
Abstract
Beta-defensins, small antimicrobial peptides, are involved in host immune responses to tumors. In this study, we used beta-defensin 2 (BD2) to explore the possible role of beta-defensins in cancer gene therapy. A recombinant plasmid expressing a secretable form of BD2 was constructed. The biological activities of BD2 in immature dendritic cells (iDCs) were tested in vitro and in vivo. The antitumor effects were investigated in three established tumor models. The secreted BD2 was detected and exhibited chemotactic activity in iDCs both in vitro and in vivo. Recruitment and activation of iDCs in tumor niches resulted in significant tumor growth inhibition. Adoptive transfer of splenocytes and depletion of immune cell subsets revealed that CD8+ T lymphocyte responses mediated the increased tumor inhibition. Furthermore, we also found that chemotactic and maturation-inducing activities in iDCs in tumor milieu contributed to enhanced local antitumor effects. Our study indicates that gene therapy with BD2 can mediate specific antitumor immunity and augment local antitumor effects. Our study also suggested that beta-defensins may merit further exploration for cancer immunotherapy as promising immunogenes.
Introduction
D
Beta-defensin 2 (BD2), a small antimicrobial peptide from the defensin family, is a ligand of both CCR6 and toll-like receptor 4 (TLR4). It recruits memory T cells, especially immature dendritic cells (iDCs), through CCR6 and induces the maturation of iDCs by TLR4 (Yang et al., 1999; Biragyn et al., 2001, 2002; Schutyser et al., 2003; Lapteva et al., 2009). Mouse BD2 (mBD2) mediates DC-targeting antigen presentation, which induces protective, T cell–dependent antitumor immunity (Biragyn et al., 1999; Wang et al., 2007). Previous studies also suggested that vaccines with acute lymphoid leukemia cells L1210 or melanoma-expressing mBD2 could generate potent antileukemia immunity (Ma et al., 2006; Mei et al., 2012). Therefore, we speculate that gene therapy with BD2 may generate direct therapeutic effects by mediating antitumor immunity.
In this study, we investigated the possible roles of beta-defensins in tumor immunity using mBD2. We used a recombinant plasmid expressing a secretable form of mBD2 to determine whether mBD2 chemoattracts iDCs to tumor site and subsequently promotes their maturation. We also analyzed whether the expression of mBD2 in situ contributes to tumor antigen presentation and further induction of specific antitumor immunity. Cationic liposome was used to enhance the efficiency of transfection.
Materials and Methods
Animals and cell lines
C57BL/6J (CD4−/−, CD8−/−, IgH−/−, and CD1−/−) knockout mice were obtained from the Jackson Laboratory. Wild-type C57BL/6J and BALB/c mice were purchased from the West China Experimental Animal Center. The Lewis lung carcinoma cell line LL/2 was cultured in Dulbecco's modified Eagle's medium, and the colon carcinoma cell line CT26 was cultured in RPMI 1640. Both were supplemented with 10% (vol/vol) fetal bovine serum. Meth A fibrosarcoma was cultured in the ascites of BALB/c mice.
Expression of biologically active mBD2 in vitro
The eukaryotic expression plasmid pSecTag/CMV with the Myc and His6 tags was used as the gene vector. The mature mBD2 gene was cloned into the pSecTag/CMV vector (Invitrogen) as described previously (Wang et al., 2007). The reconstructed plasmid pSec-mBD2 was transfected into CT26 cells by the LipofectAMINE 2000 reagent (Invitrogen), according to the manufacturer's instruction. Expression of mBD2 was detected by reverse transcription–polymerase chain reaction and by a commercially available anti-mBD2 rat monoclonal antibody (BD Biosciences). After confirming mBD2 expression at the mRNA level by reverse transcription–polymerase chain reaction, the transfectant (1×106 cells/ml) was cultured for 48 hr. The mBD2 in the supernatants was harvested by coimmunoprecipitation via His6 tag. The protein precipitates were isolated by Tricine-PAGE and analyzed by Western blot. Once mBD2 expression was confirmed, a stop codon was introduced immediately at the end of the mature mBD2 gene.
The bioactivity of mBD2 produced by CT26 cells was assayed according to the chemoattractive ability of cell supernatants to iDCs. Mouse iDCs were isolated from the bone marrow as described previously (Nair et al., 2003). To observe the effect of mBD2 on maturated DCs, iDCs were preincubated with lipopolysaccharides (LPS; 100 ng/ml, 37°C for 24 hr) to induce DC maturation. iDCs migration was assessed by a 96-well microchamber chemotaxis plate (Neuroprobe) as described previously (Wang et al., 2007). The supernatants were harvested, concentrated fourfold by centrifugation (SPD SpeedVac; Thermo), and then added to the lower compartment. iDCs that migrated to the lower surface were stained and counted microscopically in six randomly chosen high-power fields.
Tumor models and treatment with mBD2
Tumor models were established by Meth A, CT26, and LL/2 cells. In brief, 6–8-week-old female BALB/c mice or C57BL/6J mice were used. BALB/c mice were inoculated subcutaneously in the right dorsal flank with 1×106 Meth A cells or 3×105 CT26 cells. C57BL/6J mice were inoculated with 3×105 LL/2 cells. Cationic liposomes were used to increase in vivo transfection efficiency. When tumors reached a diameter of approximately 5 mm, 100 μl of cationic liposome–DNA complex (100 μg DNA:300 μg liposomes, 1:3) or 100 μl of normal saline (n.s) was administered intratumorally and around tumors once every 3 days for a total of 5 times. To detect DC infiltration at the early stage of treatment, tumors were harvested after the second injection. To detect cytotoxic T lymphocytes (CTLs), tumors were resected 1 week after the last injection. Subsequently, tumor tissues were analyzed by hematoxylin and eosin (H&E) staining and immunofluorescence assays. To observe the antitumor effects, tumor growth was monitored and the volume (V) was calculated by the formula V=½×length×width2.
