
Abstract
Efficient O6-methylguanine DNA methyltransferase (MGMTP140K)-mediated myeloprotection and in vivo selection have been demonstrated in numerous animal models and most recently in a phase I clinical study in glioblastoma patients. However, this strategy may augment the genotoxic risk of integrating vectors because of chemotherapy-induced DNA damage and the proliferative stress exerted during the in vivo selection. Thus, to improve the safety of the procedure, we evaluated a self-inactivating lentiviral MGMTP140K vector for transduction of human cord blood-derived CD34+ cells followed by transplantation of the cells into NOD/LtSz-scid/Il2rγ−/− mice. These experiments demonstrated significant and stable enrichment of MGMTP140K transgenic human cells in the murine peripheral blood and bone marrow. Clonal inventory analysis utilizing linear amplification-mediated polymerase chain reaction and high-throughput sequencing revealed a characteristic lentiviral integration profile. Among the bone marrow insertions retrieved, we observed considerable overlap to previous MGMTP140K preclinical models or the clinical study. However, no significant differences between our chemotherapy-treated and nontreated cohorts were observed. This also hold true when specific cancer gene databases and a functional annotation of hit genes by the Panther Database with respect to molecular function, biological process, or cellular component were assessed. Thus, in summary, our data demonstrate efficient and long-term in vivo selection without overt hematological abnormalities using the lentiviral MGMTP140K vector. Furthermore, the study introduces humanized mouse models as a novel tool for the pre-clinical assessment of human gene therapy related toxicity.
Introduction
H
Extensive genotoxicity analysis appears particularly warranted for gene therapy strategies exerting additional genotoxic stress on transduced cells such as in vivo enrichment or exposure to mutagenic agents. This applies specifically to myeloprotective gene therapy approaches employing the (over)expression of selectable drug-resistant genes in hematopoietic stem and progenitor cells (Lachmann et al., 2013), as here the exposure to cytotoxic and mutagenic agents and the proliferative stress exerted during the in vivo selection process associated with this approach represent additional genotoxic risk factors. Besides mutants of the dehydrofolate reductase enzyme or the multidrug resistance 1 (MDR1) gene, mutants of the O6-methylguanine DNA methyltransferase (MGMT) gene have been investigated with considerable success in this context. MGMT encodes an evolutionarily conserved DNA repair protein that removes highly toxic O6-guanine adducts from the cellular DNA and confers resistance to chemotherapeutic drugs with a high O6-methylating or -chloroethylating potential such as temozolomide (TMZ) and other triazene derivatives or chloroethylnitrosoureas such as 1,3-bis(2-chlorethyl)-1-nitrosourea (BCNU), respectively (Milsom and Williams, 2007). Transgenic overexpression of the O6-benzylguanine (BG)-resistant MGMTP140K point mutant has been demonstrated to allow for efficient enrichment of transduced hematopoietic cells by the combined application of the BG and BCNU or TMZ. These studies have been conducted in murine models (Reese et al., 1999; Ragg et al., 2000; Sawai et al., 2001; Jansen et al., 2002; Davis et al., 2003; Milsom et al., 2008; Giordano et al., 2011), including models of β-thalassemia (Persons et al., 2003; Zhao et al., 2009) and erythropoietic protoporphyria (Richard et al., 2004), humanized mice (Pollok et al., 2003; Zielske et al., 2003; Cai et al., 2006, 2008, 2010), as well as large animal models (Neff et al., 2003, 2005; Beard et al., 2009, 2010; Larochelle et al., 2009; Gori et al., 2012; Trobridge et al., 2012). MGMTP140K (henceforth referred to as MGMT) also has been utilized to select for primary lung epithelium (Reese et al., 2008) and muscle cells (Lee et al., 2009) in vivo or to enrich for transgenic and protected lymphocytes in the context of novel gene therapy approaches for AIDS (Trobridge et al., 2009; Kiem et al., 2010). Moreover, a recent clinical phase I study has demonstrated efficient myeloprotection and in vivo enrichment of genetically modified hematopoietic cells following MGMT gene therapy in a cohort of glioblastoma patients with extended survival demonstrated in individual patients (Adair et al., 2012).
To screen for potential genotoxic side effects, the clonal repertoire of hematopoiesis after MGMT-mediated selection has been investigated for hematopoietic cells after exposure to alkylatying agents in vitro (Grund et al., 2010), in murine serial transplant (Giordano et al., 2011), canine and primate models (Neff et al., 2005; Beard et al., 2009, 2010; Larochelle et al., 2009), as well as in the recent clinical phase I study (Adair et al., 2012). Reassuringly, these studies so far have failed to demonstrate any adverse effects of MGMT-mediated chemoselection on the clonal repertoire of MGMT-transduced hematopoietic cells. Nevertheless, a number of questions remain, as some of these studies were compromised by relatively short observation periods (Pollok et al., 2003; Zielske et al., 2003; Cai et al., 2006) or insufficient depth of clonal analysis (Neff et al., 2003, 2005; Beard et al., 2009, 2010; Larochelle et al., 2009). Furthermore, the majority of these studies utilized gammaretroviral vectors (Pollok et al., 2003; Zielske et al., 2003; Cai et al., 2006, 2008, 2010; Adair et al., 2012), which inherently carry a relatively high risk for insertional mutagenesis (Modlich et al., 2009). Therefore, to improve the safety of MGMT-mediated myeloprotection and in vivo chemoselection strategies, we here analyzed MGMT expression from a third-generation SIN lentiviral vector employing the truncated elongation factor 1α (i.e., elongation factor-1α short version; EFS) promoter for transgene expression. This promoter was chosen as it was previously reported to direct robust and stable MGMT expression while avoiding the very high expression levels associated with viral promoters such as the spleen focus forming virus promoter (SFFV) (Milsom et al., 2008).
Materials and Methods
Generation of the SIN lentiviral MGMT vector construct
In our study a third-generation SIN lentiviral vector with an EFS as an internal promoter was used to coexpress MGMTP140K mutant and enhanced green fluorescent (eGFP) protein coupled to internal ribosomal entry site. See Supplementary Materials and Methods (Supplementary Data are available online at
Virus production
Viral supernatant was produced by transient transfection using calcium phosphate precipitation of 293T cells as previously described (Modlich et al., 2009). See Supplementary Materials and Methods for a brief description.
Evaluation of peripheral blood for human cell engraftment in NSG mice and end analysis
Starting at 6–8 weeks post-transplantation, peripheral blood (PB) was collected from NSG mice and analyzed at regular intervals for human engraftment by flow cytometry. Mice were euthanized 24–28 weeks after transplantation and analyzed for human engraftment and the levels of GFP+ cells in the PB, spleen, thymus, and bone marrow (BM). See Supplementary Materials and Methods.
