
Abstract
The interaction between CD40 ligand (CD40L) and CD40 can directly inhibit growth of CD40-positive carcinoma cells and may indirectly inhibit tumor growth through coordination of immune responses. Many efforts in CD40L cancer gene therapy have been focused on direct CD40L gene transfer into malignant target cells. This in vivo gene therapy approach relies on high-efficiency gene transfer and could be technically challenging for the treatment of certain cancers, especially multisite metastases. We report herein an alternative means of using the tumor-homing property of neural stem cells (NSCs) to deliver CD40L molecules into tumor tissues. NSCs were derived from human induced pluripotent stem cells, transduced in vitro with a baculoviral vector encoding CD40L, and intravenously injected into immunocompetent mice with orthotopic and metastatic breast cancers. Through a bystander mechanism of intercellular transfer of CD40L from the donor NSCs to tumor target cells, the treatment impeded tumor growth, leading to prolonged survival of the tumor-bearing mice. We further showed that compared with the stem cell-based gene therapy that employed a suicide gene, the CD40L immunogene therapy did not cause liver and kidney injury in the treated mice. This new approach may be particularly valuable for metastatic cancer treatments after systemic stem cell administration.
Introduction
T
Systemic administration of stem cells with tumor-tropic migratory properties for cancer treatment is attractive, given that through circulation the systemically administered cells may home to multiple metastasis locations. These stem cells respond to cytokines, chemokines, and/or growth factors released from tumors and use them as migration-stimulatory signals for tumor homing (Jurvansuu et al., 2008; Chen et al., 2013). As cellular vehicles for the targeted delivery of therapeutic genes into cancers, mesenchymal stem cells (MSCs) and neural stem cells (NSCs) have been tested in animal tumor models (Aboody et al., 2008; Cihova et al., 2011; Binello and Germano, 2012; Shah, 2012). However, it has been recognized that MSCs, after reaching tumor stroma, can promote tumor growth by creating a niche to support cancer stem cell survival, suppressing local immune responses against tumor cells, stimulating tumor angiogenesis, and promoting cancer metastasis (Karnoub et al., 2007; Galderisi et al., 2010; Klopp et al., 2011). Thus, MSCs are viewed as a two-edged sword for cancer therapy. NSCs also effectively function as vehicles for tracking tumor cells and delivering anticancer agents (Aboody et al., 2008; Cihova et al., 2011; Binello and Germano, 2012; Shah, 2012). In particular, systemically administered NSCs may home in not only on multiple tumor foci in the brain but also on tumors in several other internal organs of the body (Tang et al., 2003; Aboody et al., 2006; Danks et al., 2007; Frank et al., 2009; Zhao and Wang, 2010; Kim et al., 2012; Yang et al., 2012; Zhao et al., 2012). Unlike with the use of MSCs for cancer therapy, protumor adverse events with the use of NSCs have not been reported in the literature.
With the increasing potential of using NSCs as cancer therapeutics, it is desirable to have a reliable and stable supply of human NSCs. In our previous studies, we have explored the feasibility of using pluripotent stem cells, such as human embryonic stem cells (hESCs) and human induced pluripotent stem cells (iPSCs), to generate cellular vehicles for cancer gene therapy and demonstrated the effectiveness of using these in vitro-generated NSCs for cancer suicide gene therapy in several different animal models (Zhao and Wang, 2010; Yang et al., 2012; Zhao et al., 2012). This suicide therapy approach is based on phosphorylation of ganciclovir (GCV) by the herpes simplex virus thymidine kinase (HSVtk) and a tumor-killing mechanism by which phosphorylated GCV stops DNA replication in proliferating tumor cells.
However, we noticed some limitations to our previous studies. After NSCs were injected intravenously into mice, most of the cells were trapped in the lungs, liver, and kidneys (Yang et al., 2012), which could be valuable for eliminating metastatic cancer cells in these organs. However, NSCs trapped in these organs may also potentially damage proliferating healthy cells there if they are used to deliver toxic compounds, for example, suicide gene products, thus worsening the patient's situation. In an effort to minimize such off-target effects, we investigated in this study whether intravenously injected induced pluripotent stem cell-derived neural stem cells (iPS-NSCs) can be used as a CD40L delivery platform to treat metastatic cancer while minimizing injury to off-target organs.
Materials and Methods
Cell culture
The mouse metastatic breast cancer cell line 4T1 and the mouse fibroblast cell line NIH3T3 were purchased from the American Type Culture Collection (Manassas, VA). 4T1 cells that stably express luciferase gene (4T1-luc) were purchased from Caliper Life Sciences/PerkinElmer (Hopkinton, MA). 4T1 and 4T1-luc cells were maintained in RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS), 2 mM
Human iPSCs were generated by means of a polycistronic lentiviral vector carrying Oct4, Klf4, Sox2, and c-Myc genes (Millipore, Bedford, MA) as described in our previous study (Yang et al., 2012). An adherent monoculture differentiation method was used to derive NSCs as previously described (Yang et al., 2012; Chen et al., 2013). Briefly, TrypLE (Life Technologies)-dissociated iPSCs were plated at a density of 106 cells per well onto a 6-well cell culture plate (Nalge Nunc, Rochester, NY) coated with 0.1% gelatin and cultured in an NSC medium (Yang et al., 2012; Chen et al., 2013). The cells reached 90% confluence after 7 days of differentiation and were split 1:2. Rosette-like structures, a typical morphology for neural differentiation, could be observed after culturing for 3–4 weeks. Homogeneous bipolar iPS-NSCs with a doubling time of approximately 3–4 days were obtained after further expansion. After the differentiation, the culture was routinely passaged at a ratio of 1:2 twice weekly. The extensive characterization of iPS-NSCs used in the current study has been reported in our previous publications, which demonstrated that these cells express important NSC markers and display the functional hallmarks of NSCs to differentiate into neurons, astrocytes, and oligodendrocytes (Yang et al., 2012; Chen et al., 2013).
Baculovirus preparation, cell transduction, and detection of transgene expression
The recombinant baculoviral vector expressing the enhanced green fluorescent protein (eGFP) reporter gene (BV-eGFP), constructed using the BAC-to-BAC baculovirus expression system (Life Technologies), carries the eGFP gene under the control of the human cytomegalovirus (CMV) early promoter (Zeng et al., 2009). The recombinant baculoviral vector expressing CD40L (BV-CD40L) was produced by homologous recombination after cotransfection of Sf9 insect cells with pBacPAK9 transfer vector carrying an expression cassette and linearized Autographa californica nuclear polyhedrosis virus (AcMNPV) viral DNA (Clontech, Mountain View, CA). The expression cassette of BV-CD40L contains the mouse CD40L gene (InvivoGen, San Diego, CA) under the control of the CMV promoter with the R segment and part of the U5 sequence of the long terminal repeat from human T cell leukemia virus type 1 (RU5) at the 5′ untranslated region (UTR) and the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) at the 3′ UTR. BV-HSVtk was generated in our previously described study (Balani et al., 2009). Recombinant baculoviruses were amplified in Sf9 cells at a multiplicity of infection (MOI) of 0.1 plaque-forming unit (PFU)/cell and the virus-containing supernatant was collected 3 days after virus infection. Viruses were pelleted at 28,000×g for 1 hr and resuspended in phosphate-buffered saline (PBS).