In situ immunofluorescence staining
Immunofluorescence staining was used to detect and analyze lymphocyte-like cells that infiltrated in the tumor site. Anti-CD11c (PE-labeled; BD Biosciences), CD40 (PE-labeled; BioLegend), and CD86 (B7.2, FITC-labeled; BD Biosciences) monoclonal antibodies were used to detect DCs. Anti-CD8 (Cy5PE-conjugated; eBioscience), anti-CD4 (FITC-labeled; BD Biosciences), and anti-CD3 (FITC-conjugated; eBioscience) monoclonal antibodies were used to identify T lymphocytes. An anti-CD19 (FITC-conjugated; BioLegend) monoclonal antibody was used to detect B lymphocytes. Tumors were snap-frozen, and 8 μm sections were prepared in Tissue Tek (Sakura Finetek) for immunofluorescence analysis. Fluorescence was visualized, and images were captured on an Olympus BX60 camera.
Adoptive transfer and CTL assays
Adoptive transfer and 51Cr release assays, which were described previously (Wang et al., 2007), were used to determine the specific cytotoxicity mediated by CTLs. Spleen cells were harvested from mice treated with pSec-mBD2, pSecTag, or n.s. Freshly isolated spleen cells (2×107 cells) were injected into recipient BALB/c mice via the tail vein on the second day after CT26 challenge. T lymphocytes were isolated from single-cell suspensions with Nylon Fiber Column T (L-Type; Wako) and used as CTL effector cells, while Meth A, CT26, and LL2 were used as target cells. The effector and target cells were seeded into a 96-well microtiter plate at various E/T ratios. The CTL activity was calculated by the following formula: % lysis=([experimental release − spontaneous release]/[maximum release − spontaneous release])×100.
Depletion of immune cell subsets in vivo
The CT26 model was established. Immune cell subsets were depleted from BALB/c mice as described previously (Wang et al., 2007). Briefly, tumor-bearing mice were injected intraperitoneally with 500 μg/kg per mouse of anti-CD4 (clone GK1.5, rat IgG), anti-CD8 (clone 2.43, rat IgG), or anti-NK (clone PK136) monoclonal antibody (mAb) on the day before the treatment and then intratumorally injected with pSec-mBD2 as described above. C57BL/6J knockout mice (CD4−/−, CD8−/−, and IgH−/−) and wild-type C57BL/6J mice were inoculated with 3×105 LL/2 cells. Tumor-bearing mice were treated as described above after the tumor diameter increased to approximately 5 mm. The tumor size was measured 20 days after treatment.
Examination of the local effects
Six- to eight-week-old female BALB/c mice were subcutaneously inoculated with 3×105 CT26 cells in both bilateral dorsal flanks. Tumor-bearing mice were divided into three groups (five mice per group). In two of the groups, the tumor on the right was injected with pSec-mBD2, while that on the left was treated with n.s or pSecTag. The third group of mice acted as the control group. The tumor size was monitored. Mice were euthanized 20 days after the first treatment and depilated with 10% sodium sulfide for photographing. Tumor tissue was analyzed by H&E staining.
Statistical analysis
Student's t-test and analysis of variance were used to evaluate the statistical significance. p-Values were considered to be statistically significant when <0.05.
Results
Expression and chemotactic effects of mBD2
mBD2 with Myc and His6 tags was harvested by coimmunoprecipitation and further verified by Western blot (Fig. 1A). It is reported that CCR6 mediates the chemotaxis of iDCs to mBD2. As mBD2 targets CCR6, which is preferentially expressed on iDCs, the physiological properties of mBD2 were determined by its ability to chemoattract iDCs accordingly to the method described previously (Biragyn et al., 2001; Wang et al., 2007). Since iDCs also exhibit chemotaxis through CXCR3, IFN-γ–inducible protein 10 (IP10), as previously described, was used as a control (Howard et al., 2005). The results showed that the supernatant from pSec-mBD2-transfected CT26 chemoattracted iDCs as IP10, whereas the supernatants from the pSecTag and untreated groups did not exhibit chemotactic activity (Fig. 1B). However, mBD2 failed to chemoattract LPS-activated DCs in vitro (Fig. 1B). These data indicated that mBD2 was secreted from tumor cells in vitro and showed chemotactic capability only to iDCs.

Characterization of biofunctional expression.
Recruitment and maturation of iDCs by mBD2 within tumors
To determine the expression and effects of mBD2 in vivo, CT26 models were established. Tumor-bearing mice were divided into three groups, which received two intratumoral injections of pSec-mBD2, pSecTag, and n.s in 2 days, respectively. Tumors were harvested from the euthanized mice on the third day. H&E and immunohistochemical staining indicated that mBD2 could be effectively expressed and secreted from tumor cells in vivo, and was accompanied by the infiltration of monocyte-like cells (Fig. 2A).

Recruitment and activation of iDCs through mBD2 in vivo. Intratumoral mBD2 promotes the recruitment and activation of iDCs. Expression of mBD2 was detected with an anti-mBD2 antibody after receiving two injections. The expression of mBD2 was detected within tumor tissue from pSec-mBD2-treated mice. Expression and secretion of mBD2 was accompanied with increased lymphocyte-like cell infiltration (arrows). The presence and status of DCs was determined with anti-CD11c antibody, anti-CD86 antibody, and anti-CD40 antibodies. Immunofluorescence analysis revealed that these infiltrated lymphocyte-like cells are primarily CD11c, CD86, and CD40 copositive cells. The quantitative data of increased DCs appeared within the pSec-mBD2-treated tumors (*p<0.01). Tumor-draining lymph nodes were analyzed for the presence of CD11c+ CD86+ DCs by flow cytometry (*p<0.01). The data are expressed as mean±SD. Columns, mean of three tumors per group; bars, SD.
