
Abstract
Nonintegrating gene delivery vectors have an improved safety profile compared with integrating vectors, but transgene retention is problematic as nonreplicating episomes are progressively and rapidly diluted out through cell division. We have developed an integration-deficient lentiviral vector (IDLV) system generating mitotically stable episomes capable of long-term transgene expression. We found that a transient cell cycle arrest at the time of transduction with IDLVs resulted in 13–45% of Chinese hamster ovary (CHO) cells expressing the transgene for over 100 cell generations in the absence of selection. The use of a scaffold/matrix attachment region did not result in improved episomal retention in this system, and episomes did not form after transduction with adeno-associated viral or minicircle vectors under the same conditions. Investigations into the episomal status of the vector genome using (1) linear amplification-mediated polymerase chain reaction followed by deep sequencing of vector–genome junctions, (2) Southern blotting, and (3) fluorescent in situ hybridization strongly suggest that the vector is not integrated in the vast majority of cells. In conclusion, we have developed an IDLV procedure generating mitotically stable episomes capable of long-term transgene expression. The application of this approach to stem cell populations could significantly improve the safety profile of a range of stem and progenitor cell gene therapies.
Introduction
G
Integration-deficient lentiviral vectors (IDLVs) exploit the natural tendency of lentiviruses to form episomal circles as an intermediate or by-product during infection (Bukrinsky et al., 1991). The occurrence of these circular forms can be greatly increased by impairing the ability of the virus to integrate. The most efficient way of achieving this is through the use of genetic mutants with changes in the catalytic site of integrase (IN), the enzyme that catalyzes the integration of the viral cassette into the host genome. These class I IN mutants, of which D64V is the most common variant, are only defective in catalyzing integration and retain other IN functions such as reverse transcription and nuclear import of the genome, and thus do not result in reduced levels of viral DNA. In transduced cells, IDLV genomes form circular, nonreplicating nuclear episomes (Engelman et al., 1995; Leavitt et al., 1996).
The ability of single-stranded adeno-associated viral (AAV) vectors to form episomal circular structures is well documented (Straus et al., 1976; Yang et al., 1999), and the most common in vivo forms of self-complementary AAV genomes have been found to consist of 1–2 genomes circularized through homologous recombination between the ITRs (Choi et al., 2005). Another class of vectors demonstrably capable of long-term episomal transgene expression are minicircle vectors, plasmid-derived units that have been through an intramolecular recombination process resulting in a vector entirely lacking in prokaryotic backbone sequences (Chen et al., 2003; Mayrhofer et al., 2009).
Although safer than integrating systems, vectors that exist as nuclear episomes are usually lost during repeated cell division, thus limiting gene therapy application mainly to transduction of stable postmitotic tissues (Philippe et al., 2006; Yáñez-Muñoz et al., 2006; Wanisch and Yáñez-Muñoz, 2009; Peluffo et al., 2013). Mitotically stable replicating episomes would overcome this limitation as well as safety concerns posed by integrating vectors. Scaffold/matrix attachment regions (S/MARs) are structural elements of chromosomal DNA involved in anchoring the chromatin to the nuclear matrix and have been described as capable of conferring mitotic stability to episomes (Piechaczek et al., 1999; Giannakopoulos et al., 2009; Broll et al., 2010; Wong et al., 2011). Furthermore, prokaryotic sequences seem to promote the clearance or silencing of introduced episomes, whereas selection pressure and prolonged exposure to the nucleoplasm appear to favor the establishment of replicating episomes (Piechaczek et al., 1999; Broll et al., 2010). We thus hypothesized that prolonged exposure to the nucleoplasmic environment, with or without the presence of S/MAR elements, might facilitate retention and replication of viral nuclear episomes.
We developed IDLV and AAV vectors containing or lacking a ∼700 bp truncated miniMAR variant of the human β-interferon S/MAR, which originally arose during long-term culture of cells transfected with S/MAR-containing minicircle vectors (Broll et al., 2010). The protocol used here included a prolonged initial exposure of the vector to the nuclear environment by inducing the transduced cells to undergo a transient cell cycle arrest using methionine- and serum-depleted (MSD) culture medium. We additionally investigated the mitotic retention of non-S/MAR-containing minicircles in cells subjected to a transient cell cycle arrest.
The main aim was to increase the frequency of episomal establishment, thus avoiding the need for antibiotic or surface marker selection to enrich the transgene-expressing population (Piechaczek et al., 1999). This would remove one of the main hurdles for clinical development. Using our system, stable transgene expression from IDLV episomes without S/MAR element was retained in polyclonal and clonal populations for over 100 generations.