For transduction, iPS-NSCs were cultured with baculoviral vectors at an MOI of 100 PFU/cell overnight. A full medium change was carried out the next day to stop the transduction. GFP expression was detected by fluorescence microscopy and flow cytometric analysis. To confirm CD40L gene expression, reverse transcriptase-polymerase chain reaction (RT-PCR) and flow cytometric analysis using anti-CD40L antibody (BD Biosciences, San Jose, CA) were performed. The forward and reverse primers used for CD40L RT-PCR analysis were 5′-TCCCCCAGATCCGTG GCAACT-3′ and 5′-GGCCGACGATGAATGGGCGT-3′. PCR products were electrophoresed on a 1% agarose gel.
NSC migration analysis
Migration assays of iPS-NSCs toward tumor cells were performed in Opti-MEM (Life Technologies), using 24-well plates with Boyden chambers containing BD Falcon HTS FluoroBlok cell culture inserts (BD Biosciences) (pore size, 8 μm). The chambers contain a transmembrane with a cell culture insert in each well allowing cells to move from the top to the bottom chamber through the membrane. 4T1 or 4T1-luc breast cancer cells were seeded into the lower receiver well, at 50,000 cells per well, 1 day before the assay and used as attractants. Baculoviral vector-transduced iPS-NSCs or nontransduced iPS-NSCs were seeded into the upper chamber, at 50,000 cells per well. After 6 hr, migratory NSCs on the bottom of the transmembrane were fixed with 100% methanol for 3 min and stained with 4′,6-diamidino-2-phenylindole (DAPI). Pictures were then taken at ×100 magnification for each well and used for cell counting. Cell migration toward Opti-MEM alone without tumor cells in the lower chamber was included for measurement of the basal migration rate of iPS-NSCs.
Cytokine antibody array and cell viability assay
To profile the cytokine production of 4T1 breast cancer cells under CD40L treatment, the BD Falcon HTS FluoroBlok 24-multiwell insert system (pore size, 8 μm) was used to coculture 4T1 cells with CD40L-expressing NSCs. 4T1 cells were seeded at a density of 1×105 cells per well in 24-well companion plates. After the 4T1 cells were attached, the medium was replaced with 800 μl of Opti-MEM and 24-multiwell cell culture inserts were placed into the 24-well companion plates. BV-CD40L-transduced NSCs and BV-eGFP-transduced NSCs, as vial transduction control, were seeded into multiple inserts at a density of 5×104 cells per insert. After 24 hr of coculture, the supernatants of the bottom wells were harvested. The cytokine profiles in the supernatants were measured in triplicate with the RayBio mouse cytokine antibody array 2 (RayBiotech, Norcross, GA). The RayBio analysis tool (RayBiotech) was used to correlate the average signal intensities to relative expression levels of cytokines.
Cell viability assays were also performed with the BD Falcon HTS FluoroBlok 24-multiwell insert system to evaluate the in vitro bystander cytotoxic effects of CD40L. 4T1 cells, which are CD40 positive, and NIH313 cells, as CD40-negative control, were seeded at a density of 5×104 cells per well in 24-well companion plates. After the cells were attached, 24-multiwell cell culture inserts were placed into the 24-well companion plates. CD40L-expressing NSCs and eGFP-expressing NSCs were seeded into multiple inserts at a density of 5×104 cells per insert. After 48 hr of coculture, cell viability assays were performed with the CellTiter 96 AQueous assay (Promega, Madison, WI), and caspase-3/7 activity was measured with the Caspase-Glo 3/7 assay system (Promega).
Animal studies
BALB/c immunocompetent mice (weight, 20 g; age, 6–8 weeks) were used. To generate an orthotopic breast cancer model, mouse 4T1-luc breast cancer cells were injected subcutaneously into the mammary fat pad (1×105 cells in 50 μl of PBS). To generate a breast cancer lung metastasis model, 4T1-luc cells were injected intravenously via the tail vein (1×104 cells in 200 μl of PBS).
To examine the in vivo tumor tropism of iPS-NSCs in the mice with breast cancer lung metastasis, an orange-red fluorescent dye (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate [DiL]; Life Technologies was used to label the cells. Labeled iPS-NSCs (1×106 in 200 μl of PBS) were injected into mice via the tail vein. Tissue distribution of DiL-labeled iPS-NSCs was examined under a fluorescence microscope.
To evaluate in vivo immune responses and apoptosis induction, lungs were collected, 28 days after tail vein injection of CD40L-expresssing iPS-NSCs and other control cells, from the mice with breast cancer lung metastasis. Using ProteoJET mammalian cell lysis reagent (Thermo Fisher Scientific, Waltham, MA) supplemented with pepstatin, leupeptin, and aprotinin (2 ml/g tissue), tissues were homogenized and then sonicated on ice. Supernatants were collected after centrifugation at 13,000×g for 20 min at 4°C. Interferon (IFN)-γ and TNF-α release in lung tissues was determined with a mouse IFN-γ ELISA kit and a mouse TNF-α ELISA kit (BD Biosciences). Caspase activity in lung tissues was measured with the Caspase-Glo 3/7 assay system (Promega).
To evaluate therapeutic effects in the orthotopic breast cancer model, tumor-bearing mice were randomly divided into four groups (n=10 per group) 3 days after tumor inoculation for tail vein injection: (1) PBS, 200 μl; (2) 1×106 NSCs; (3) 1×106 NSCs transduced with BV-eGFP (NSC-eGFP); and (4) 1×106 NSCs transduced with BV-CD40L (NSC-CD40L). Baculoviral transduction was performed 12 hr before the tail vein injection of NSCs. On day 28, animals received an intraperitoneal injection of
To evaluate therapeutic effects in the breast cancer lung metastasis model, tumor-bearing mice were randomly divided into four groups (n=10 per group) 7 days after tumor inoculation. The second tail vein injection was performed with PBS, NSCs, NSCs expressing eGFP, or NSCs expressing CD40L at 1×106 cells in 200 μl per injection. Animal survival was monitored until day 42.