To examine whether DCs were first recruited to the tumor site and induced maturation in response to mBD2, the infiltrated monocyte-like cells were analyzed by three antibodies that were directed against the DC markers CD11c+, CD86, and CD40. Significantly more CD11c+ cells were found in both pSec-mBD2-treated tumors. Upregulation of costimulatory molecules CD86 and CD40 was observed on the surface of most CD11c+ cells (Fig. 2). However, mBD2 also failed to chemoattract LPS-activating DCs in vitro (Fig. 1B). A rational explanation for this is that mature DCs are not chemoattracted by mBD2 in vivo, whereas iDCs are attracted into the tumor site and subsequently mature in response to intratumoral mBD2 secreted from the tumor cells. Moreover, pSec-mBD2 treatment resulted in an approximately threefold increase of DCs in tumor-draining lymph nodes (TDLNs) compared with other groups (Fig. 2), suggesting enhanced antigen presentation.
Antitumor effects of mBD2
Currently, there is no evidence that mBD2 can directly inhibit tumor growth or induce apoptosis of tumor cells. In vitro, we first investigated the direct antitumor effects of mBD2 by transfecting tumor cells with pSec-mBD2 or pSecTag. Flow cytometry and MTT assay results indicated slightly increased apoptosis and decreased proliferation in both pSec-mBD2- and pSecTag-transfected CT26 cells. However, no significant difference was observed between pSec-mBD2- and pSecTag-transfected CT26 cells (data not shown). Since the liposome–DNA complex also exhibits cytotoxicity to tumor cells (Khazanov et al., 2006; Nguyen et al., 2007), the findings do not support that mBD2 exerts direct cytotoxicity on tumor cells.
Subsequently, the in vivo effects of mBD2 were examined in the Meth A, CT26, and LL/2 tumor models. Tumor-bearing mice were treated with pSec-mBD2 (100 μg), pSecTag (100 μg), or n.s, respectively. Significant inhibition of tumor growth was observed in pSec-mBD2-treated mice compared with other groups (Fig. 3A–C). In the Meth A model, it was notable that the tumors were almost eradicated after intratumoral injection of pSec-mBD2 (Fig. 3A). The pSec-mBD2 groups also