Isolation and transduction of CD34+ cells
Human umbilical cord blood (CB) was collected from the donors after their informed written consent and after approval by the Hannover Medical School institutional ethics committee and the Norddeutsches Knochenmark- und Stammzellspender Register (Hannover, Germany). The CB was processed within 4 hr of collection, and human CD34+ cells were isolated according to the Miltenyi human CD34+ isolation kit protocol (Bergisch Gladbach, Germany). Isolated CD34+ cells were prestimulated for 48 hr in StemSpan medium (StemCell Technologies, Cologne, Germany) supplemented with 2 mM glutamine and 1% penicillin/streptomycin (PAA, Coelbe, Germany) and 100 ng/ml rhSCF, 100 ng/ml rhFLT3L, and 50 ng/ml rhTHPO (all from PeproTech GmBH, Hamburg, Germany). Subsequently, the cells were transduced (multiplicity of infection 20) with either EFS.MGMT.GFP or EFS.GFP viral supernatant on Retronectin-coated (10 μg/cm2; Takara, Otsu, Japan) dishes and using spinoculation at 2,000 rpm for 45 min at 4°C. Cells were cultured for a day further before transplantation into NSG mice.
Transplantation of NSG mice
A breeding colony of NSG was established and maintained at Hannover Medical School. Mice were kept in IVC racks (Allentown Inc., Moebris, Germany) in specific pathogen-free conditions. All animal experiments were approved by the local animal welfare committee and performed according to their guidelines. For the in vivo experiments 8–10-week-old NSG mice received a sublethal dose of 300 cGy total body irradiation by the PRIMUS linear accelerator (Siemens, Berlin, Germany) and were transplanted within 24 hr. The drinking water was supplemented with Ciprobay (Bayer, Leverkusen, Germany) for the first 3 weeks after irradiation to protect the mice from potential infections.
Administration of chemotherapy
Nine weeks after transplantation, the mice infused with the EFS.MGMT.GFP-transduced CD34+ cells were either treated with chemotherapy or left unexposed (non-treated group; NT). The treatment included weekly administration of 20 mg/kg BG (Sigma, Seelze bei Hannover, Germany) and 5–10 mg/kg of BCNU (Bristol Meyer Squibb GmBH & Co KGaA, Munich, Germany) 1 hr later for 2 consecutive weeks. BG and BCNU were prepared as described (Pollok et al., 2003). The mice transplanted with the EFS.GFP-transduced CD34+ cells (n=3), henceforth referred to as control, were not treated and monitored similar to the MGMT non-treated animals.
Linear amplification-mediated polymerase chain reaction
Vector insertion sites were amplified by linear amplification-mediated polymerase chain reaction (LAM-PCR) using 500 ng of DNA isolated from PB and BM samples using the QIAamp DNA mini Blood kit (Qiagen GmBH, Hilden, Germany). The LAM-PCR was performed as described previously (Schmidt et al., 2007) with the following modifications. After the double-strand synthesis step, each sample was equally divided into three parts for restriction digestion with different enzymes, namely, Tsp509I, HaeIII, and TaqαI (all from New England Biolabs, Ipswich, MA). Separate ligation reactions were carried out using enzyme-specific linkers (sequences in Supplementary Table S1) and were pooled for the denaturation step before the nested end-point PCRs. The PCR products were separated on a 2% agarose gel to visualize the bands and validate the PCR run. In total, we performed LAM-PCR on pretransplant (pre-TX; an aliquot of hCD34+ cells before transplantation), PB (collected before and after chemotherapy), and BM samples for mice from all groups.
High-throughput sequencing and integration-site analysis
For the 454-pyrosequencing run, LAM-PCR samples were digested with the SacI restriction enzyme (New England Biolabs) after the first exponential PCR at 37°C for 2 hr, followed by heat inactivation at 65°C for 20 min. This step removed most of the vector internal control bands. The digested DNA was diluted 1:30 with water, and 2 μl was used for the second exponential PCR along with the barcoded primers (Eurogentec, Seraing, Belgium). This final PCR product was purified via the QIAquick PCR Purification Kit (Qiagen GmBH), and the concentration was determined with a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, Germany). Equal amounts of amplicons were used to create a library with a total of 500 ng DNA. The library was dispatched to GATC Biotech AG (Konstanz, Germany) for the 454 high-throughput sequencing with the Roche GS FLX+Titanium platform. The resulting sequences were sorted for their DNA barcode, using a custom Bioperl script. A sequence was considered valid only if it contained the part of the SIN lentiviral LTR along with the nested LTR primer and linker sequences with >95% identity. This step was performed using cross_match (Phil Green, University of Washington;
Locus-specific PCR and VCN determination
To validate individual vector integrations, reverse primers specific for the genomic region near the 3′ vector boundary were designed. The LTR primer (lvLTRIII) from the nested PCR step during the LAM-PCR served as the forward primer for validation of all integration sites. At first, conventional PCR using the Taq polymerase (PEQLAB Biotechnologie GmBH, Erlangen, Germany) was performed specifically on the sample from which the particular integration site was retrieved to confirm its presence and also check the functionality of the primers. Subsequently, real-time TaqMan PCR (Life Technologies GmBH, Frankfurt, Germany) was carried out on all the PB samples collected at different time points using a StepOnePlus (Applied Biosystems, Carlsbad, CA). Additionally, the VCN was determined using primers detecting the woodchuck posttranscriptional element (PRE) and polypyrimidine tract-binding protein 2 (PTBP2) as an internal reference. All quantitative PCRs (qPCRs) were performed in triplicates with standard deviations in cycles ranging from 0.02 to 1.23 (with signals obtained in a range of 24–36 cycles). A plasmid standard harboring sequences for the residual part of the LTR, PRE, and PTPB2 was used for the quantification. The list of all primers is provided in Supplementary Table S1.
Statistical tests
Statistical significance was determined by nonparametric t-test, Fisher's exact test, chi-square test, and two-way ANOVA test, and p-values≤0.05 were considered statistically significant.
Results
In our xenograft model, human CB-derived CD34+ were transduced with a third-generation SIN lentiviral construct expressing MGMT from an internal EFS promoter (EFS.MGMT.GFP; Fig. 1A) and transplanted into immunodeficient NSG mice. For control, NSG mice were transplanted with (EFS.GFP)-transduced cells. PB and BM samples from these animals were procured for integration-site analysis. In all our experiments, the progenies of 1–2×105 CD34+ cells were transplanted per mouse with transduction efficiencies ranging between 10% and 35% (n=4 transplants). Subsequent to hematopoietic reconstitution, mice were subjected to combined BG/BCNU chemotherapy (Fig. 1B).