To assess liver and kidney toxicities after the HSVtk/GCV regimen, three mice with 4T1 breast cancer lung metastases were injected with NSCs transduced with BV-HSVtk (NSC-HSVtk, 1×106 cells in 200 μl) 7 days after tumor inoculation via the tail vein. Baculoviral transduction was performed 12 hr before the tail vein injection of NSCs. The mice were given a daily intraperitoneal injection of GCV (50 mg/kg body weight) for 6 days. One week after NSC injection, these three mice were used together with mice in the four groups (three mice per group) used in the previously described experiment evaluating the therapeutic effects of CD40L therapy in the breast cancer lung metastasis model for blood collection. Blood (100 μl per mouse) was collected from the orbital sinus to measure the concentrations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) as liver damage indicators and of creatinine and blood urea nitrogen (BUN) as kidney damage indicators, using MaxDiscovery ALT, AST, BUN enzymatic assay, and creatinine assay kits (Bioo Scientific, Austin, TX).
Animal experiments were performed according to the Guidelines on the Care and Use of Animals for Scientific Purposes issued by the National Advisory Committee for Laboratory Animal Research, Singapore.
Statistical analysis
All data are represented as means±SD. The statistical significance of differences was determined by two-factor analysis of variance with replication followed by Fisher least significant difference post hoc analysis. The statistical analysis of survival data was performed using the log-rank test followed by the Holm–Sidak method for pairwise multiple comparison tests. p<0.05 was considered to be statistically significant.
Results
Baculoviral transduction-mediated CD40L expression in iPS-NSCs
We demonstrated previously the tumor tropism of NSCs derived from human iPSCs (Yang et al., 2012; Chen et al., 2013). Successful loading of the CD40L gene into NSCs and maintenance of transgene expression long enough in these cells are the prerequisites for their use as a CD40L delivery platform for cancer therapy. Because baculoviral transduction is effective in mediating transgene expression in many different types of NSCs (Zeng et al., 2009; Zhao and Wang, 2010; Lee et al., 2011; Yang et al., 2012), we constructed a recombinant baculoviral vector encoding CD40L (BV-CD40L) and tested it for gene transfer into iPS-NSCs. Two viral posttranscriptional regulatory elements, RU5 and WPRE, were included in the CD40L expression cassette to improve transgene expression efficiency (Du et al., 2010). As shown in Fig. 1A, iPS-NSCs could be effectively transduced by a baculoviral vector. Approximately 99% of cells expressed high levels of CD40L in the first 2 days posttransduction. Although the percentage of CD40L-positve cells decreased over time, possibly because of dilution of the nonreplicative and nonintegrative baculoviruses by cell division, CD40L mRNA (Fig. 1B) and protein (Fig. 1A) expression lasted for at least 14 days in iPS-NSCs.

CD40 ligand (CD40L) expression in induced pluripotent stem cell-derived neural stem cells (iPS-NSCs) after baculoviral vector (BV) transduction. Flow cytometric analysis
Because the tumor tropism of NSC vehicles is decisive for successful anticancer gene therapy applications of these cells, we further tested whether baculoviral transduction-mediated CD40L expression would impede the migratory capacity of iPS-NSCs toward mouse 4T1 breast cancer cells. In vitro Transwell cell migration assays were performed with Boyden chambers. 