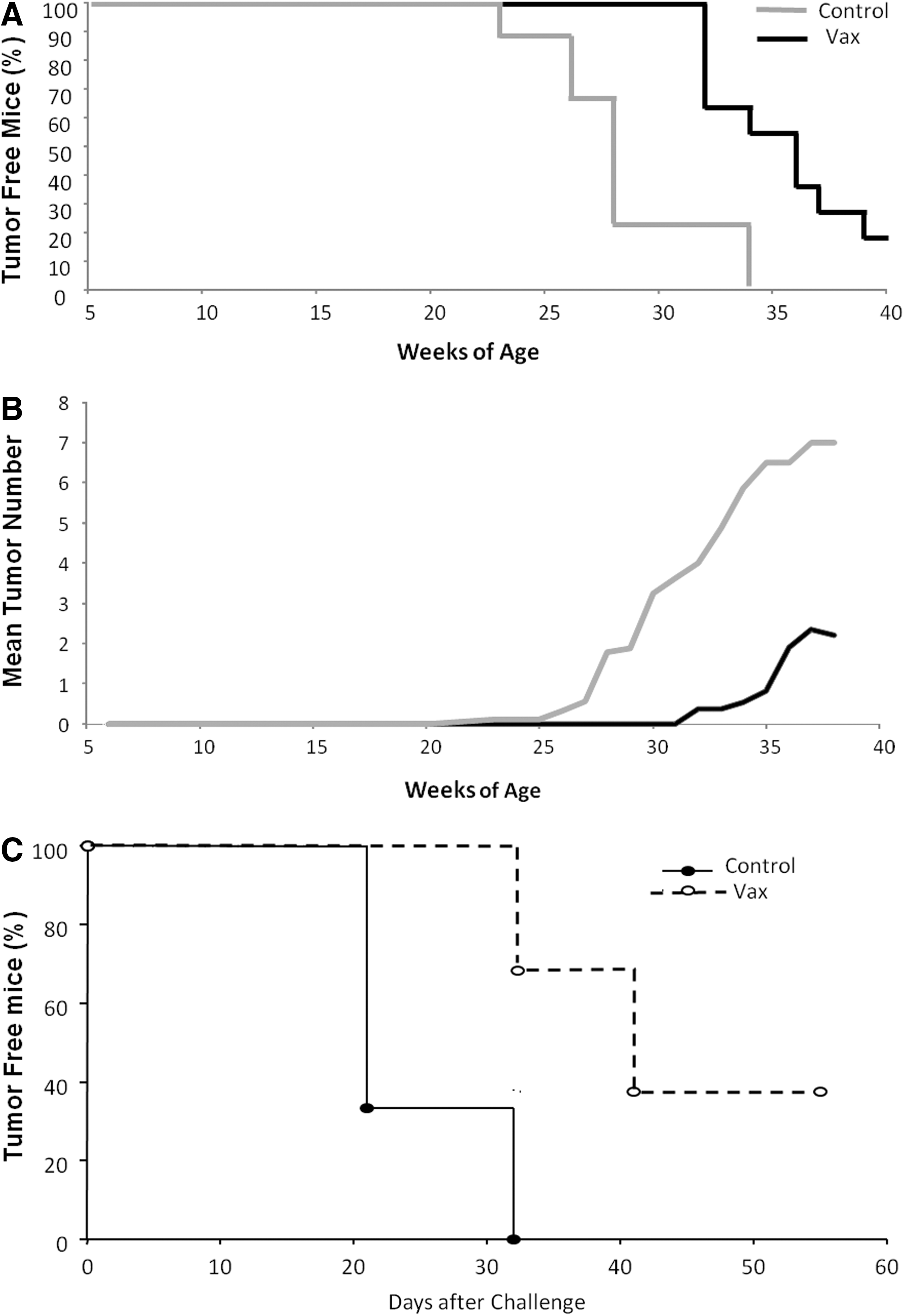
Antitumor effects of dual-target/DNA-EGT and adenovirus. Groups of 10 CEA-NeuT female mice were vaccinated starting at week 10 of age as described in text and monitored once per week for tumor development. Tumor assessment was performed by palpation of the mammary glands.