Materials and Methods
Plasmids
HIV-1 vector plasmids pMDLg/pRREintD64, pMDLg/pRRE, pRSV.REV, pMD2VSV.G, and the pRRL-type self-inactivating transfer plasmids have been previously described (Naldini et al., 1996b; Dull et al., 1998; Yáñez-Muñoz et al., 2006). The truncated 733 bp S/MAR element was present in plasmid pEPI-SV40MC-M18 (Broll et al., 2010). The S/MAR-containing lentiviral transfer plasmid pRRLsc-SV40-eGFP-mMAR was made by digesting pRRLsin-PPT-ISceIT with EcoRV and ligating to a 1861 bp, end-filled XhoI/BamHI fragment (SV40-eGFP-mMAR) from pEPI-SV40MC-M18. The control lentiviral plasmid pRRLsc-SV40-eGFP without the S/MAR element was made by ligating EcoRV-cut pRRLsin_PPT_ISceIT to a 1130 bp, end-filled XhoI/AseI fragment (SV40-eGFP) from pEPI-SV40MC-M18. The pscAAV CMV GFP plasmid was kindly donated by Amit Nathwani (Department of Clinical Haematology, UCL Cancer Institute, London). To clone the pscAAV CMV GFP miniMAR, the pscAAV CMV GFP plasmid was linearized with SbfI and the pEPI-SV40MC-M18 plasmid was linearized with KpnI. The digested ends were blunted using T4 DNA polymerase. Both plasmids were then digested with BsrGI, and the 769 bp miniMAR fragment was ligated into the pscAAV backbone. The minicircle vector and the control plasmid pCMV GFP were purchased from PlasmidFactoryGmbH, McBox product PFBox102.
Cells and culture media
CHO-K1 cells were maintained in proliferating culture medium: Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/l glucose (PAA/GE Healthcare), with added L-proline at 0.02 g/l (Sigma Aldrich), 10% fetal bovine serum (FBS; PAA/GE Healthcare), 100 units/ml penicillin, and 100 μg/ml streptomycin. Cell cycle arrest was induced with methionine-free DMEM (Gibco/Life Technologies) supplemented with 2% FBS, 0.02 g/l L-proline, 100 units/ml penicillin, and 100 μg/ml streptomycin. It has been estimated that L-methionine concentration in bovine serum is around 15 μM, so the final concentration of L-methionine in our nominally L-methionine-free experiments would be around 0.3 μM (Martinez-Chantar et al., 2003).
Cell cycle analysis
CHO-K1 cells were prepared for cell cycle analysis by staining nuclei with propidium iodide, as follows: approximately 1×106 cells were trypsinized and spun down at 200×g for 4 min. The supernatant was discarded and the cells were resuspended in the remaining drops of medium by gently flicking the tube. About 1 ml of Solution I (10 mM NaCl, 1 mg/ml tri-sodium citrate, 0.06% NP40, and freshly added 25 μg/ml propidium iodide and 10 μg/ml RNase) was added into each tube while shaking, and cells were incubated at room temperature in the dark for 30 min. About 1 ml of Solution II (15 mg/ml citric acid, 85.6 mg/ml sucrose, and freshly added 40 μg/ml propidium iodide) was then added into each tube while shaking and incubated at 4°C for 1–2 hr. Data from the stained nuclei were acquired with a FACSCantoII flow cytometer using FACSDiva software, and analyzed using FlowJo to determine the cell cycle profile.
Lentivector manufacture
Lentivectors pseudotyped with the vesicular stomatitis virus glycoprotein G envelope were produced by transient transfection of 293T cells and titrated as previously described (Yáñez-Muñoz et al., 2006). For eGFP titration, HeLa cells were transduced with serial dilutions of vector stock in the presence of 8 μg/ml polybrene. eGFP-positive cells were scored using flow cytometry 72 hr posttransduction and titers calculated as transducing units/ml. The titers of IDLV-SV40-eGFP and IDLV-SV40-eGFP-miniMAR were 8.6×107 and 3×107 TU/ml, respectively.