4T1 cells, as well as 4T1-luc cells, seeded in the bottom chamber were used as attractants. After counting the number of migratory iPS-NSCs attached to the bottom side of the membrane, we observed that the percentages of migratory iPS-NSCs in the presence of tumor cells were approximately 20%, whereas the percentage of cells migrating toward plain Opti-MEM cell culture medium was about 5% (Fig. 1C). There was no difference in migration capacity between iPS-NSCs and iPS-NSCs transduced with BV-eGFP or BV-CD40L. To examine the in vivo migration capacity of these transduced cells toward tumors, we generated a breast cancer lung metastasis model in immunocompetent BALB/c mice by intravenous injection of mouse 4T1 breast cancer cells via the tail vein. One week later, iPS-NSCs transduced with BV-CD40L were labeled with the orange-red fluorescent dye DiL and injected intravenously into the tumor-bearing mice. On days 1, 3, and 7 after NSC injection, we examined DiL signals in tissue sections of the lung with tumor nodules by fluorescence microscopy. On day 1, some DiL signals could already be detected in the lungs. On day 3, DiL signals became stronger and obvious cell accumulation could be observed in the tumor nodules with a high density of Hoechst-stained nuclei (Fig. 1D), indicating the migration of iPS-NSCs toward tumors in the lungs. There was no sign of decrease in DiL signals in the tumor regions on day 7. Thus, baculoviral transduction and CD40L expression did not display any noticeable effects on the tumor tropism of iPS-NSCs.
CD40L from BV-CD40L-transduced iPS-NSCs stimulates cytokine release from 4T1 cells and induces cell death in vitro and in vivo
Given that the CD40L–CD40 interaction can provide strong immunostimulatory effects, we investigated the effects of CD40L-expressing iPS-NSCs on the release of cytokines from CD40-positive tumor cells. CD40 expression on the surface of 4T1 breast cancer cells was confirmed by flow cytometric analysis with an antibody against CD40 (Supplementary Fig. S1A; supplementary data are available online at
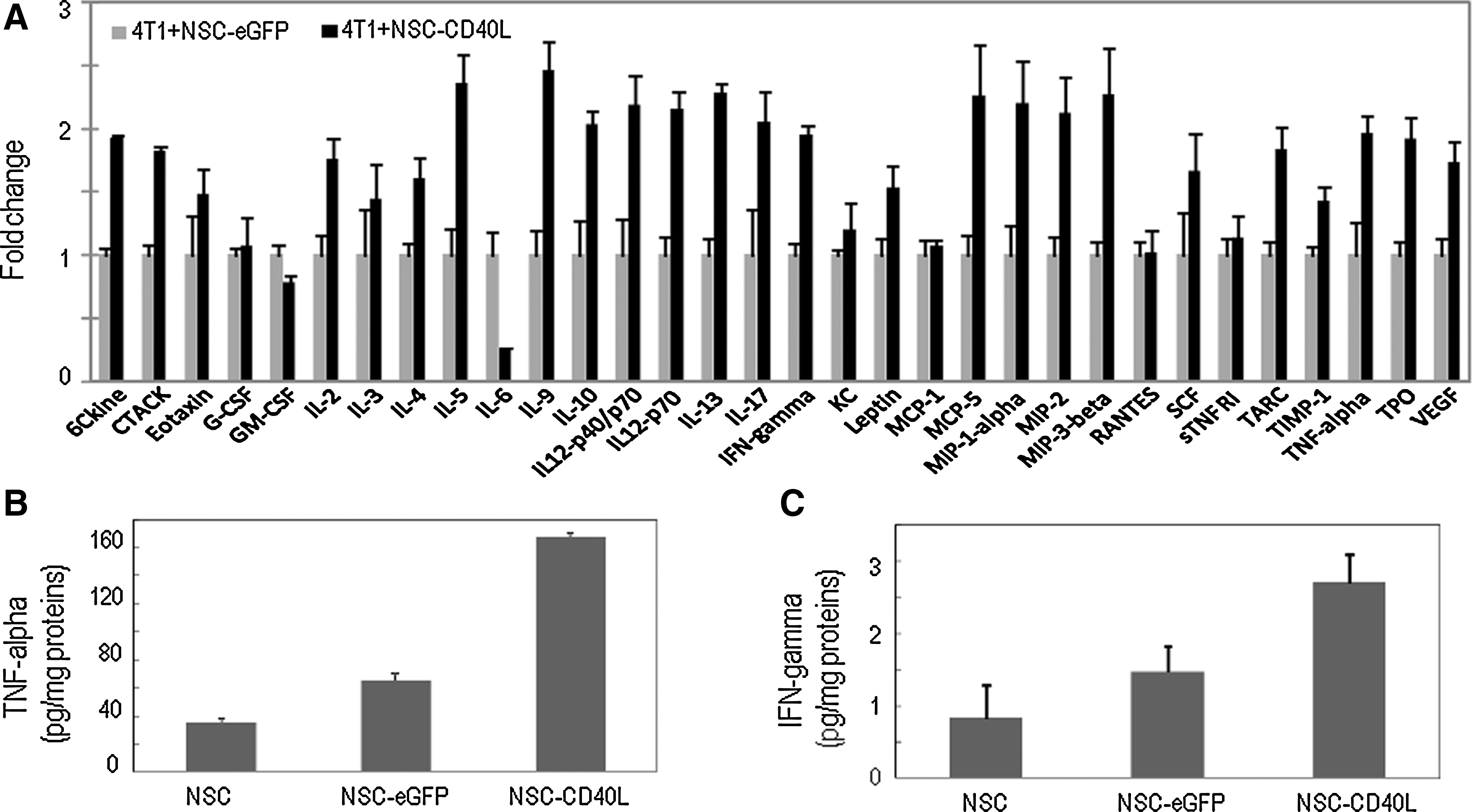
Release of cytokines from 4T1 breast cancer cells after coculture with CD40L-expressing iPS-NSCs.
In view of the broad stimulatory effects of CD40L-expressing iPS-NSCs on the release of multiple cytokines, including proapoptotic cytokines TNF-α and IFN-γ, we then evaluated the bystander effects of iPS-NSCs on the induction of cell death and apoptosis in 4T1 cells. To show that the effects originated from the CD40L–CD40 interaction between CD40L molecules released from iPS-NSCs and CD40 on 4T1 cells, instead of direct cell interaction, assays were performed by the Boyden chamber method as described previously. The mouse fibroblast cell line NIH3T3, a CD40-negative cell line, was introduced in this assay as a negative control. After coculture for 48 hr, cell viabilities of 4T1 and NIH3T3 cell cultures in the bottom wells were measured in MTS assays. Caspase-3/7 activity levels were also measured with a luminogenic caspase-3/7 substrate. We observed that the viability of 4T1 cells was reduced by more than 50% (Fig. 3A) and that the caspase activity in 4T1 cells was significantly increased to 160% of control (Fig. 3B), demonstrating that apoptosis was induced in these cells. On the other hand, cell viability and caspase activity in CD40-negative NIH3T3 cell cultures were not significantly changed. Thus, CD40L-expressing iPS-NSCs provide a strong bystander cell-killing effect on CD40-positive tumor cells, but not on CD40-negative cells. We also detected significantly increased caspase-3/7 activities in tissue homogenates of lungs collected from mice with breast cancer lung metastasis and treated with CD40L-expressing iPS-NSCs (Fig. 3C), indicating the induction of apoptosis after delivery of CD40L into the lungs by these stem cells.
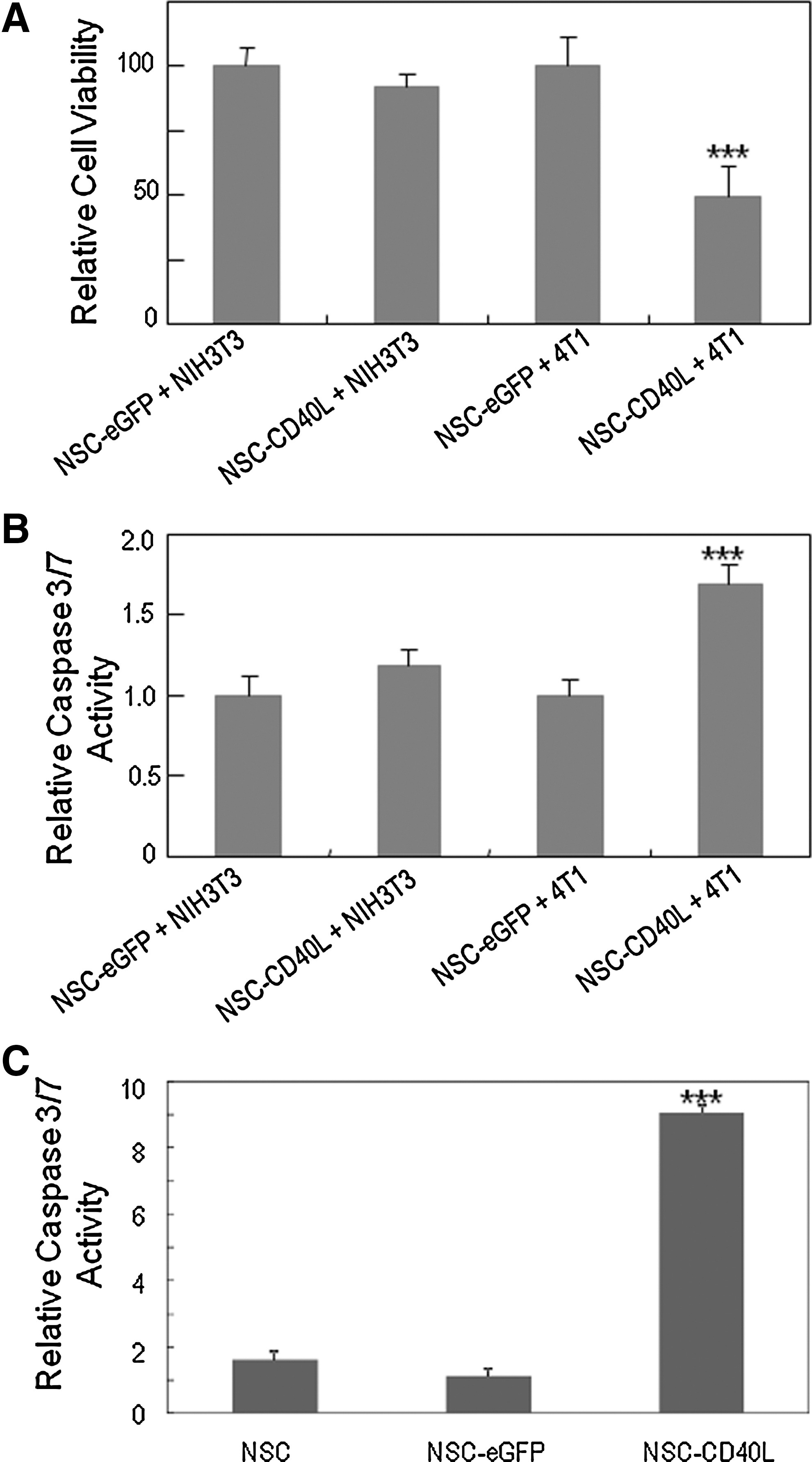
Cell-killing effects of CD40L-expressing iPS-NSCs on 4T1 breast cancer cells.