Because mammary tumors developing in CEA-NeuT double-transgenic mice do not express CEA (data not shown), to assess the impact of immunity against CEA groups of CEA-NeuT mice were vaccinated as described previously and then challenged with MC38-CEA (H-2b) colon adenocarcinoma cells. As shown in Fig. 3C, all control mice developed palpable tumors by day 32 after implantation, whereas 40% of vaccinated mice remained tumor free for the entire course of the experiment. In addition, the adenocarcinoma growth rate in vaccinated tumor-bearing mice was noticeably slower than in control mice whereas there was no therapeutic impact on parental MC38 (CEA-negative) tumors (data not shown). These data indicate that the immune response elicited by CEA-rat HER2 vectors was indeed antigen specific and able to interfere with tumor progression.
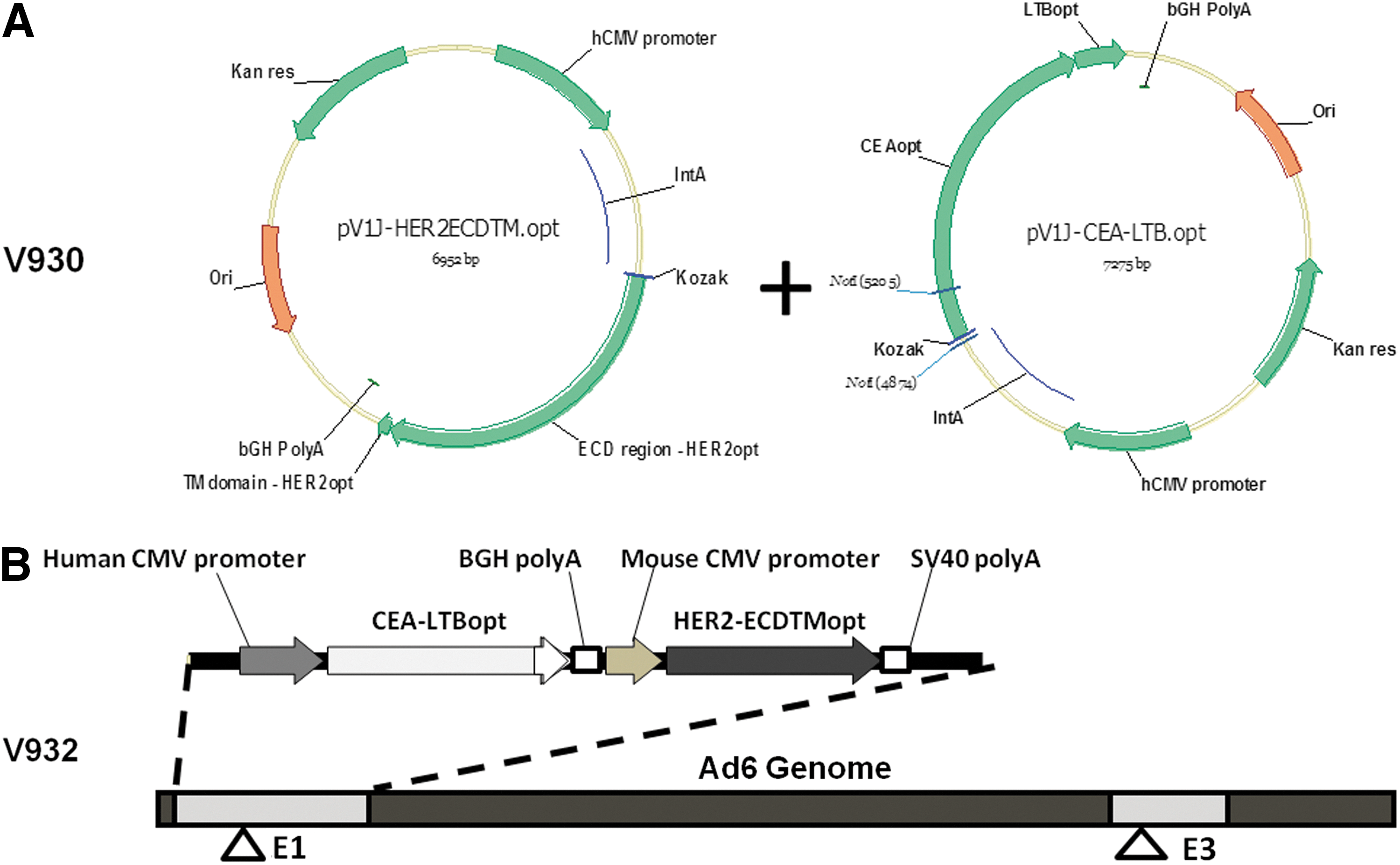
V930 and V932 immunogenicity and antitumor efficacy in wild-type mice
The mixture of plasmids described previously, expressing CEA-LTB and human HER2-ECD.TM, was designed as V930 (Fig. 4A). For clinical translation, the use of two adenoviral vectors can represent an industrial and regulatory challenge. For this reason, we sought to generate a single Ad6-based dicistronic vector (V932; Fig. 4B). This vector contains the CEA-LTB expression cassette consisting of (1) the immediate-early gene promoter from human cytomegalovirus, (2) the coding sequence of CEA fused at its C terminus with the subunit B of E. coli heat-labile enterotoxin, and (3) the bovine growth hormone polyadenylation signal sequence. The CEA-LTB cassette