exhibited a survival advantage over other groups (Fig. 3D and E). These results indicated that the expression and secretion of mBD2 within tumors generated antitumor effects in vivo. To exclude the possibility that mBD2 or recruited DCs directly triggered tumor cells' death (Taieb et al., 2006; Nicolas et al., 2007), we performed the same procedures in CT26-bearing nude mice. No difference was observed between pSec-mBD2- and pSecTag-treated mice (data not shown), which also indicated that mBD2 did not generate direct cytotoxicity and that the recruited DCs within tumors did not produce obvious direct antitumor effects. These findings indicated that the antitumor effects of mBD2 may depend on the subsequent immune responses.
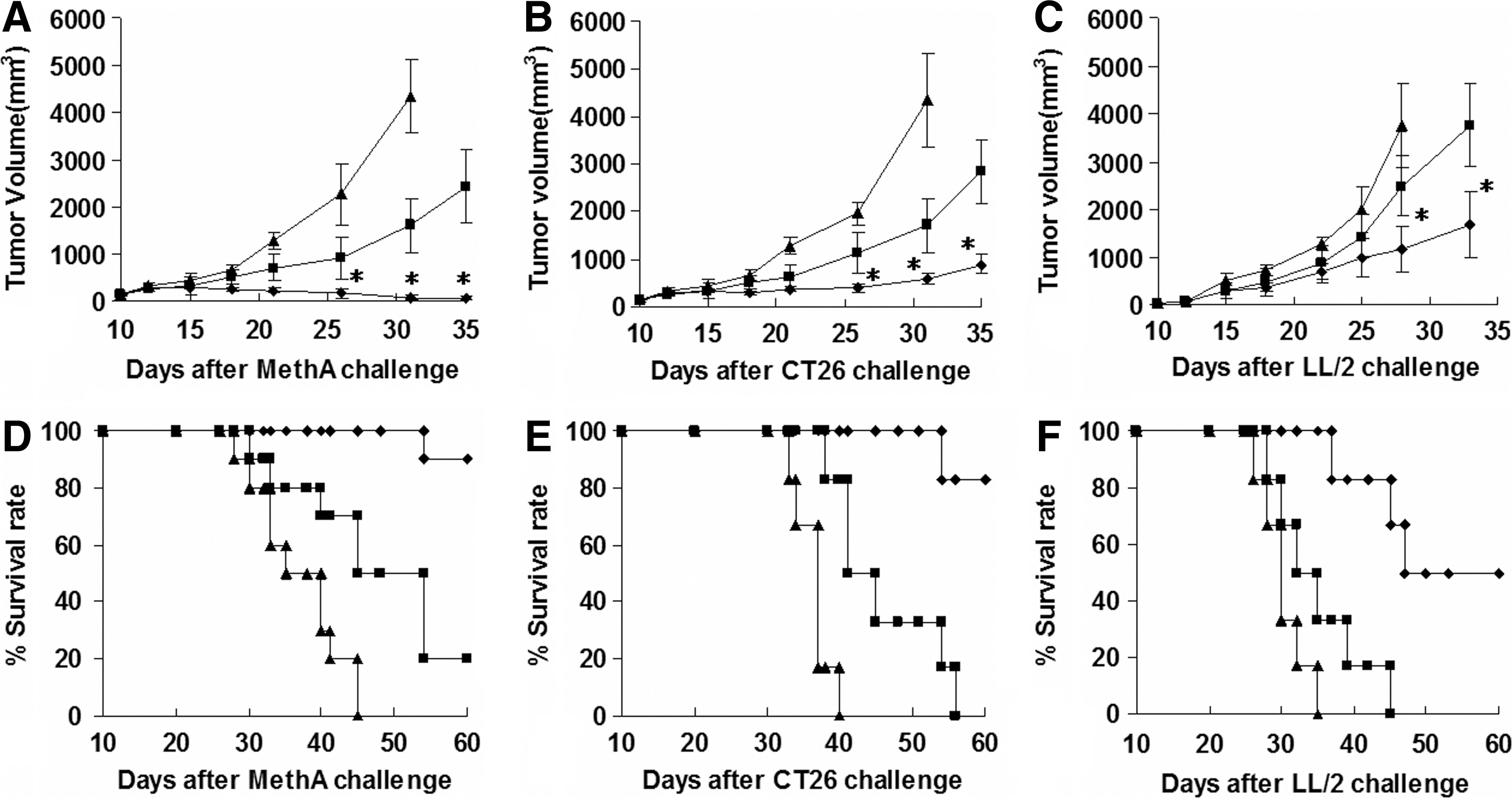
Inhibition of tumor growth through intratumoral injection of pSec-mBD2. Rhombus, square, and triangle indicate pSec-mBD2 group, pSecTag vector group, and n.s group, respectively.
Mediation of specific antitumor immunity by mBD2
An abundance of lymphocytes, accompanied by a relatively lower density of tumor cells, were observed in the margin and interspaces of tumor tissues from pSec-mBD2-treated mice (Fig. 4). The results from the immunostaining of anti-CD8 and anti-CD3 mAbs revealed that these infiltrated lymphocytes were mainly CD8+ T lymphocytes (Fig. 4). In addition, few CD4+ or CD19+ cells were detected in pSec-mBD2-treated tumors (Supplementary Fig. S1; Supplementary Data are available online at
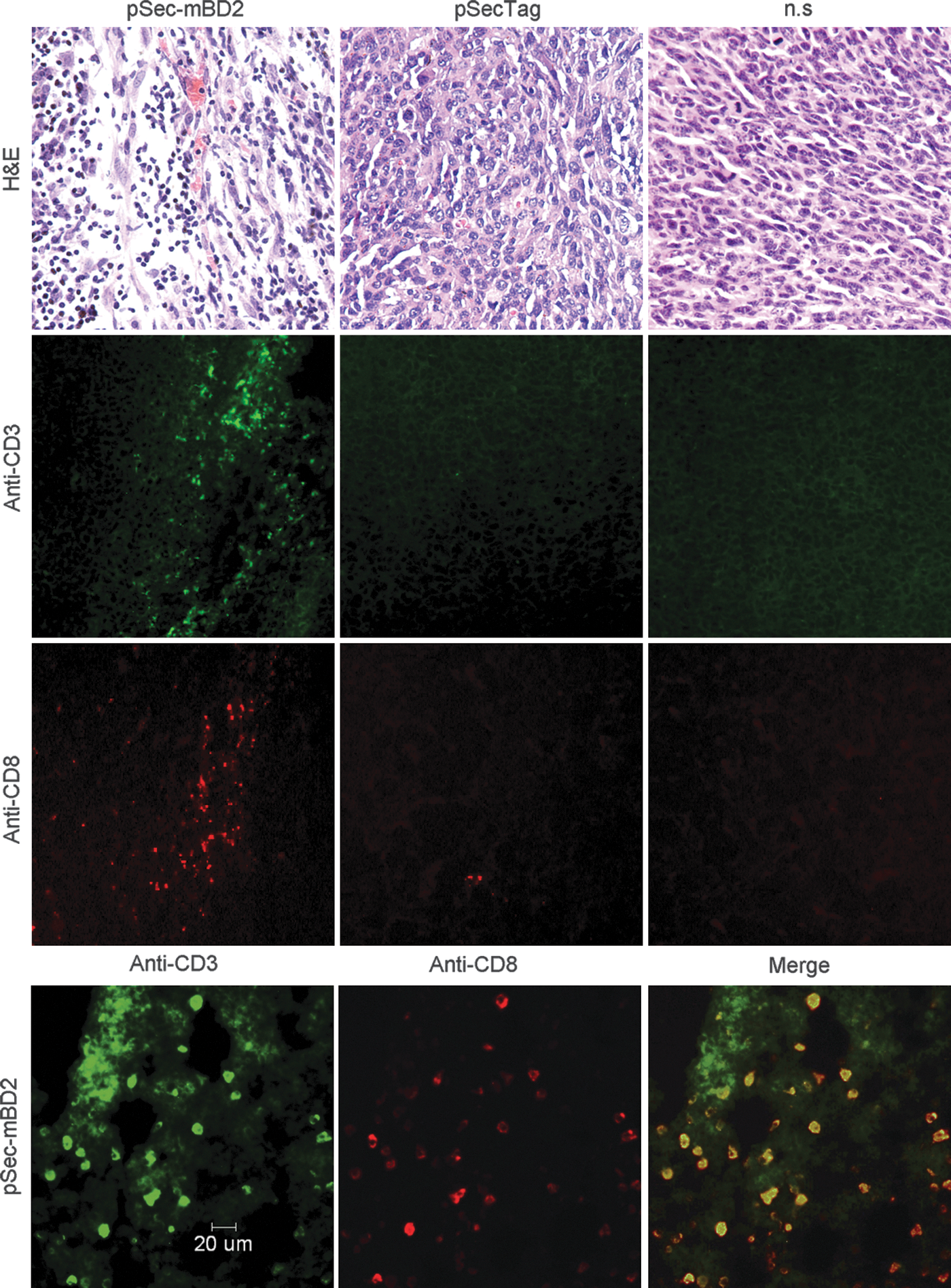
Increased infiltration of CD8+ TL in tumor via administration of pSec-mBD2. Tumor-bearing mice were treated as described in the Materials and Methods section. Tumors were resected 1 week after the last injection and subsequently analyzed by hematoxylin and eosin staining and immunofluorescent detection with anti-CD3 and anti-CD8 antibodies. There is an increased infiltration of lymphocytes in the margin and interspaces of tumor tissues from pSec-mBD2-treated mice. Shown is a representative from the CT26 model. The immunofluorescent analysis revealed that these accumulated lymphocytes are mainly CD3 and CD8 copositive cells.