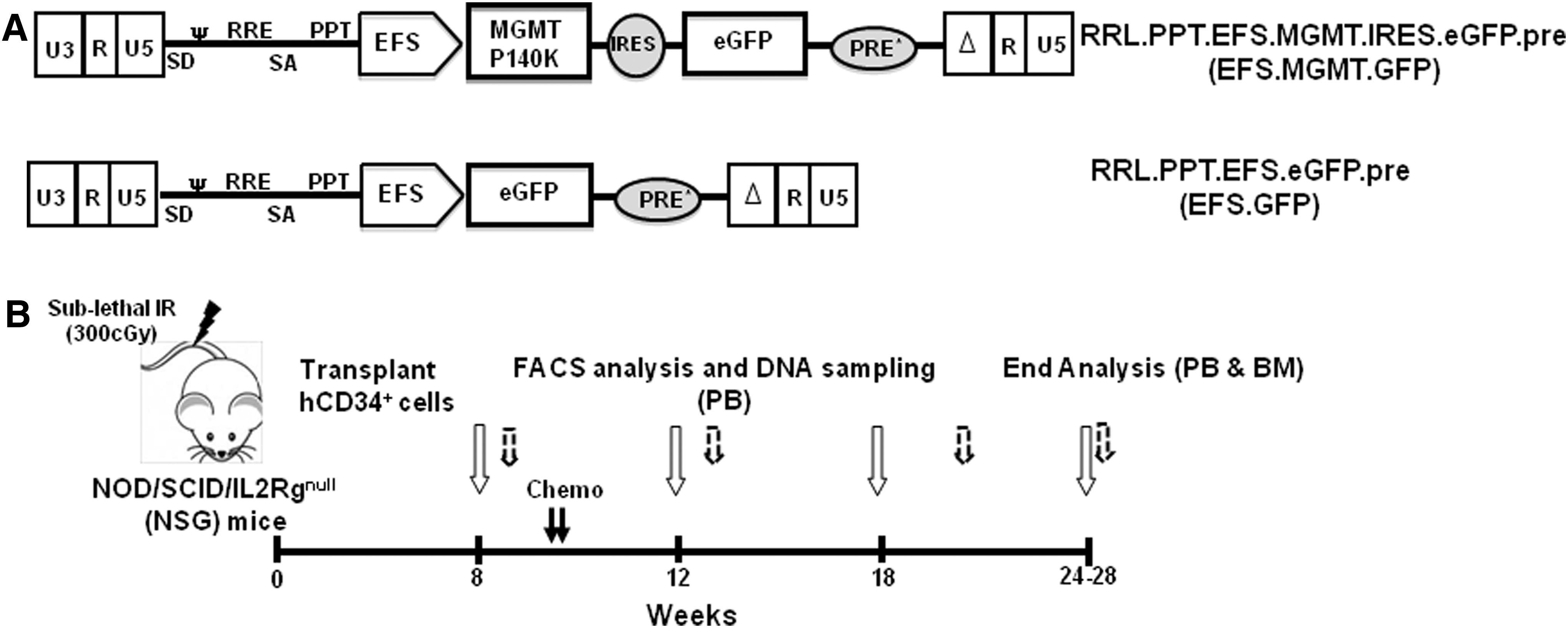
Schematic representation of the lentiviral vectors and the xenotransplant model.
High-dose chemotherapy allows efficient enrichment of transgenic human cells but is toxic to NSG mice
In our initial experiment, four animals were treated with a BCNU dose of twice 10 mg/kg (a week apart). The pretransplant transduction efficacy was 18.8%. This regimen resulted in a profound granulocyto- and thrombocytopenia with nadir values of <0.1×103/μl and 112×103±28×103/μl, respectively, measured 4 days after chemotherapy application (Fig. 2A and B). Within 8–21 days after treatment, three out of the four animals succumbed to severe pancytopenia. The only surviving animal was monitored for another 9 weeks (i.e., 17 weeks post-transplantation) with PB counts recovering to normal values 4–8 weeks after chemotherapy. Chemotherapy resulted in profound loss of human cells in the PB of all four NSG mice within 7 days of treatment (Fig. 2C). However, a significant enrichment of transduced human cells from 2.7%±0.8% before to 65%±8.8% one week after treatment (p=0.005; n=4) was observed in the human CD45+ PB population, indicating effective selection (Fig. 2D). Animals were analyzed for the engraftment of human cells in the BM immediately after death (two animals) and at the end of the experiment for the only long-term survivor. At these time points, the human cells in the BM mainly were composed of myeloid and B-cells (Fig. 2E). High levels of GFP+ human cells were detected in all hematopoietic compartments of the BM (Fig. 2F). Thus, the high-dose BG/BCNU treatment schedule clearly resulted in selection of MGMT-transduced cells, but at the expense of lethal myelotoxicity in the majority of animals. To circumvent this fatality and protect the murine hematopoietic system, we applied the same chemotherapy dose schedule but combined it with the transplantation of 1×106 freshly isolated NSG whole BM cells 24 hr after the second dose of BCNU. While this approach substantially reduced the pancytopenia and led to survival of all animals until the end of the experiment, the percentage of human cells in the PB of these animals dropped to negligible levels (<0.01%) within 4–5 weeks after the murine cell transplant (Supplementary Fig. S1). However, when these animals were treated for a second time with a reduced chemotherapy regimen of 2×20 mg/kg BG/7.5 mg/kg BCNU given a week apart, transduced human cells were efficiently recovered and even enriched within the hCD45+ PB and BM populations.