is directly followed by the HER2-ECD.TM expression cassette consisting of (1) the immediate-early gene promoter from mouse cytomegalovirus, (2) the coding sequence of a truncated form of the human HER2/neu protein that encompasses the extracellular and transmembrane domain (ECD.TM), and (3) the simian virus 40 early polyadenylation signal sequence. The codon usage in the CEA-LTB and HER2-ECD.TM open reading frames was optimized for expression in human cells (Facciabene et al., 2007).
To verify the ability of V932 to elicit immune responses to both target antigens two studies were carried out. In the first one, groups of BALB/c and C57BL/6 mice were immunized with different doses of V932 on days 0 and 14. Two weeks later, the immune response to human CEA and human HER2/neu was analyzed by IFN-γ enzyme-linked immunospot (ELISPOT) assay. As shown in Fig. 5A, significant immune responses against CEA, LTB, and HER2/neu were detected in both mouse strains. In BALB/c mice the immune response was directed mainly against the N-terminal region of CEA and HER2/neu as shown by the higher degree of immunoreactivity of the Neu-1 and CEA-1 peptide pools. In contrast, the immune response in C57BL/6 mice was targeted mainly to the C-terminal region of CEA (CEA-2). In C57BL/6 mice both HER2/neu peptide pools showed some reactivity, albeit to a lower extent than was observed in BALB/c mice with the Neu-1 peptide pool. LTB reactivity was observed in both mouse strains.
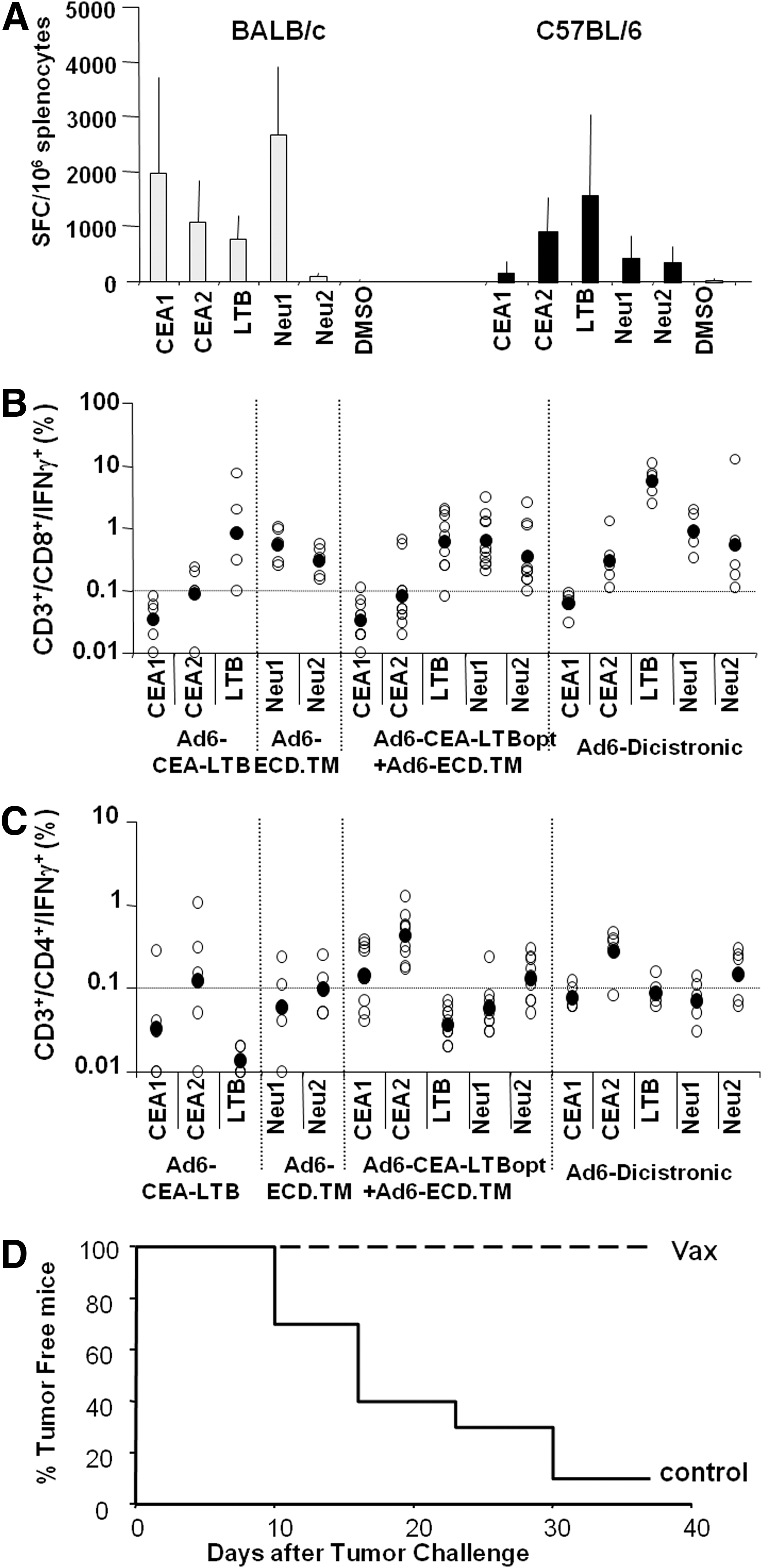
Immunogenic potency and prophylactic efficacy of Ad6/CEA-LTB-HER2-ECD.TM.