Systemic delivery of iPS-NSCs expressing CD40L inhibits tumor development and prolongs survival of mice bearing breast cancer lung metastases
To assess the prospect of CD40L-expressing iPS-NSCs as a cellular therapeutic for cancer treatment, we examined in vivo effects of the cells on tumor development in two mouse breast cancer models in immunocompetent BALB/c mice: a breast cancer lung metastasis model generated by intravenous injection of luciferase-expressing mouse 4T1 breast cancer (4T1-luc) cells via the tail vein and an orthotopic breast cancer model generated by subcutaneous injection of 4T1-luc cells into the mammary fat pad.
We first tested the effects on tumor growth at the inoculation site and on metastasis in the orthotopic breast cancer model (Fig. 4A). On day 3 after tumor inoculation, tumor-bearing mice were randomly divided into four groups for tail vein injection with PBS, iPS-NSCs, iPS-NSCs expressing eGFP, or iPS-NSCs expressing CD40L. On day 28 after tumor inoculation, all mice were killed and nine organs, including the lungs, liver, spleen, kidneys, heart, stomach, brain, femurs, and spinal cord, and the mammary fat pad with the major tumor body, were collected for ex vivo bioluminescence imaging of 4T1-luc cells. We observed smaller sizes of primary tumors in the mammary fat pad in the NSC-CD40L group compared with those in the control groups (Fig. 4B). Furthermore, ex vivo organ imaging showed that there were no tumor metastases in the examined organs from the NSC-CD40L group, whereas there were 5, 13, and 4 organs with metastases in the PBS, NSC, and NSC-eGFP groups, respectively (Fig. 4C and Supplementary Fig. S2). These findings indicate that NSC-CD40L not only inhibits primary tumor growth, but also inhibits tumor metastasis.
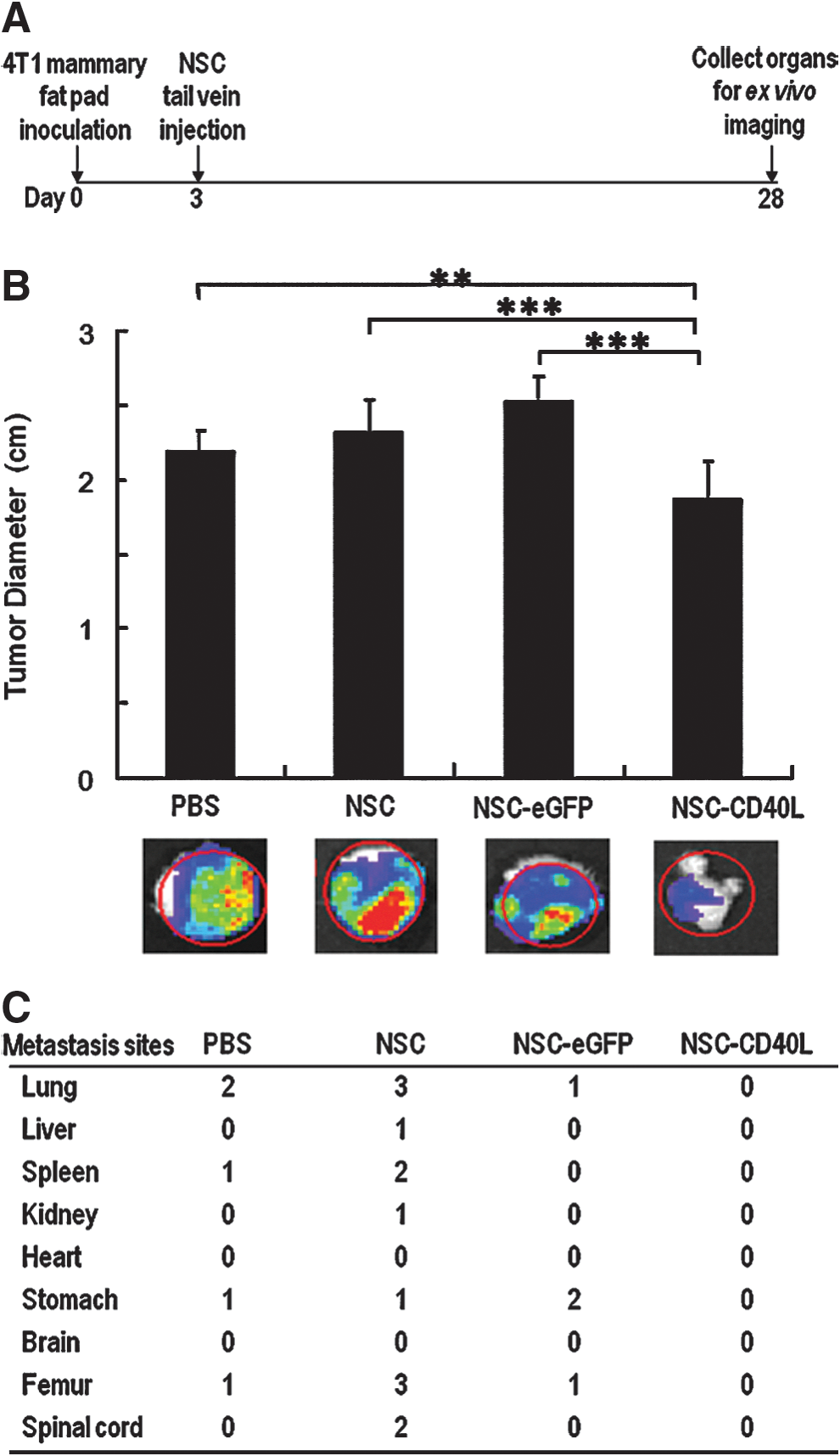
In vivo effects of CD40L-expressing iPS-NSCs on orthotopic tumor growth and metastasis of 4T1 breast cancer.