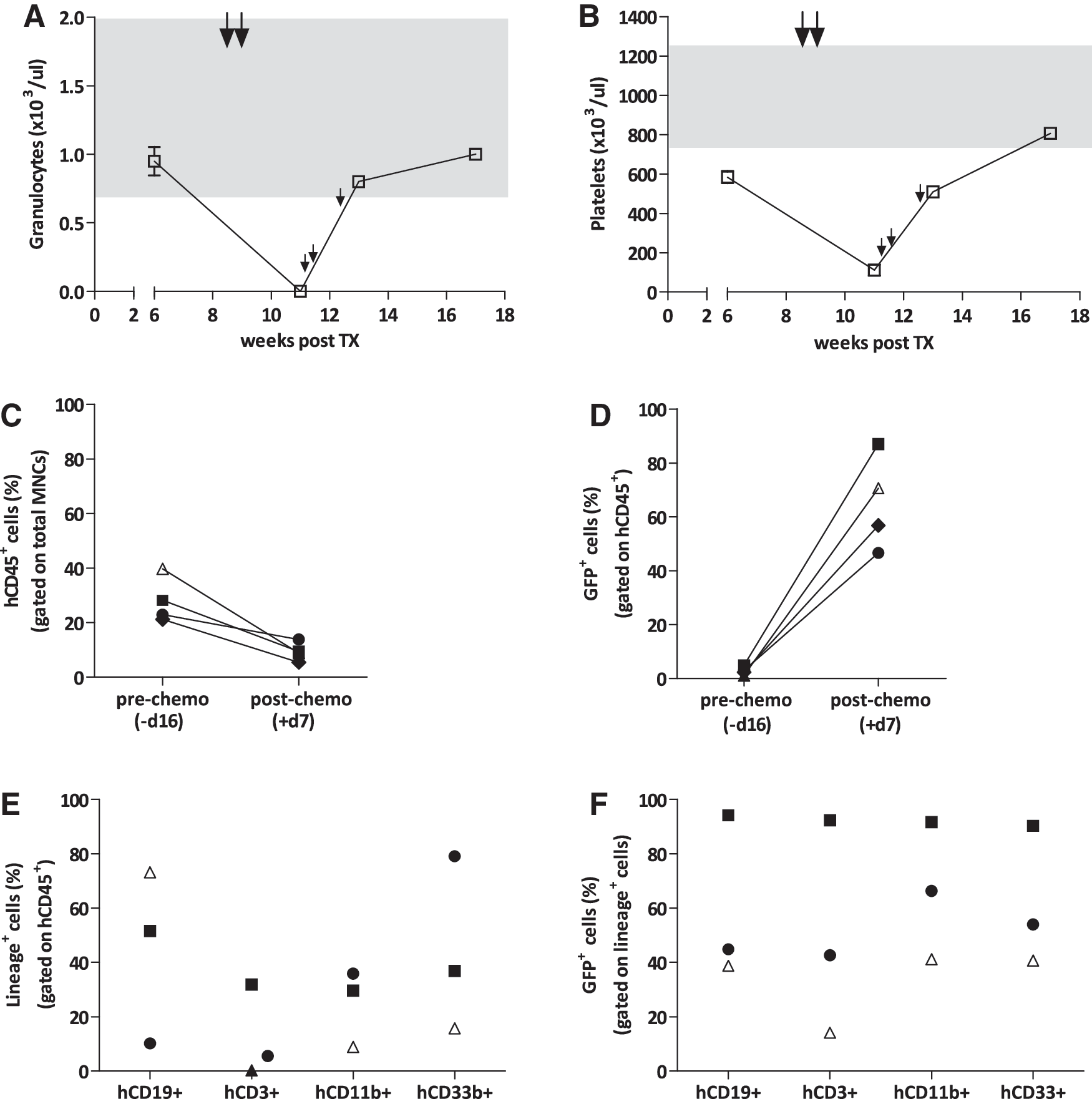
High-dose chemotherapy allows effective chemoselection of transduced human cells but also lethal myelosuppression. Mice were administered two doses of 20 mg/kg 6-BG/10 mg/kg BCNU 8 weeks after transplant (large arrows). PB values for
Efficient in vivo selection of MGMT-transduced human cells with dose-reduced chemotherapy
On the basis of these results, we reduced the dose of BCNU to 7.5 mg/kg followed by 5.0 mg/kg (intermediate dose) or 5.0 mg/kg twice (low dose). Mice tolerated these chemotherapy regimens relatively well, although severe granulocytopenia (<0.1×103/μl) was observed for both groups 4 days after chemotherapy application. Thrombocytopenia, however, was only moderate (652×103±55×103/μl for the intermediate and 580×103±158×103/μl for the low-dose regimen), and granulocyte as well as platelet values recovered to normal levels within 3 and 8 weeks of chemotherapy application, respectively (Fig. 3A and B). By the end of the experiment at 28 weeks, granulocyte counts dropped considerably in all animals (including non-treated controls; data not shown), which could be caused by long-term chemotherapy toxicity as well as the prolonged observation period. In both treatment groups, two out of five animals were lost 5–12 weeks post-transplant, although the cause of death appeared unrelated to the chemotherapy. Contribution of human cells in the PB is depicted in Fig. 3C, showing significant decrease in both the treated groups (p=0.008, medium dose, and p=0.0381, low dose, compared with the non-treated group, NT), 2 weeks after chemotherapy application. Engraftment levels in both the treated groups improved over time but remained well below the values of the NT cohort. For both the dose groups, chemotherapy resulted in considerable enrichment of the transduced human GFP+ population in the PB, peaking approximately 12 weeks after chemotherapy application (Fig. 3D). We detected an increase of GFP+ cells within hCD45+ PB cells from 2.3%±1.3% to 21.1%±7.2% (p=0.02; n=3) in the intermediate and from 0.89%±0.47% to 18.4%±10.4% (p=0.26; n=3) in the low-dose group. In the intermediate-dose group, these values were maintained until the end of the experiment (19.6%±2.3%), whereas selection was partially lost in the low-dose group (7.6%±4.7%; at week 26). In contrast, percentages of GFP+ cells remained low in non-chemotherapy-treated animals and even decreased throughout the experiment from 2.1%±0.42% to 0.5%±0.24% (p=0.02; n=6), a loss most likely reflecting the lower transduction rates that can be expected for primitive long-term SCID-repopulating cells (SRCs) in comparison to more short-term active stem cell clones.
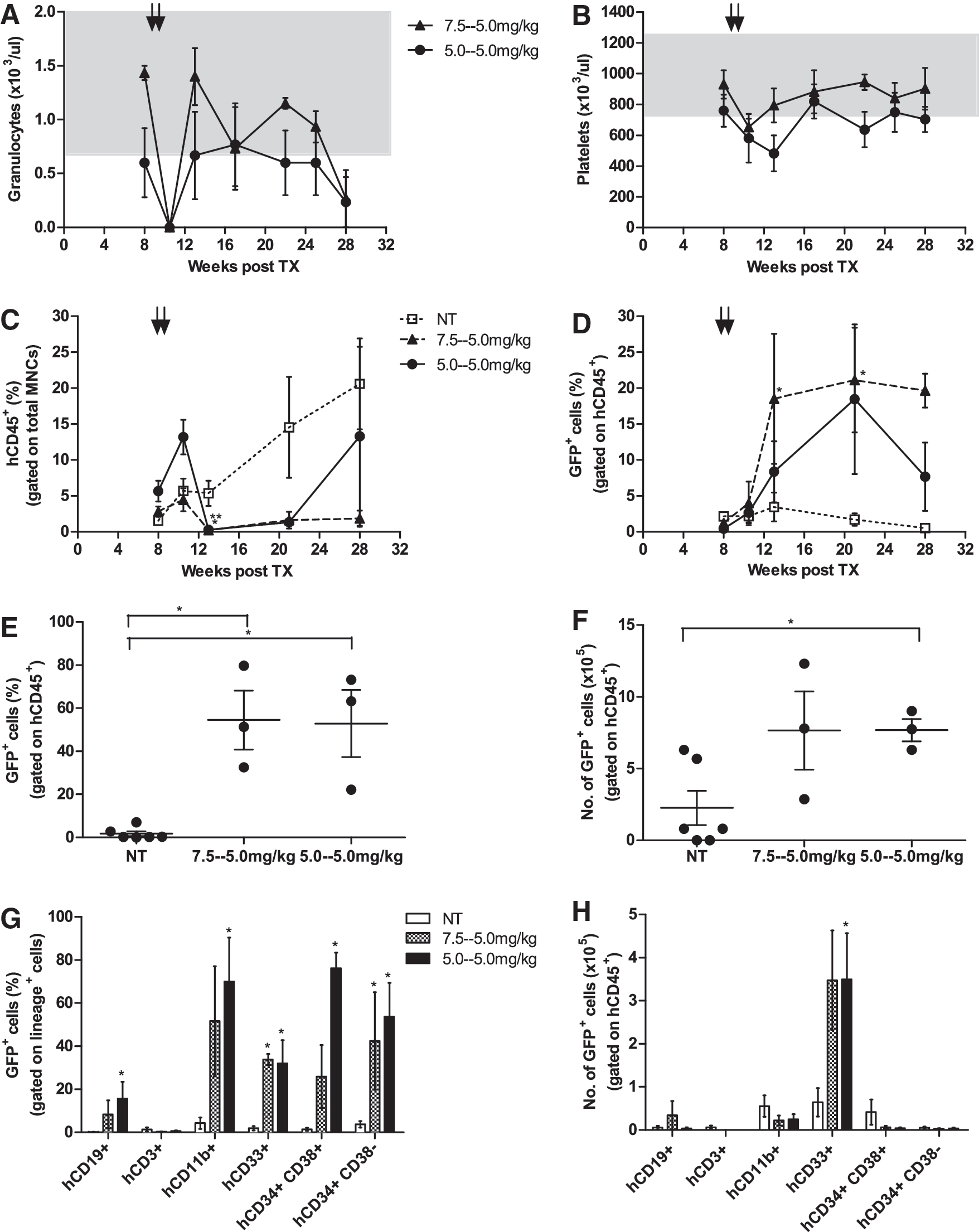
Reduced doses of BG/BCNU avoid myelosuppression and efficiently select MGMTP140K-positive human cells. Mice were treated with two cycles of chemotherapy (black arrows) 8 weeks posttransplant. One group received 20 mg/kg BG/7.5 mg/kg BCNU and 20 mg/kg BG/5.0 mg/kg BCNU (intermediate dose; triangle), and the other cohort received two times 20 mg/kg BG/5.0 mg/kg BCNU (low dose; circle). The nontreated animals (NT; open square) served as controls. PB values for
Furthermore, effective in vivo enrichment of EFS.MGMT.GFP-transduced cells was confirmed in our model when BM cells were isolated from animals euthanized at the end of the experiment 18 weeks after BG/BCNU application (Fig. 3E). Here, the percentage of transduced GFP+ cells within the human cell population was significantly increased in the intermediate- and the low-dose treatment groups compared with NT animals (54.5%±13.7%, p=0.02, and 52.9%±15.6%, p=0.02 [n=3], respectively, vs. 1.8%±1.1% [n=6]). Substantially increased counts in comparison with NT controls (2.3±1.2×105) were also detected regarding absolute numbers of transgenic human cells in the murine BM, with 7.7±2.7×105 cells (p=0.09; n=3) for the intermediate-dose group and 7.7±0.8×105 cells (p=0.03; n=3) for the low-dose group (Fig. 3F). Significant enrichment of transgenic B cells (hCD19+), mature myeloid (hCD11b+) and myeloid progenitor cells (hCD33+), as well as more differentiated (hCD34+ hCD38+) and primitive (hCD34+ hCD38−) stem/progenitor cells after BG/BCNU application also was observed when the data were broken down to distinct hematopoietic BM subcompartments (Fig. 3G). The expansion of transgenic cells was achieved primarily in the hCD33+ myeloid progenitor compartment (Fig. 3H; representative flow cytometry plots for a treated and NT animal are depicted in Supplementary Fig. S2). Although T (hCD3+) cell engraftment was negligible in the BM, T cells comprised 50–70% of human cells in the spleen and 70–75% in the thymus (see Supplementary Fig. S3A), and we observed higher percentages of transduced T cells in the chemotherapy-treated groups (21.8%±18.1% and 13.9%±5.6%) versus the non-treated (6.9%±5.4% and 0.24%±0.08%) cohort in the thymus and the spleen, respectively (data not shown). Thus, these results demonstrate that moderate doses of chemotherapy regimen can significantly reduce hematotoxicity while still allowing for the efficient and stable enrichment of transgenic GFP/MGMT-positive human cells in the major hematopoietic compartments for the entire observation period of up to 18 weeks after chemotherapy.
Characteristic lentiviral integration profile observed in all groups
As a next step we sought to assess the effect of chemoselection on the clonal repertoire of transduced human cells engrafted in these NSG employing LAM-PCR followed by high-throughput pyrosequencing for integration-site analysis. After removal of the linker- and vector-specific sequences, 486 unique sequences were mapped to the human genome using the HISAP (Arens et al., 2012) and assigned to the following samples: 231 in 2 pretransplant samples (pre-TX; from reduced chemotherapy and GFP control experiments), 144 in treated (10 BM and 20 PB; samples from all dose groups), 54 in NT (4 BM and 8 PB), and 57 in control (3 BM and 6 PB) samples. Irrespective of the vector or treatment, all groups exerted similar insertion-site profiles (Fig. 4). In accordance with previous studies employing lentiviral vectors (Mitchell et al., 2004; Laufs et al., 2006; Grund et al., 2010), in our experiments we found most of the integrations within gene-coding regions; 76.5% in pre-TX samples, 67.4% in treated samples, 60.5% in NT samples, and 74% in control BM samples. Furthermore, among these insertions, 65.4%±4% occurred in introns. In line with a typical lentiviral insertion pattern, no preference for insertions in the vicinity of transcription start sites (TSS) was observed. Also, overall ∼20% of hits were found±10 kb around CpG regions with no significant differences between the groups. These results indicate a representative insertion profile of SIN lentiviral vectors that was not significantly altered by chemotherapy application or MGMT expression. An important technical limitation regarding PB samples after xenotransplantation is the low absolute number of human cells in these samples. Combined with a low chimerism in some animals, these are suboptimal conditions for LAM-PCR and 454-sequencing. Consequently, we mapped either no or only very few (≤5) insertions in the PB samples of our animals. Thus, we mainly focused on the BM-derived insertions for further clonality assessment (Supplementary Table S2). We compared insertion sites retrieved from chemotherapy-treated, NT, and GFP control vector-transduced BM samples to the pre-TX samples using the retroviral insertion sites published in previous MGMT studies (Beard et al., 2009, 2010; Grund et al., 2010; Giordano et al., 2011; Adair et al., 2012). Moreover, we matched the genes closest to lentiviral integrations found in our model with the available cancer databases [D'Antonio et al. (2012) and
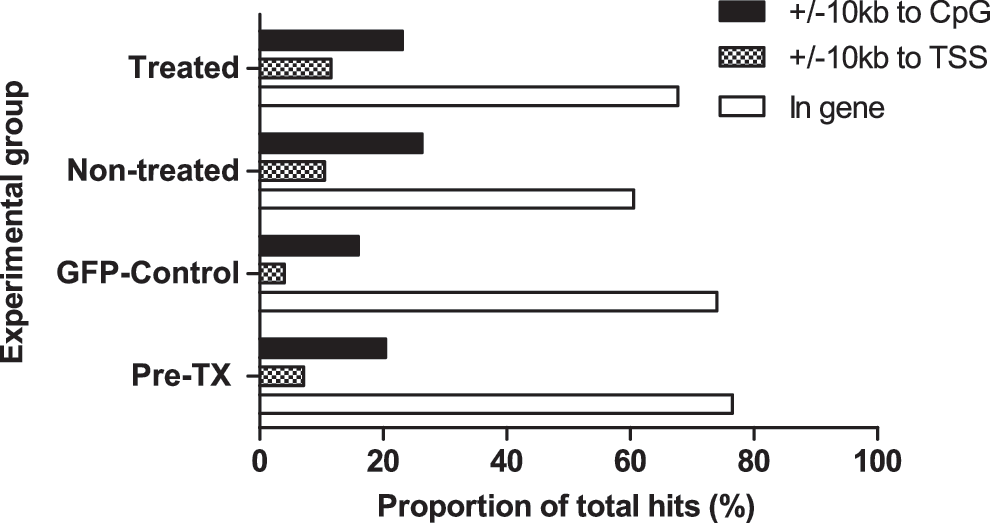
Retroviral integration-site analyses. Insertion sites from different samples of control, treated, non-treated, and pre-transplant (pre-TX) were analyzed for their location with respect to the CpG islands (±10 kb distance; black column), transcription start sites (TSS;±10 kb distance; checkered column), and hits within genes (white column). The chi-square test revealed no significant differences between the groups in any of the categories.