To further explore the possible mechanism by which antitumor activity is elicited by pSec-mBD2, spleen cells were isolated from the CT26 model and then transferred intravenously into syngenic mice that had been inoculated with CT26 cells on the previous day. Significant tumor growth inhibition and increased survival were observed in the group that received T cells from pSec-mBD2-treated mice (Fig. 5A and B). Spleen T lymphocytes were isolated to detect CTL activity by a standard 51Cr release assay. T lymphocytes from pSec-mBD2-treated mice in the CT26 model exhibited increased cytotoxicity to CT26 cells compared with other groups (Fig. 5C). Similar results were observed in the Meth A and LL/2 models (data not shown). Furthermore, a mixture of cells consisting of CT26, Meth A, and LL2 cells (1:1:1) treated with mitomycin c was used to re-stimulate isolated T lymphocytes from pSec-mBD2-treated CT26-bearing mice. The resulting T lymphocytes showed more intense cytotoxicity to CT26 cells but not to Meth A or LL2 cells (Fig. 5D). These findings indicated that specific cellular immunity was evoked against the tumors.
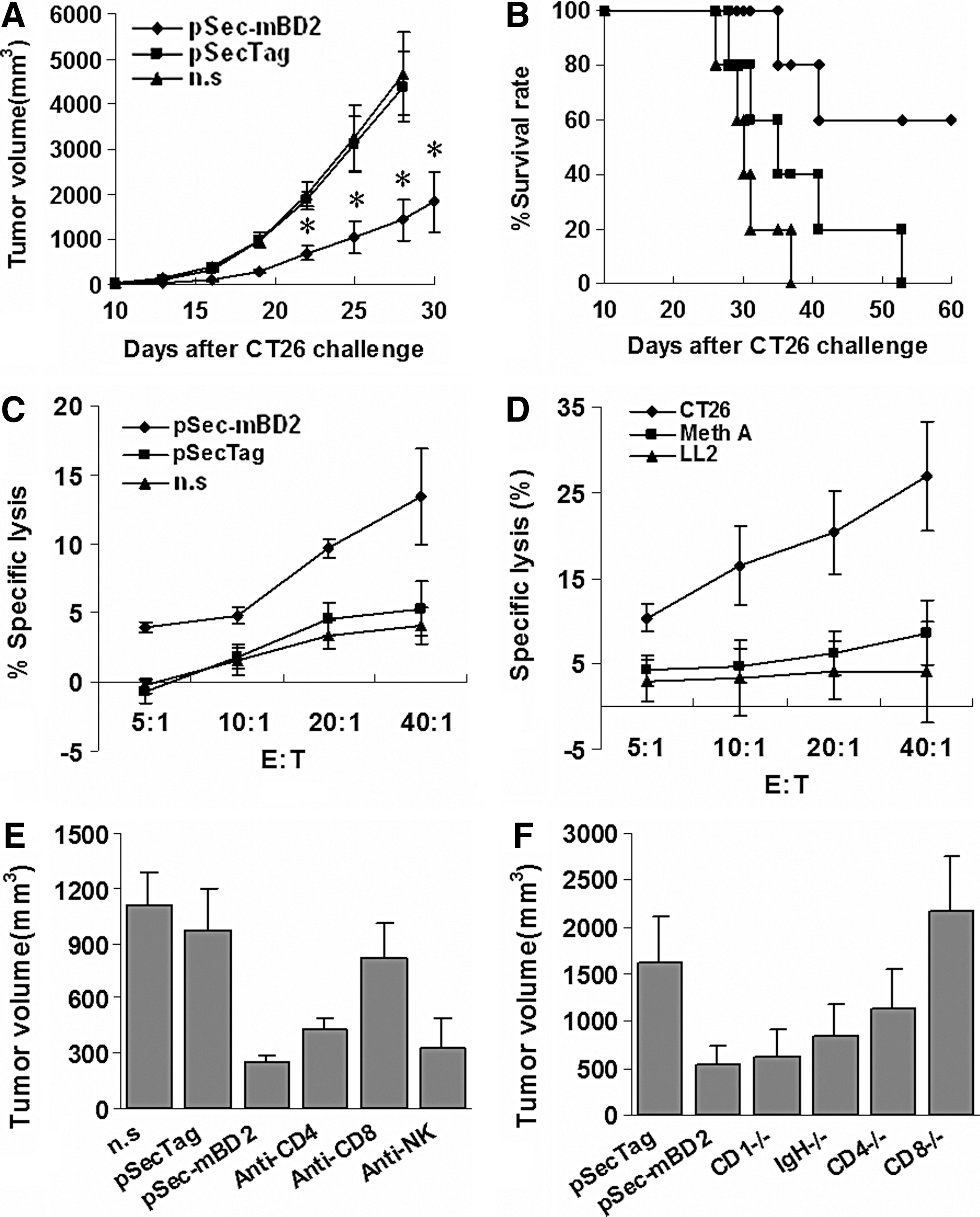
mBD2 mediates specific antitumor immunity.