To further characterize the immunogenic potency of V932 and assess whether the two expression cassettes in the same backbone may interfere with each other, groups of C57BL/6 mice were immunized with V932, individual monocistronic vectors (Ad6-hCMV-CEA-LTB or Ad6-mCMV-hHER2-ECD.TM), or their combination. The immune response was determined by intracellular staining for IFN-γ. As shown in Fig. 5B, the mixture of the two adenoviral vectors was equally effective in the induction of a CD8+ response. Notably, the amplitude of the response obtained with V932 was even higher, especially against LTB. Similar results were observed for a CD4+ response, albeit the responses were not significantly different among the groups (Fig. 5C). Therefore, these data demonstrate that V932 is immunogenic and can elicit a CMI response to both target antigens in different mouse strains.
To verify the antitumor efficacy of the combined DNA-EGT/Ad immunization regimen, 5 μg of V930 plasmid was injected into C57BL/6 mice. Two weeks later, mice were treated with 108 VP of V932. These doses were selected as previously shown to be efficacious, using the CEA monocistronic vectors in this nontolerant mouse model (data not shown). Two weeks later, mice were challenged with a subcutaneous injection of 106 MC38-hCEA/hHER2 tumor cells. This syngeneic colon carcinoma cell line was engineered to express both human target antigens. As shown in Fig. 5D, all vaccinated mice were completely protected from tumor progression. Thus, these data demonstrate that the V930/V932 prophylactic vaccination protocol is able to protect mice from tumor growth.