Oligoclonal repopulation in NSG recipients is not affected by chemotherapy application
The number of reads resulting from pyrosequencing for a particular insertion site is commonly used to estimate the abundance of a clone, thus providing an assessment of a sample's clonality (Biffi et al., 2011). The pre-TX samples consisted of various clones harboring integrations without any obvious overrepresentation, as 99.4%±0.5% of the detected insertions were represented by low (≤5) reads (Fig. 5). In contrast, in the treated, NT, and GFP control BM samples, 32.7%±6.6%, 43.0%±6.0%, and 25.1%±1.6% of insertions with low read counts were detected (p<0.01; two-way ANOVA). At the same time, high percentages of insertions with 101–1,000 (25.7%±6.3%, 12.9%±4.3%, and 35.4%±0.8%) and >1,000 (21.6%±9.4%, 13.2%±4.8%, and 11.7%±0.5%) reads were retrieved for treated, NT, and control BM samples, respectively. Statistical analysis using the chi-square test, however, revealed these differences between the three experimental cohorts as not significant. As the presence of insertion sites with high read counts (e.g., >1,000 reads) can be an indication of clonal imbalance, it is important to stress that we did not observe any overt clonal expansion in our animals (with the potential exception of animal RP10; see below). This and the fact that insertions with high read counts were detected in all three experimental cohorts (treated, non-treated, and GFP controls) seem to suggest that the chemotherapy and the selection process as such did not affect the clonality in the treated cohort. The overall reduced clonality in the BM samples of all groups suggests that only a few clones contributed to the in vivo repopulation repertoire over a prolonged period.
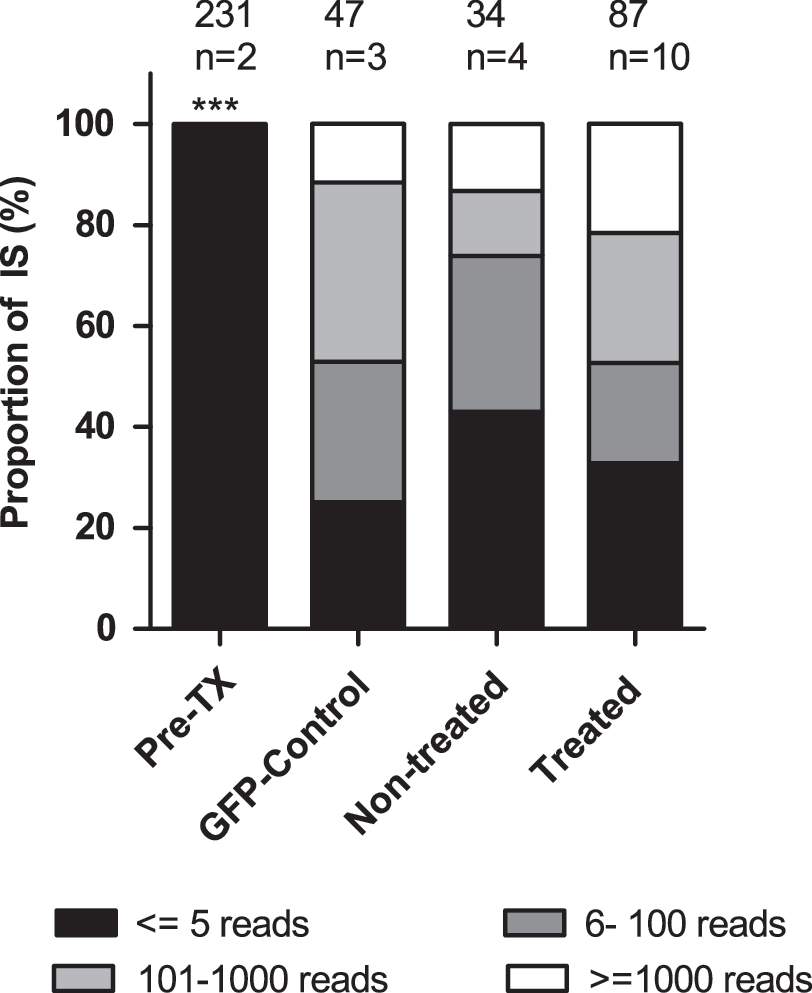
Clonal repertoire reduces in vivo after repopulation. Every integration site within a sample was ranked according to the pyrosequencing reads recovered. Percentages of insertions with ≤5 reads (black area), 6–100 reads (dark-gray area), 101–1,000 reads (light-gray area), and ≥1,000 reads (white area) are depicted for the pre-TX, GFP control, nontreated, and treated BM samples. The value above each column indicates the number of integration sites retrieved from the corresponding samples; n is the number of BM samples analyzed. Two-way ANOVA was used to compare pre-TX and BM samples (***p<0.01).