To confirm the role of immune cell subsets in the antitumor activity elicited by mBD2, CD4+ or CD8+ T lymphocytes or NK cells were depleted. Mice that were depleted of CD8+ T lymphocytes and treated with pSec-mBD2 showed decreased protection against CT26 challenge in comparison with wild-type BALB/c mice. Depletion of CD4+ T lymphocytes in pSec-mBD2-treated mice resulted in a mild but insignificant decrease in protection when challenged by CT26 cells. In contrast, treatment with mAbs against NK cells had no discernable effect on the antitumor activity (Fig. 5E). Moreover, C57BL/6J knockout mice (CD4−/−, CD8−/−, and IgH−/−) and wild-type C57BL/6J mice were also adopted. The antitumor effects of pSec-mBD2 were dramatically impaired in CD8−/− mice compared with wild-type C57BL/6J mice (Fig. 5F). CD4 knockout only slightly attenuated the antitumor effects induced by mBD2. No significant changes were observed in IgH−/− mice. Furthermore, we collected the sera to detect possible antibodies. Increased but weak antibody responses were observed in pSec-mBD2-treated mice in the CT26 model (Supplementary Fig. S2). These findings indicated that mBD2-mediated antitumor immunity primarily depended upon CD8+ T lymphocytes, partially depends on CD4+ T lymphocytes, and may be independent of antibody responses.
Increased local antitumor effects in situ
It has been reported that mBD2 can chemoattract both naïve and memory T lymphocytes through CCR6 and that DCs at early stages of maturation can generate high frequencies of CTLs (Kaiser et al., 2003). This raises the possibility that intratumoral mBD2 may enhance local antitumor effects. To test this hypothesis, BALB/c mice were inoculated subcutaneously with CT26 cells (3×105) in both bilateral dorsal flanks. Tumor-bearing mice were divided into three groups. One group was treated with pSec-mBD2 (right tumor) and pSecTag (left tumor), another group was administered with pSec-mBD2 (right tumor) and n.s (left tumor), and the third group was left untreated and served as the control group. The results showed that the volumes of the tumors from pSec-mBD2-treated mice were significantly smaller than those from the untreated mice. Interestingly, the right tumors treated with pSec-mBD2 were smaller than the left tumors treated with pSecTag or n.s. (Fig. 6). Since mBD2 may induce specific active immunity against tumors, which would generate systemic antitumor effects, the tumor volumes of both bilateral tumors were expected to be similar, especially in pSecTag- and pSec-mBD2-treated mice, after excluding the cytotoxicity of liposome–DNA complexes. Furthermore, there was no evidence that mBD2 could directly induce apoptosis of tumor cells both in vitro and in vivo (data not shown), but there was a significant difference between pSecTag- and pSec-mBD2-treated tumors. These findings suggest that the intratumoral expression of mBD2 not only elicits systematic antitumor immunity, but also enhances local antitumor effects in situ.
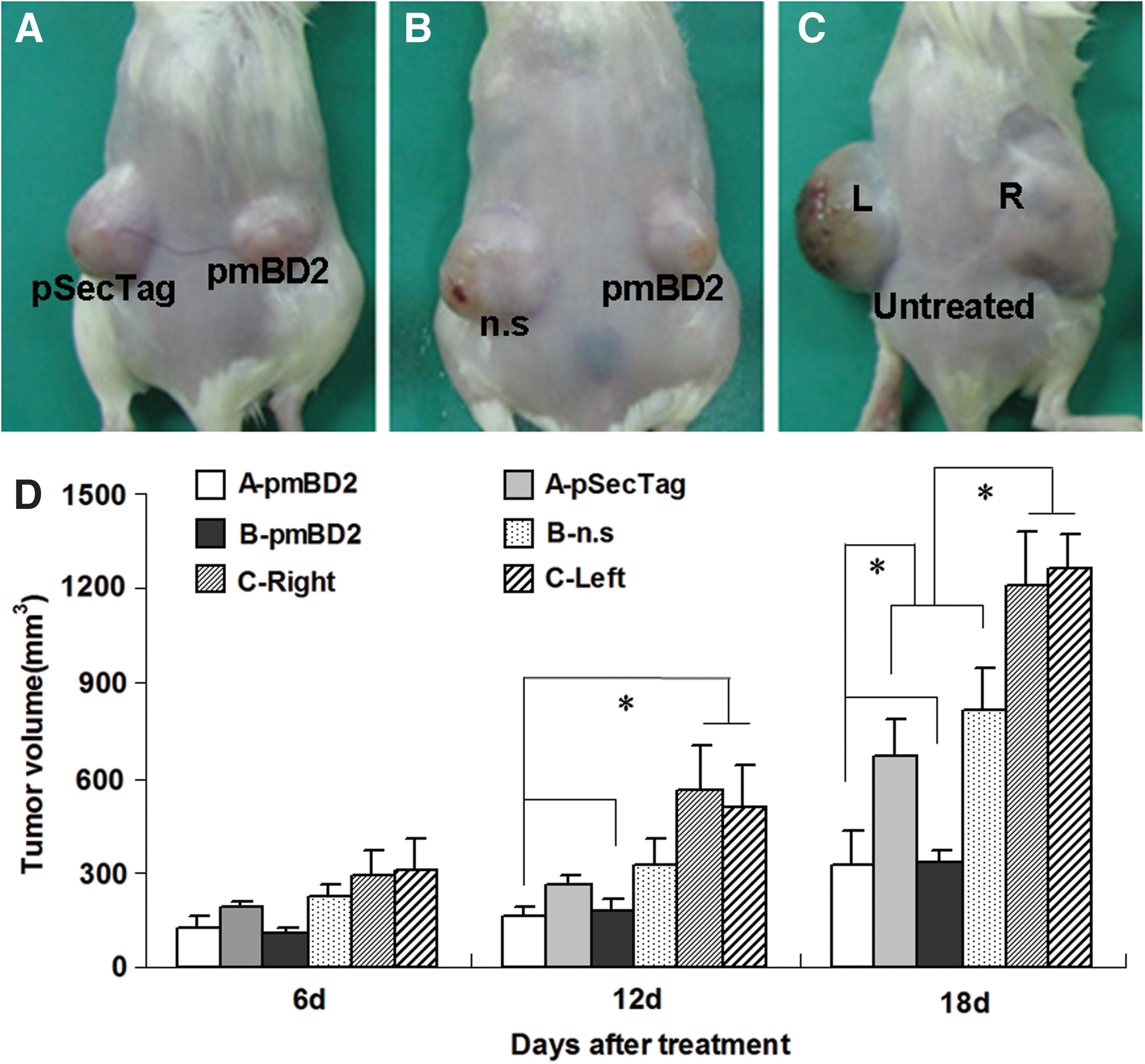
mBD2 enhanced antitumor effects in situ. BALB/c mice were inoculated with 3×105 CT26 cells in their bilateral flanks. Tumor-bearing mice were divided into three groups (n=5). One group was treated with pSec-mBD2 (right) and pSecTag
Discussion