Immunogenicity and antitumor efficacy of V930/V932 in double-transgenic human CEA/HER2 model
To further characterize the immunogenic potency of V930/V932 in a tolerant setting, human HER2 and human CEA double-transgenic (hCEA-HER2) mice were generated (see Materials and Methods) and used as the animal model for an immunization study. A group of hCEA-HER2.Tg mice received four weekly injections of V930 (50 μg) and 15 days later received two biweekly intramuscular injections of 1×1010 VP of the V932 vector. Fifteen days later, a subgroup of mice was killed and splenocytes were analyzed by intracellular staining for HER2/neu and CEA-specific IFN-γ production. As shown in Fig. 6A, significant immune response was measured against CEA and HER2/neu peptide pools, particularly against the C-terminal region of CEA (CEA-2) and HER2-ECD.TM (Neu-2). Response against LTB was similarly high. Two weeks later, mice were challenged with a subcutaneous injection of 106 MC38-hCEA/hHER2 tumor cells as described previously. As shown in Fig. 6B, the control group developed palpable tumors by day 80 postchallenge whereas 50% of V930/932-vaccinated mice were completely protected (p<0.0001).
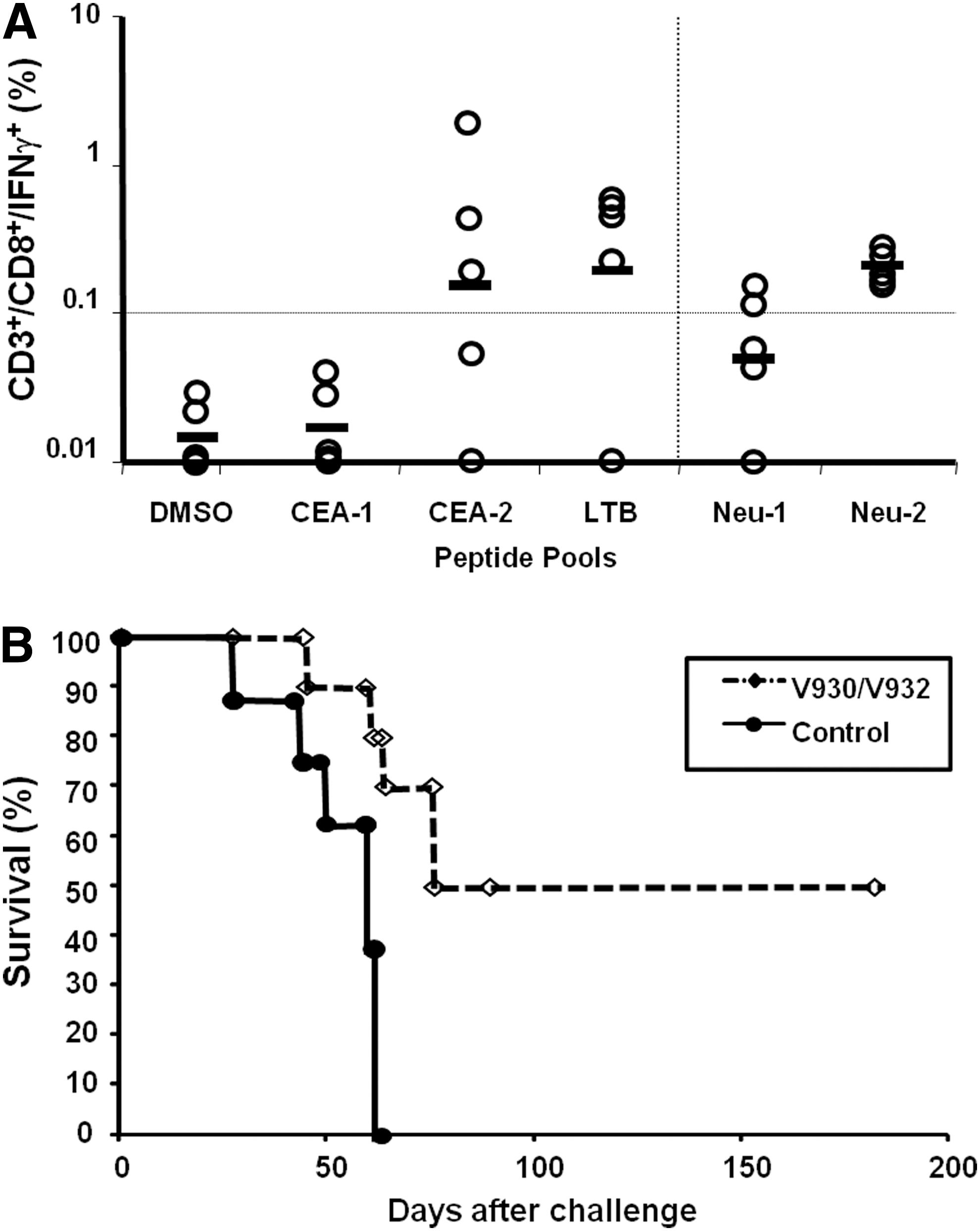
V930/V932 regimen breaks tolerance to both target antigens and confers significant antitumor effects.