Verification of top clones reveals discrepancies in read counts and quantification by qPCR
Among the many insertions detected by the high-throughput sequencing for each animal (see Supplementary Table S2), the 10 most abundant integrations (top 10 clones) were designated based on their contribution to the total read counts of a specific sample. Similar to quantitative reverse transcription-PCR in validation of microarray experiments, ls-qPCR can be used to verify 454-read counts and to measure the contribution of a particular integration among all insertions in the PB and BM samples (Fig. 6A–E). As depicted, the proportion of a particular insertion varied at different time points, supporting the theory that different clones may contribute to the hematopoietic clonal repertoire at a given time in vivo. In our data, specific clones tended to increase over time, which in some cases correlated with the higher number of transduced human cells in PB samples. These data do not, however, allow for differentiation between single clones with several insertions or several clones with a single integration. Along this line, we could validate insertions close to the genes IFITM2, DNM2, JSRP1, FAM59A, and CCNB3, for the treated cohort and RAPGEF6, ARHGEF17, CAMSAP2, and TRAIP in the NT group (representative graphs in Fig. 6A–C). For one animal of the high-dose-treated cohort, the 454-results implied the presence of only one insertion close to TRGJP2 (Supplementary Table S2). As we observed >90% GFP expression in this animal, we cannot rule out that this represents a clonal amplification. However, we were unable to verify this insertion by ls-qPCR or to determine the VCN because of the very low amount of sample DNA available 7 days after chemotherapy when the animal died from myelosuppression. We here certainly cannot rule out clonal dominance, but given the discrepant results, the limited material available for analysis, the fact that the clone was not at all detected 3 weeks earlier (i.e., 1 week before chemotherapy), and also other explanations such as physiological clonal fluctuations in hematopoiesis have to be considered.
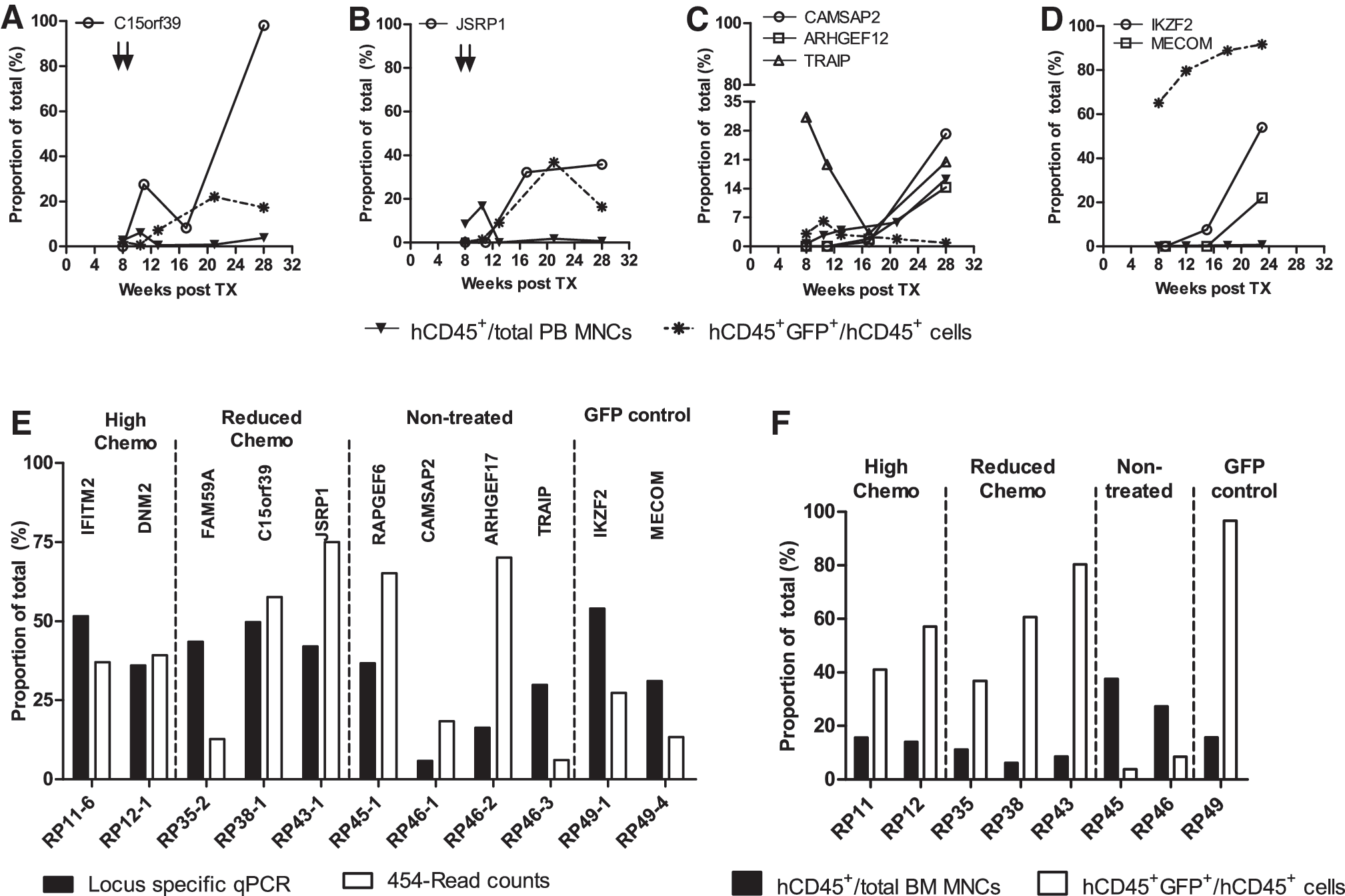
Validation of individual integration sites in the PB and BM. Locus-specific quantitative PCR (ls-qPCR) for the abundant integrations was performed on PB samples collected at different time points. Representative graphs indicate contribution of specific insertion to the total integrome (open symbols) along with the percentage of human cells (closed inverted triangle) and GFP+ human cells (asterisk) over time for individual insertion clones of
Apart from animal RP10, we did not detect overt clonal imbalance in our mice. However, insertions around +65.2 kb of the IKAROS family zinc finger 2 (IKZF2) gene and +95.2 kb of the MDS1 and EVI1 gene complex (MECOM) were found in a control animal (RP49; Fig. 6D). IKZF2 is known to encode for a hematopoietic-specific transcription factor essential for lymphocyte development and is mutated in leukemias and lymphomas (Huret et al., 2000). MECOM, on the other hand, is a transcriptional regulator and is associated with esophageal and ovarian cancers, hematological malignancies, and so on (Metais and Dunbar, 2008). Furthermore, in the X-CGD clinical trial, insertions in this locus lead to clonal expansion and myelodysplasia in both the patients (Stein et al., 2010). As seen in Fig. 6D, the animal harboring these integrations showed an increased proportion of the IKZF2 insertion in the PB sample at 23 weeks, and at this point it also represented as the major insertion in the BM sample (Fig. 6E). In addition, in four of five methylcellulose colonies derived from BM cells of this animal, both insertions IKZF2 and MECOM were detected by ls-qPCR, suggesting that a single clone harbored both the insertions. This example clearly demonstrates that with the help of integration-site analysis and deep-sequencing technology, clinically relevant insertions can be detected in the humanized xenograft model. Furthermore, when comparing the clonal contribution in BM samples either quantified by ls-qPCR or 454-sequence read counts, we frequently observed substantial discrepancies in the representation by the two technologies (Fig. 6E). This incongruity was irrespective of the chimerism detected in the BM sample, arguing against insensitive detection as the basis of the observation (Fig. 6F). Furthermore, in few samples we observed discrepancies in the contribution of individual clones in BM and PB (see Supplementary Fig. S3B).