Possible effects of CD40L-expressing iPS-NSCs on tumor development were further investigated in the breast cancer lung metastasis model after tail vein injection of cells (Fig. 5A). Taking advantage of the quick animal death in this model, we focused on survival of tumor-bearing animals in this experiment. We observed that the survival of treated mice was significantly prolonged. On day 30, whereas all the mice in the control groups had died, nearly 40% of the mice in the NSC-CD40L group remained alive (Fig. 5B). The median survival time increased from not more than 26 days for the three control groups to 30 days for the NSC-CD40L group (Fig. 5C and Supplementary Table S2). Statistical analysis revealed that in all cases, there was a significant difference between the NSC-CD40L group and the control groups (Supplementary Table S2).
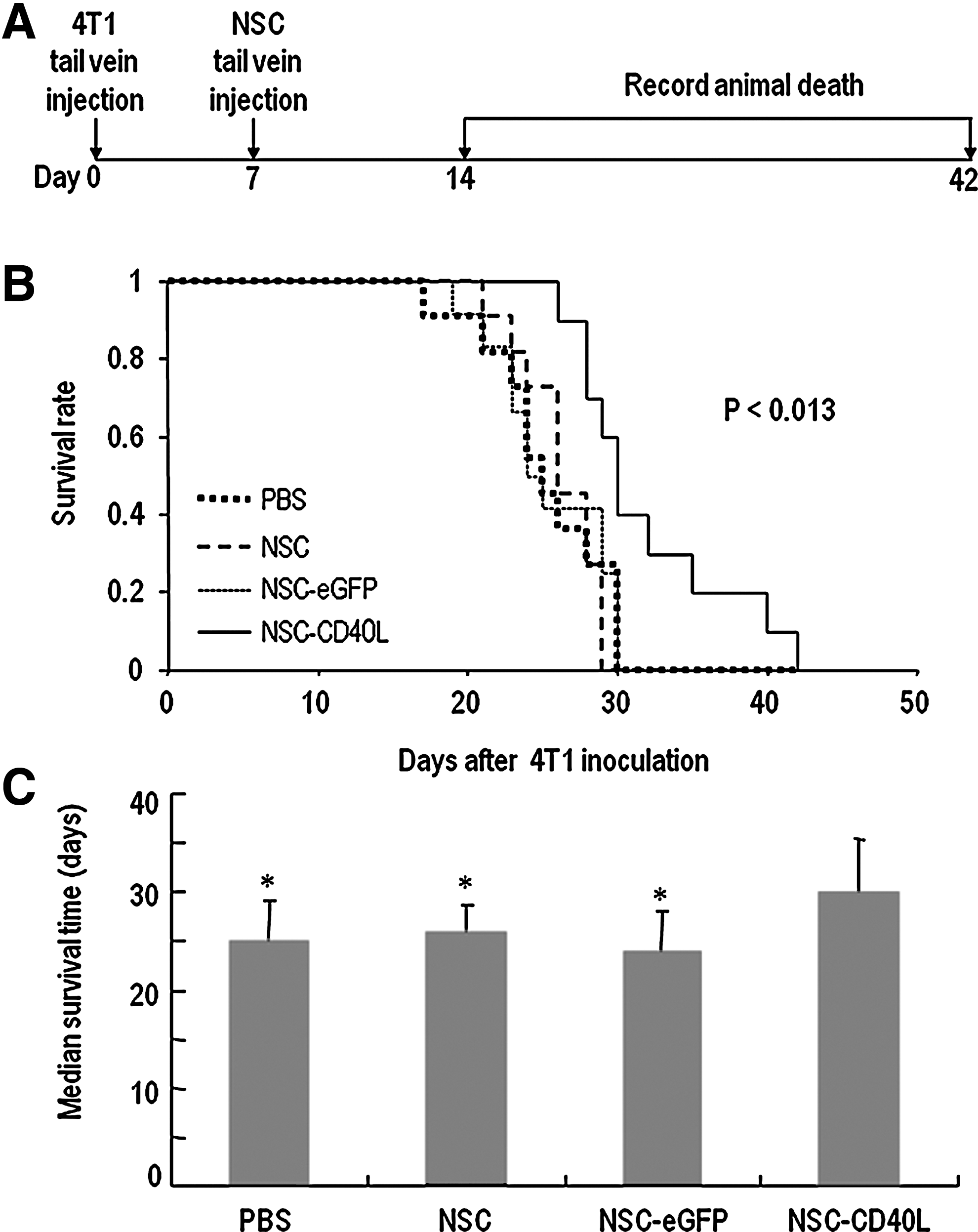
Therapeutic effects of CD40L-expressing iPS-NSCs in the 4T1 breast cancer lung metastasis model.
Comparison of side effects between NSC-based CD40L and HSVtk/GCV therapies
After initial pulmonary trapping, tail vein-injected iPS-NSCs will redistribute from the lungs to other organs, including the liver and kidneys (Yang et al., 2012). To determine whether the off-target distribution was associated with any obvious side effects, we analyzed several biochemical markers of liver and renal injury in the previously described mice used to test therapeutic effects in the 4T1 breast cancer lung metastasis model. We also treated three mice with 4T1 breast cancer lung metastases with the NSC-based HSVtk/GCV therapy regimen (NSC-TK) in order to compare side effects between the suicide gene therapy approach and the CD40L immunogene therapy approach. One week after NSC tail vein injection, mouse blood was collected by orbital sinus blood sampling to examine concentrations of the liver damage indicators ALT and AST and the kidney damage indicators creatinine and BUN. Figure 6 shows that the blood concentrations of ALT, AST and creatinine in the NSC-TK group were approximately 2- to 3-fold higher than in the control groups. In the NSC-CD40L group, on the other hand, their levels were not significantly changed. These results demonstrate that compared with the NSC-TK approach the NSC-CD40L approach is less toxic to off-target organs.