These data demonstrate that the V930/V932 regimen is indeed able to break tolerance to both TAAs and provides therapeutic effects against a tumor cell line expressing both antigens.
V932 is immunogenic in NOD/scid-DR1 mice engrafted with the human immune system. Next, we decided to assess whether V932 was able to induce an immune response in mice with a humanized immune system. Groups of NOD/scid-DR1 mice that received intrahepatic injection of HLA-A*0201+ cord blood mononuclear cells (CBMNCs) were immunized intramuscularly with two injections of V932 (1010 VP), 2 weeks apart. Fourteen to 21 days postimmunization, mice were killed and splenocytes were prepared for intracellular staining for human IFN-γ and incubated with CEA peptides for a cytotoxicity assay. Figure 7 shows a representative experiment on three mice engrafted with HLA-A*0201+ CBMNCs from donors. As previously observed in this chimeric model (Camacho et al., 2007), the engraftment of human T cells was variable (10.3–30.5% CD3+) and generally higher for CD4+ (70.2±9.5%) than for CD8+ (20.8±5.9%). As shown in Fig. 7A, vaccinated mice developed a strong and specific T cell response against the TAAs. Both mouse 1 and mouse 2 (same CBMNC donor) developed both a CD8+ and a CD4+ response against CEA (CEA-1 plus CEA-2 pools) and HER2/neu (Neu-1 plus Neu-2 pools). On the other hand, mouse 3 (different CBMNC donor) mounted mainly a strong CD8+-specific response against both antigens.

V932 is immunogenic in chimeric DR1-scid-hu mice. DR1-scid-hu mice were engrafted intrahepatically at 5 days of age with CBMNCs as described in Materials and Methods. Six weeks later, mice were immunized intramuscularly with two injections of V932 (1010 VP).
To determine whether the elicited T cells were endowed with cytotoxic properties, a CTL assay was performed using JY cells, an HLA-A*0201+ EBV-transformed cell line, as target. Splenocytes were incubated with CEA or HER2/neu peptide pools. When loaded with CEA and HER2/neu peptides, JY cells were efficiently lysed by effectors. Similarly, HLA-A*0201-selected CEA peptides (Conforti et al., 2009; Fridman et al., 2012) were recognized and significant lytic activity was observed (Fig. 7B).
These data indicate that V932 was able to induce an in vivo TAA-specific immune response in the presence of the human immune system.
Discussion
Cancer immune therapy and its translation to the clinic need the development of efficient vaccine technologies and delivery systems. In addition, these novel strategies must be evaluated in appropriate preclinical models. In this study, we have developed a novel heterologous prime–boost genetic vaccination strategy cotargeting two well-known tumor antigens, CEA and HER2/neu, and tested its immunogenicity and therapeutic efficacy in immune-tolerant mice.
CEA.Tg mice express CEA with a tissue distribution similar to that of humans (Eades-Perner et al., 1994) and provide a particularly relevant model to critically verify the potential efficacy of a human CEA-based vaccine (Hance et al., 2005). Similarly, BALB/NeuT mice allow assessment of active cancer immunotherapy against HER2/neu. In this model, overexpression of the activated (V664E) form of the rat HER2/neu oncogene, under the control of the mouse mammary tumor virus promoter, leads to progressive development of invasive mammary adenocarcinoma in all 10 mammary glands (Lucchini et al., 1992). Both models have been extremely useful for evaluating vaccine therapies targeting individually HER2/neu or CEA, respectively. Induction of immune responses to self-antigens can be monitored in these mice, along with overt toxicities connected with them. Furthermore, vaccine efficacy on spontaneous tumor development can be examined in BALB/NeuT mice.