Immunogene therapy, especially by targeting DCs, has proven to be an effective strategy to break down tumor immune tolerance. An ideal strategy is to recruit iDCs to the tumor site and promote their activation to initiate antigen recognition and presentation. In this study, we report that gene therapy with BD2 can recruit iDCs to the tumor niche, promote their maturation, and finally elicit enhanced local antitumor immunity, ultimately resulting in tumor rejection.
Normally, DCs are widely distributed throughout tissues and blood in an immature form and express no or low levels of costimulatory molecules (Shortman and Liu, 2002; Gabrilovich, 2004; Shin et al., 2008). iDCs respond to most signals from pattern recognition receptors, such as TLRs, by undergoing maturation, turning on the production of specific sets of cytokines, and promoting antigen presentation (Roncarolo et al., 2001). Previous studies have shown that the location of DCs and the status of DCs in tumor niches determine different host immune responses to tumors. The accumulation of iDCs leads to tumor immune tolerance and even promotes tumor growth because of inadequate expression of costimulatory molecules and cytokines (Dhodapkar et al., 2001; Bonnotte et al., 2004). However, the recruitment and subsequent maturation of iDCs can trigger specific antitumor immunity (Furumoto et al., 2004; Stary et al., 2007). In this study, we found that mBD2 was expressed and secreted from tumor cells when its gene was introduced into tumor cells; mBD2 exerted chemotactic effects on iDCs in vitro. The expression and secretion of mBD2 was also detected in vivo and resulted in the recruitment and activation of iDCs at the tumor site. Increased levels of DCs were also found in TDLN, indicating enhanced antigen presentation. Subsequent in vivo experiments showed that tumor growth was significantly inhibited in pSec-mBD2-treated mice, and significantly higher numbers of lymphocytes infiltrated in the tumor tissue from pSec-mBD2-treated mice. The antitumor effects could be transferred to untreated mice by spleen cell transfer, indicating that intratumoral administration of pSec-mBD2 elicits specific antitumor immunity. The antitumor immunity was further verified by the 51Cr release assay and depletion of immune cell subtypes. It is known that host defensins such as human alpha defensins (HNP1–3) are directly cytotoxic to tumor cells, probably as a result of damage to the cellular membrane and DNA (Lichtenstein et al., 1986; Gera and Lichtenstein, 1991; Lichtenstein, 1991; Aarbiou et al., 2006). However, mBD2 was not directly cytotoxic to tumor cells. Thus, the antitumor effects mediated by mBD2 are dependent on specific cellular immunity and exclude the nonspecific cytotoxicity of mBD2 toward the tumor in host mice. Beta-defensins may promote angiogenesis by their effects on DCs that cooperate with VEGF-A to promote tumor angiogenesis (Conejo-Garcia et al., 2004). Therefore, we performed a CD31 immunochemical staining to determine blood vessel density. However, we did not observe significant difference when pSec-mBD2 was administered (data not shown). Since beta-defensin-mediated angiogenesis is dependent on endothelial-like specialization of DC precursors, probably in existence of VEGF-A, we believe that mBD2 alone does not promote tumor angiogenesis.