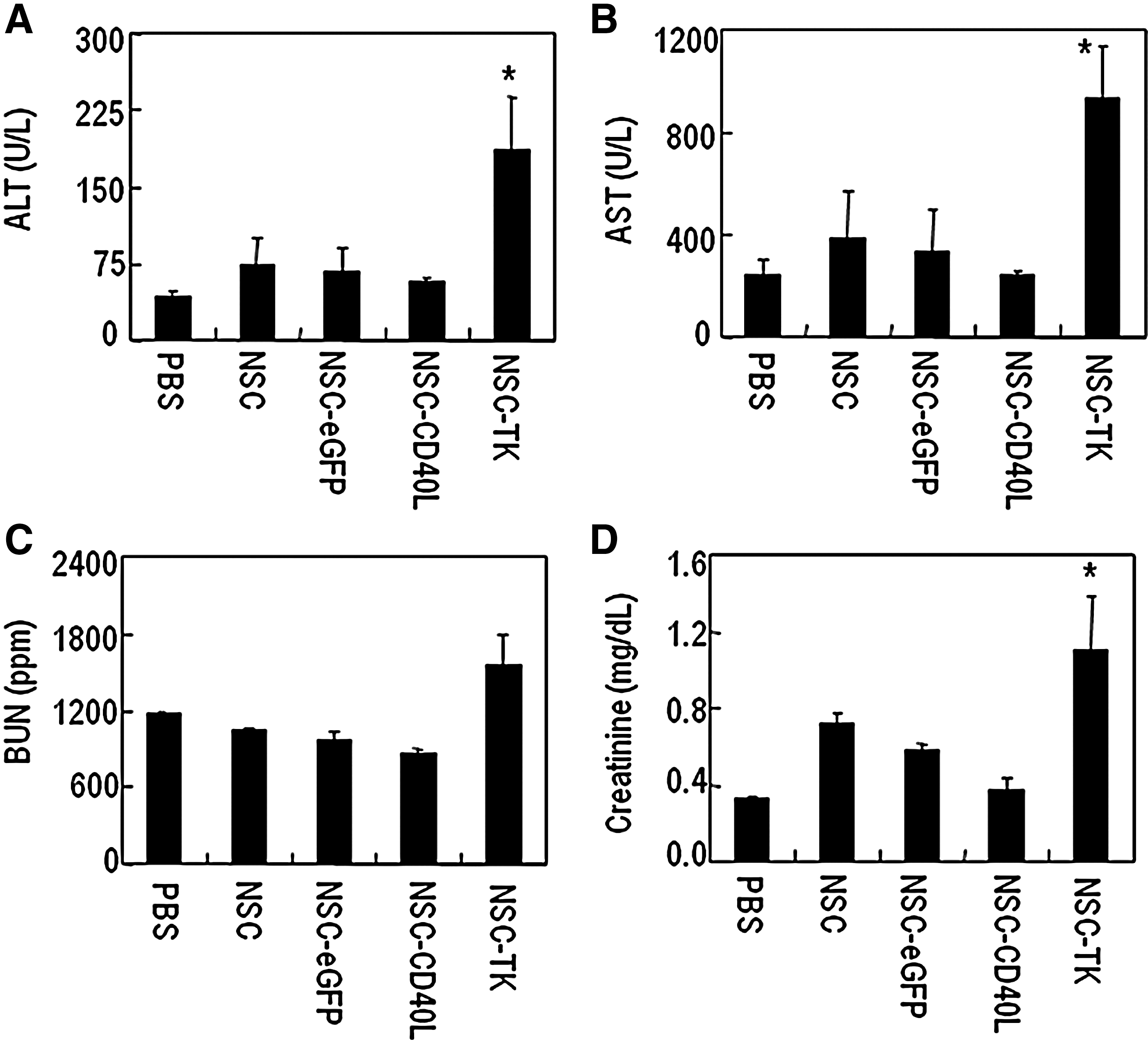
Hepato- and nephrotoxicities: NSC-CD40L versus NSC-TK/GCV treatment.
Discussion
Approximately 90% of human malignancies arise from epithelial tissue. CD40 is selectively expressed in many epithelial cancers including breast, lung, ovarian, cervical, bladder, and colon carcinomas, whereas most normal, nonproliferating epithelial tissues are CD40 negative (Tong and Stone, 2003). Ligation of CD40 on these tumor cells generates direct growth-inhibitory effects without obvious side effects on their normal counterparts (Tong and Stone, 2003). CD40/CD40L ligation can activate antigen-presenting cells such as dendritic cells, stimulating their maturation to present antigens to T cells, as well as secretion of cytokines. CD40L stimulation may also abrogate regulatory T cell suppression. While being an immune activator, CD40L can directly induce apoptosis and/or cell cycle blockage in tumor cells. Hence, a National Cancer Institute immunotherapy agent workshop held in 2007 has ranked CD40L as the No. 4 agent with high potential for use in cancer therapy (Cheever, 2008). Given their potent antitumor effects in experimental cancer animal models, therapeutic interventions targeting CD40L are currently under intense investigation in cancer therapy clinical trials (Loskog and Totterman, 2007; Ullenhag and Loskog, 2012).
The current study presented a new approach for CD40L-based cancer therapy and demonstrated the feasibility of delivering CD40L into tumors by systemically injected tumor-tropic stem cells to inhibit tumor development. In contrast to the strategy of local CD40L gene transduction of tumor cells, our approach makes use of a cellular vehicle preloaded by baculoviral CD40L transduction and the tumor tropism property of the cells after systemic delivery to activate local CD40L–CD40 interactions within tumors. Because the systemic delivery of CD40L-loaded NSCs can target multiple metastases at various sites through the circulation, our approach is attractive in enabling efficient therapy for metastatic cancer. As suggested in our in vitro study, tumor growth inhibition could occur through a bystander mechanism involving transfer of CD40L protein from donor iPS-NSCs to nearby tumor cells in association with the cell-killing effects mediated by Th1-type cytokines. It has been demonstrated previously that CD40L protein transfer to tumor cells occurs in transduced fibroblasts, epithelial cells, and bone marrow stromal cells (Biagi et al., 2003). Our in vivo experiments further demonstrated an increase in TNF-α and IFN-γ concentrations and caspase-3/7 activity in lungs collected from tumor-bearing animals treated with CD40L-expressing iPS-NSCs. In an ongoing study, we have tested human iPSC-derived endothelial progenitor cells that were genetically modified to express CD40L to treat 4T1 metastasis in immunocompromised BALB/c nude mice and again observed the anticancer effects of CD40L (our unpublished observation). BALB/c nude mice lack T cells but still acquire B cells and innate immune cells, such as natural killer (NK) cells, and a complement immune system. Thus, upregulation of Th1 cytokines and induction of apoptosis, but not induction of T cell responses, might play more important roles in the observed antitumor action in the current study.