To determine the efficacy of plasmid DNA and/or adenovirus vaccination against both CEA and HER2, we generated double-transgenic mice through crossing of BALB/NeuT and CEA.Tg mice. These mice are immunologically tolerant to both target antigens and suitable for antitumor studies. A heterologous prime–boost DNA-EGT/Ad regimen with a mixture of vectors encoding CEA-LTB and rat HER2 was able to break immune tolerance (Figs. 1 and 2). Both a cell-mediated immune response and high antibody titers were detected. Most importantly, the vaccine was able to confer a therapeutic effect in spontaneous rat HER2+ addicted mammary tumors (Fig. 3A and B) and CEA+ colon tumors implanted subcutaneously (Fig. 3C). These data prompted us to generate vectors able to elicit an immune response against the human antigens. In particular, DNA and adenoviral vectors encoding human HER2 ECD.TM were generated and a dicistronic Ad6 vector was designed to minimize the amount of injected viral particles and simplify the scale-up procedure (Fig. 4). The Ad6 serotype was chosen because it belongs to the subgroup C human adenoviruses; like the more common serotype Ad5, Ad6 has the same elevated degree of immunogenicity, but its seroprevalence in the human population is significantly lower (Colloca et al., 2012). V932 was able to induce a strong T cell immune response against both TAAs in wild-type mice, and its immunogenicity was comparable to that elicited by the two monocistronic vectors (Fig. 5A–C). In this setting, V932 was able to confer complete tumor protection (Fig. 5D). To assess the immunogenic potency of V930/V932 in a tolerant environment, human HER2 and human CEA double-transgenic mice were generated and used as the animal model for an immunization study. These mice are derived from CEA.Tg mice crossed with a human HER2 transgenic line. The latter mice carry the full-length wild-type cDNA of human HER2 under the control of the whey acidic protein (WAP) promoter. HER2/neu protein expression is detected in the secretory mammary epithelia during pregnancy and lactation and expressed constitutively in the Bergman glial cells within the molecular layer of the cerebellum. No neoplastic transformation is detected in any tissue (Piechocki et al., 2003), and thus this model needs tumor implantation for therapeutic assessment. Again, a significant immune response and antitumoral effects were observed in this model (Fig. 6). Finally, the immunogenicity of V932 was also evaluated in an NOD/scid-DR1 model engrafted with the human immune system. This model, albeit complex, provides an excellent tool with which to evaluate vaccine potency and characterize immunogenic epitopes. The strong T cell immune response and the cytotoxic activity indicated that V932 was strongly immunogenic in this surrogate model (Fig. 7). Unfortunately, we were not able to assess the potency of V930. It would be of interest to test the heterologous prime–boost strategy as well as the antitumoral effects by using HLA-A*0201+, CEA+, and/or HER2/neu + tumor xenografts in this mouse model.
The findings of our study provide the basis for evaluation of the DNA-EGT/Ad combination in human clinical trials. We have reported the results of two phase 1 trials aimed at evaluating the safety/tolerability and immunogenicity of V930 with EGT alone and followed by V932 (Diaz et al., 2013). Importantly, the immunization regimens were safe and well tolerated. However, only the immune response to the bacterial portion of the vector (LTB) was detected and none of the vaccinated patients had detectable cell-mediated responses to either CEA or HER2/neu. It must be pointed out that a rather heterogeneous population of patients at different clinical stages and with different cancer diagnoses was enrolled in this trial. Such heterogeneity likely prevented any meaningful conclusions. Therefore, this points to the consideration that encouraging results from preclinical testing can be dampened by the overwhelming difficulty of clinical testing. Because V930/V932 was indeed safe and immunogenic (at least against LTB), it would be of great interest to evaluate the vaccine in patients with cancer at the same clinical stage and tumor type who have completed curative intent therapy and have a high risk of recurrent disease.
Footnotes
Acknowledgments
Work performed by L.A. and G.C. was supported in part by grants AIRC IG10507 and IG10334, respectively. The authors thank Cinzia Roffi for editorial assistance.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.