Recent studies suggest that defensins are involved in host response to tumor in situ (Muller et al., 2002; Hubert et al., 2007). A previous investigation also supports the concept that mBD2 may mediate antileukemia immunity by activating innate and adaptive immunities (Ma et al., 2006). However, the involvement of DCs in mBD2-mediated antitumor responses is not well known, especially when DCs are recruited to solid tumors by mBD2. Our study shows that in vivo mBD2 exerts potent chemotactic and maturation-inducing effects on iDCs within tumors, and this triggers specific antitumor immunity. On the other hand, the potent capacity of DCs to activate naïve T cells (Banchereau and Steinman, 1998; Banchereau et al., 2000) and the outcome of the interaction between T cells and DCs are critically influenced by the maturational stage of the DCs (Mapara and Sykes, 2004). Moreover, DCs at early stages of maturation are able to generate high frequencies of CTLs (Kaiser et al., 2003). Thus, the recruitment and activation of iDCs by mBD2 may be another important event by which T lymphocytes are recruited into tumor tissue and are activated to generate more CD8+ CTLs. In this study, specific antitumor immunity was confirmed. Moreover, increased tumor inhibition was observed in pSec-mBD2-treated tumor in the bilateral tumor model. Since mBD2 can also recruit CD8+ effector memory T lymphocytes by CCR6, which produce cytokines, the increase of CTLs may be attributed to the activation of both iDCs and CD8+ effector memory T lymphocytes. This may increase the extent of the subsequent antitumor effects. Indeed, clinical benefits may not be achieved unless the tumor milieu can be altered to enable CD8+ T cell efficacy, even if effective tumor-specific CD8+ T cells are induced in the circulation (Appay et al., 2006). Our findings indicate that changes in the tumor milieu, locally increased chemotactic effects and iDC-inducing activity in the tumor site, may enhance the local antitumor effects of immunotherapy.
Furthermore, mBD2 also functions as a ligand of TLR4 to activate iDCs. TLR4 can modulate both innate and adaptive immunities. Stimulation of TLR4 pathways through their ligands could not only enhance immune response to vaccines by acting as an adjuvant (Andreani et al., 2007; Kanzler et al., 2007), but also mediate host anticancer immune response (Lapteva et al., 2007). Since both the number and activation state of intratumoral DCs are critical factors in host response to tumors, the high density of iDCs in tumor site is not a sufficient condition to induce an immune response (Dhodapkar et al., 2001; Bonnotte et al., 2004; Furumoto et al., 2004). CCL20, a potent chemokine for iDC, when transfected into a rodent tumor cell line, increased intratumoral iDC infiltration; however, tumor growth was enhanced and immunogenicity decreased (Bonnotte et al., 2004). On the contrary, Fushimi et al. (2000) demonstrated that the intratumoral injection of adenovirus-mediated gene transfer of CCL20 suppressed tumor growth via mediating antitumor immunity. In that experiment, however, the maturation of iDCs that were attracted by CCL20 could be activated by viral infection-mediated local inflammation. Indeed, viral infection-induced IFN-α production was demonstrated to cause DC maturation (Blanco et al., 2001). Biragyn and colleagues (2007) also reported that CCL20-based vaccines induced protective and therapeutic antitumor responses similar to mBD2. In that study, however, CCL20 was fused with a tumor antigen to help tumor antigen internalization into APCs, which actually contribute to antigen presentation. The authors also suggest that CCL20 may promote cross-talk between various inflammatory cells, though not activate iDCs (Biragyn et al., 2007). In our investigation, the direct maturation-inducing effects of mBD2 may depend on TLR4 (Biragyn et al., 2002). In addition to recruited iDCs, tumor-infiltrated DCs may also be activated by TLR4, while activated tumor-infiltrated DCs could also effectively direct antitumor immunity (Preynat-Seauve et al., 2006). In addition, two investigations have suggested that some tumor cells constitutively express TLR4, and that activated TLR4 signaling upregulates the level of cytokines, including those of MIP1α, IP10, and MCP1. TLR4-activated tumor cells can then directly induce an antitumor response, even in TLR4-deficient mice (Gatti et al., 2006; Andreani et al., 2007). In this study, we performed the same treatment with pSec-mBD2, pSecTag, and n.s in LL/2-loaded TLR4 knockout mice (n=5), and found that the antitumor effects were deleted completely (Supplementary Fig. S3). Therefore, the activation of the TLR4 signal by mature mBD2 is required for mBD2-mediated antitumor responses and the locally enhanced antitumor effects.
In this study, the three tumor models used had different prognosis, which may be attributed to differences in the innate antigenicity of the tumor cells rather than mBD2 expression level. The weak antigenicity of tumors and defective antigen processing and presentation of immune cells are thought to be the primary reasons for immune escape of tumors. Changing the antigenicity of tumors by modifying tumor cells and enhancing tumor antigen presentation by modifying APCs have proven to be two effective strategies for tumor immunotherapy (Chiodoni et al., 1999; Fushimi et al., 2000; Kirk et al., 2001; Eguchi et al., 2005). It is not easy to manipulate the antigenicity of tumor cells, and it will certainly not be successful without effective antigen presentation. In fact, activation of DCs that mediate tumor antigen presentation has proven to be an inspiring immunotherapy strategy. DCs pulsed with weak tumor antigens are also able to induce specific antitumor immunity (Fong and Engleman, 2000). In this study, the mBD2 gene was introduced into some tumor cells. Local expression of mBD2 attracted and activated endogenous iDCs to the tumor site and subsequently initiated antitumor immunity. Furthermore, the strategy may potentially increase the immunogenicity of tumor cells. This further supports the concept that enhancing the effective antigen presentation by introduction of immunogenes may play a very important role in tumor immunotherapy and in extending its clinical application prospects.
In summary, our study opens up new avenues for understanding the role of host defensins in antitumor immunity. mBD2 and most human defensins have similar biological effects on iDCs and T cells. Our results also indicate that host beta-defensins, which are similar to mBD2, should be explored as immunogene candidates in cancer gene therapy.
Footnotes
Acknowledgments
We thank Ms. Hannah Madson (Middlebury College) for editing the language of the article. This work was supported by National Major Project of China (2013ZX09301003-005) and National Natural Science Foundation of China (81272523).
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.