In the present study, we tested NSCs derived from human iPSCs for CD40L-mediated cancer therapy. If the NSCs to be used are progeny differentiated from iPSCs that were generated by reprogramming of the patient's own cells, the treatment is viewed as autologous cell therapy and the likelihood of immune rejection of NSCs after transplantation will be significantly reduced. If the NSCs are derived from an iPSC line selected from a bank of human leukocyte antigen (HLA)-typed human iPSCs, they can be used as semiallogeneic differentiated cells sharing some HLA alleles with the recipients. It is also possible to use iPSC-derived NSCs as allogeneic cells for therapeutic applications that do not require long-term survival of transplanted cells, such as cancer therapy. In contrast to regenerative medicine technologies using allogeneic stem cells that require long-term immunosuppressive treatment, using iPSC-derived cells as exogenous cellular vehicles for cancer treatment may not need immune suppression because the therapy does not really require stem cell engraftment. Using a real-time qPCR method that analyzes HLA-A short tandem repeat (STR) #54, we assessed the fate of intravenously injected human iPS-NSCs in the lungs, liver, and spleen of immunocompetent BALB/c mice (Yang et al., 2012). An approximately 51% decrease in iPS-NSCs was detected in the three organs over the 4-week period. Thus, we would expect that allogeneic cells derived from iPSCs will ultimately be eliminated from a patient's body.
Several reasons justify the use of allogeneic cells derived from iPSCs as cellular vehicles for cancer therapy. First, developments in genetic engineering methods, especially those based on targetable nucleases that are capable of introducing transgenes at specific sequences, have opened the possibility for generation of human iPSCs with therapeutic genes in “safe harbor” loci. These genetically modified iPSCs can then be used as a master cell line for the production of cell therapeutics. Second, allogeneic cell therapeutics can be produced at a large scale, allowing multiple-site clinical trials using the same batch of cells and repeated cell therapy in the same patient. Large-scale mass production can also simplify the logistics of cell culture operations, thus significantly increasing cost-effectiveness. For commercial purposes, large-scale cell therapeutic production is necessary to prepare “off-the-shelf” cryopreserved cells in a ready-to-go format. Third, the allogeneic cell approach makes standardized manufacture easy. This is useful in eliminating quality variability of cell therapeutics, thus allowing reliable comparative assessment of treatment outcomes in a multiple-site clinical trial.
We observed in this study the accumulation of iPS-NSCs in the lungs, liver, and kidneys after intravenous administration in mice. Although such an accumulation can be valuable for cancer therapy in these organs, CD40 ligation-based therapy there could potentially damage CD40-positive healthy cells. One example is apoptosis in CD40-positive hepatocytes after CD40 activation by anti-CD40 agonist monoclonal antibodies (Afford et al., 1999). CD40 ligation on endothelial cells also triggers complex stimulating effects on the secretion of cytokines, chemokines, coagulation factors, and adhesion molecules (Karmann et al., 1995; Thienel et al., 1999). Several genetic engineering strategies can be adopted to minimize such off-target side effects and enhance the tumor-specific treatment. For example, tumor-specific promoters, such as the hypoxia response elements, the proendothelin-1 promoter, and the survivin promoter, can be used to restrict therapeutic gene expression in the tumor environment (Dong and Nor, 2009; Keung et al., 2013). Given the fact that the vesicular stomatitis virus glycoprotein (VSV-G) can act in a pH-dependent manner to kill cells through syncytium formation, we have genetically engineered VSV-G to generate a mutant that is capable of stimulating the formation of multinucleated syncytia specifically at the tumor extracellular pH. Injection via the tail vein of iPS-NSCs expressing the VSV-G mutant can selectively kill tumors in mice with metastatic breast cancer in the lungs, without detectable hepato- and nephrotoxicities in off-target organs (D. Zhu et al., 2013).
To employ NSCs as donor cells to carry the CD40L gene or other therapeutic genes for cancer therapy, the cells need to be manipulated genetically in a safe and effective way to express transgenes long enough to achieve therapeutic goals. Our previous studies demonstrated that insect baculoviral vectors are effective in transducing human NSCs for transgene expression. Advantages associated with baculoviral transduction include large gene cloning capacity, ease of virus generation, low cytotoxicity to transduced cells, and the absence of risks of virus replication and infection in human cells (Airenne et al., 2013). Commonly used replication-defective gene therapy vectors are derived from human viruses such as adenovirus, retrovirus, and adeno-associated virus. Some of them may carry the risk of recombination with wild-type viruses, resulting in replication-competent infectious viruses. Also, preexisting immune responses in the host against adenovirus raise the concern that the use of adenovirus-based vectors will be restricted or compromised (Jooss and Chirmule, 2003; Bessis et al., 2004). Using human blood samples, it has been demonstrated that baculovirus is not targeted by preexisting immunity in humans (Strauss et al., 2007). However, the nonintegrating nature of baculovirus permits only transient transgene expression, which will limit its efficacy in applications that need long-term expression of a transgene. To overcome this limitation, we have developed several baculoviral transduction-based approaches using either recombinase-mediated cassette exchange (RMCE), zinc finger nuclease (ZFN) technology, or transcription activator-like effector nuclease (TALEN) technology for site-specific integration of a transgene in human pluripotent cells without random integration (Ramachandra et al., 2011; Phang et al., 2013; Tay et al., 2013; H. Zhu et al., 2013). We are in the midst of establishing iPSC lines to achieve stable expression of transgenes in the differentiated progeny of the iPSCs, including NSCs.
Baculoviruses are commonly found in nature. As insect-specific pathogens, baculoviruses have been used as biopesticides to protect forest and crops against injurious insects. Clinical evaluation of volunteer human subjects in feeding tests using occluded Heliothis zea baculovirus revealed no signs of inflammation, allergy, or side effects, demonstrating the safety of the viruses (Heimpel and Buchanan, 1967). Recombinant baculoviruses are currently widely applied in the biotech industry for protein production in insect cells. The U.S. Food and Drug Administration (FDA) has so far approved two protein therapeutics manufactured using baculovirus–insect cell systems. Cervarix, a vaccine for the prevention of human papillomavirus (HPV)-associated cervical cancer, is produced in a baculovirus–insect cell system expressing the L1 major capsid protein of HPV. Provenge is a therapeutic cancer vaccine for advanced prostate cancer, which uses patients' dendritic cells pulsed with a recombinant protein containing prostatic acid phosphatase fused with granulocyte-macrophage colony-stimulating factor (Lesch et al., 2011). The fused protein is produced with a baculovirus–insect cell system. The European Medicines Agency (EMA) has also approved human use of Cervarix. The approvals of these two products that make use of proteins manufactured in baculovirus–insect cell systems set important precedents for future baculovirus therapies to gain regulatory endorsement (Airenne et al., 2013).
In conclusion, cancer gene therapy using CD40L-expressing stem cell vehicles provides a promising means to stimulate the immune system to combat cancer. When iPSC derivatives are used, this approach provides the possibility of ex vivo gene therapy to use the progeny of iPSCs generated by reprogramming of the patient's own cells, thus holding potential for translation to clinical application.
Footnotes
Acknowledgments
This research was supported by the Singapore Ministry of Health's National Medical Research Council (NMRC/1284/2011), the Singapore Ministry of Education (MOE2011-T2-1-056), and the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology, and Research, Singapore).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.