Discussion
We here report the use of a SIN lentiviral vector to express MGMT in human hematopoietic cells and successfully enriched these cells in vivo by the combined application of BG and BCNU. Recapitulating earlier studies by other groups in murine as well as xenotransplant models (Reese et al., 1999; Ragg et al., 2000; Sawai et al., 2001; Jansen et al., 2002; Pollok et al., 2003; Zielske et al., 2003; Cai et al., 2006, 2010; Milsom et al., 2008; Giordano et al., 2011), our study documents robust and stable chemoselection of transgenic human cells over extended periods in a model employing the xenotransplantation of MGMT-transduced CB-derived CD34+ cells. Insertion-site analysis by LAM-PCR coupled with high-throughput sequencing revealed a typical lentiviral integration pattern. An overall oligoclonal repertoire was detected in all experimental groups at the end of the 24–28-week observation period without enrichment of clones with integrations in or near-specific gene classes.
In the past, the majority of studies on MGMT-mediated in vivo selection, including the recent clinical trial, primarily used gammaretroviral vectors (Pollok et al., 2003; Zielske et al., 2003; Cai et al., 2006, 2008, 2010; Adair et al., 2012). As these vectors have a preference to integrate near the TSS of genes and carry a substantial risk of gene activation, we here evaluated a SIN lentiviral vector in order to reduce this risk. To direct MGMT expression, we specifically chose the EFS promoter, which in the context of the SIN gammaretroviral vector has been shown to allow for stable and functional MGMT expression while avoiding the toxicity associated with very high expression levels (Milsom et al., 2008). To assess the efficacy and safety of our lentiviral MGMT vector in a relevant in vivo model, we exploited the xenotransplantation of human CB-derived CD34+ cells into NSG mice (McDermott et al., 2010; Parekh and Crooks, 2012). Initial experiments with two cycles of 20 mg/kg BG/10 mg/kg BCNU resulted in effective selection of gene-marked human cells but also fatal myelotoxicity in the non-protected murine hematopoietic system. Infusion of 1×106 fresh murine BM cells 24 hr after high-dose BCNU application prevented this toxicity, but also resulted in a loss of the vast majority of engrafted human cells. Similar observations have been described by other investigators and in part may be related to the excess of murine hematopoietic cells competing with their human counterparts for the murine BM niche (Cai et al., 2006). In contrast, reduced chemotherapy regimens employing 5–7.5 mg/kg BCNU allowed for effective selection of MGMT-transduced human cells without fatal myelotoxicity and resulted in significant enrichment of transgenic myeloid, B, and primitive CD34+ cells in the BM as well as selection of T cells in the spleen and thymus. These results are in accordance with previous findings stressing the importance of repetitive chemotherapy application to recruit dormant HSCs into an active cell cycle for effective MGMT-mediated chemoselection in murine transplant models (Jansen et al., 2002; Lin et al., 2011). In comparison to former xenotransplant studies (Cai et al., 2006), however, we now have considerably prolonged the observation period in the NSG recipients, enabling us to gauge the effectiveness of our selection strategy on the level of bona fide SRCs (McDermott et al., 2010). In this model, EFS-driven MGMT expression from our SIN lentiviral vector allowed for sustained in vivo selection over the entire observation period of up to 18 weeks after chemotherapy.
Insertion-site analysis by LAM-PCR demonstrated the well-known lentiviral integration pattern (Mitchell et al., 2004), characterized by a majority of hits (55–75%) in intronic sequences of genes and less hits in the vicinity of the TSS or CpG islands. This profile was detected in all experimental groups, ruling out a major influence of the experimental conditions associated with the in vivo selection process. However, when pre-TX and BM samples were compared, a substantial decrease in clonal diversity became obvious in all experimental groups. This is not surprising as only a minority of the originally transduced CD34+ cells have long-term engraftment capacity. Given that we transplanted progeny of 1–2×105 hCD34+ cells and based on a frequency of 1–5 SRCs per 104 CB CD34+ cells (Wang et al., 1997; McDermott et al., 2010) with a transduction efficiency of approximately 35–65%, we expect roughly 8–60 MGMT-positive SRCs in individual NSG mice. This correlates well with the results obtained from the BM samples of our BG/BCNU-treated animals (median 8 insertions, range 1–22).
In almost all animals, high read integrations were detected by end-point and quantitative PCR. When we analyzed the contribution of “top clones” to PB samples, we observed high variations with a strong trend for increased contributions to final samples (Fig. 6A–D). This observation clearly reflects substantial fluctuation in the contribution of individual insertion clones to the PB over time. The high-level contribution observed at the end of the experiment does not necessarily result from progressive clonal dominance but merely may reflect the fact that these clones initially were selected as “top clones” in the BM at even this time point. Furthermore, despite the successful validation of various insertions over time, we encountered substantial problems to quantify the contribution of a specific clone in a sample based on the pyrosequencing read counts. This was especially true for the PB samples and could not be overcome by reanalysis of the same sample. This technical limitation reflects the current concerns associated with clonality assessment based on sequential PCR-based techniques (Giordano et al., 2011; Brugman et al., 2013; Rittelmeyer et al., 2013).
We did not detect significant differences with regard to gene classes hit by insertions in the different cohorts, utilizing the Panther database as a reference (Mi et al., 2012). Importantly, this was also true for genes listed in cancer gene databases [D'Antonio et al. (2012) and
Thus, in summary, our study convincingly demonstrates the potential of SIN lentiviral vectors in MGMT-based chemoprotection and in vivo selection strategies while at the same time avoiding HSC toxicity. Our xenotransplantation model combined with integration analysis allowed to detect and to monitor long-term the clonal fate of clinically relevant insertions. Nevertheless, the limited size of our experimental cohorts clearly calls for additional studies utilizing more animals and potentially for further evaluation of the construct specifically in the context of TMZ application, given the relevance of this agent in the treatment of glioblastomas.
Footnotes
Acknowledgments
We thank Axel Schambach for providing all vector backbones and Nico Lachmann for his help in generating MGMT vector constructs. We thank Doreen Lüttge for her excellent technical assistance and Rena Struβ for her help during mouse transplants. We appreciate Jannik Daudert for his help in developing scripts to evaluate integration analysis data. We are thankful to Sandra Zilz and the team of the medical school pharmacy for preparing cytotoxic drugs, and to Jörg Frühauf for his assistance during irradiation of mice (all from Hannover Medical School). This work was supported by grants from the Deutsche Forschungsgemeinschaft: Cluster of Excellence REBIRTH (Exc 62/1; T.M.) and MO 886/4–1 (T.M.), SPP1230 grant MO 886/3–1 (U.M. and T.M.) and European Union (Cell-PID, MR and UM). R.P. was partially supported by a stipend from Hannover Biomedical Research School.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.