Lentivector transduction
IDLV transduction (multiplicity of infection [MOI] 0.5) of CHO cells was carried out 24 hr after seeding the cells, with polybrene at a final concentration of 8 μg/ml. About 24 hr posttransduction, the proliferation medium of the designated cell populations was changed to methionine-free and serum-depleted medium to induce a transient cell cycle arrest. This medium was replaced daily for 5 days, and on day 7 posttransduction the cells were returned to proliferation medium. For the control cells not undergoing cell cycle arrest, transduction was done as above, and the cells were split upon reaching confluency at 4 days posttransduction. From day 7, all cells were allowed to proliferate normally with routine passaging for a period up to 100 days. The positive and negative control cell populations for fluorescent in situ hybridization (FISH) and linear amplification-mediated polymerase chain reaction (LAM-PCR) were generated by transducing CHO-K1 cells with IPLV-SG or IDLV-SG at MOI 2 about 24 hr after seeding. The cells were harvested 24 hr after transduction and either fixed for FISH or used for DNA extraction using Qiagen Blood & Tissue kit.
AAV manufacture
AAV vectors were produced by transient transfection of 293T cells and titrated using dot blot as previously described (Koo et al., 2011). Recombinant pseudotyped ssAAV2/8 vector stocks were kindly donated by Helen Foster & Keith Foster. The titers of ssAAV CAG GFP and ssAAV8 CAG GFP miniMAR were 3.31×1012 and 4.89×1012 vg/ml, respectively. The titers of scAAV9 CMV GFP and scAAV9 CMV GFP miniMAR were 3.92×1011 and 3.83×1011 vg/ml, respectively.
AAV transduction
AAV in vitro transduction of CHO cells was carried out 24 hr after seeding at an MOI of 105 using serum-free media. An equal volume of standard DMEM with 20% FBS was added 5 hr posttransduction. Cell cycle arrest and analysis of transduced cells were done as described above for IDLV vectors.
Minicircle transfection
CHO cells were transfected with minicircle and plasmid using 1 μg DNA/105 cells and Lipofectamine 2000 (Invitrogen/Life Technologies) with a Lipofectamine-to-DNA ratio 1:2. The transfection media were removed and replaced with normal media after 6 hr. The cell cycle arrest was induced 24 hr after transfection, and analysis and culture of transfected cells was done as described above for IDLV vectors.
Dilution cloning
IDLV-transduced polyclonal CHO cell populations were allowed to proliferate for 100 days, and then subjected to dilution cloning by seeding an average of 0.7 cells per well in 96-well plates. After 14 days, the eGFP+ clones from each vector were evaluated by flow cytometry and three expanded to establish cell lines. The IPLV-transduced polyclonal population was allowed to proliferate for 40 days, and then subjected to dilution cloning as above. After 14 days, the clones were evaluated and three expanded for further investigations.
Isolation of high-molecular-weight DNA from mammalian cells
About 5×107–1×108 cells were washed twice with phosphate buffered saline (PBS), detached using trypsin, neutralized with medium, centrifuged at 1000 rpm for 5 min, and washed twice with 5 ml Tris-buffered saline. The cells were resuspended in TE (pH 8.0) at a concentration of 5×107 cells/ml, 10 ml of extraction buffer (10 mM TrisCl [pH 8.0], 0.1 M EDTA [pH 8.0], 0.5% SDS, 20 μg/ml RNAse] was added for each ml of cell suspension, and the mixture was incubated for 1 hr at 37°C. Proteinase K was then added to a final concentration of 100 μg/ml, and the suspension incubated for 3 hr at 50°C. The solution was cooled and poured into a centrifuge tube, an equal volume of phenol equilibrated with 0.5 M TrisCl (pH 8.0) was added and the two phases gently mixed by turning the tube end over end for 10 min. The two phases were separated by centrifugation at 5000×g for 15 min. The viscous aqueous phase was transferred to a clean centrifuge tube and the extraction with phenol was repeated twice.
After the third extraction with phenol, the aqueous phases were transferred to a fresh centrifuge tube and 0.2 volumes of 10 M ammonium acetate and 2 volumes of 100% ethanol were added at room temperature, and the tube was swirled until the solution was thoroughly mixed and the DNA precipitated. Since we were interested in retaining the total DNA including also the smaller DNA molecules such as episomes, the precipitated DNA was collected by centrifugation at 5000×g for 5 min at room temperature in a swinging-bucket rotor. The precipitate was washed twice with 70% ethanol, and the DNA collected by centrifugation as described above, dried, and resuspended in 1 ml TE buffer.
Quantitative real-time PCR
The quantification of episomes in transduced CHO cells was carried out by real-time PCR using the Roche LightCycler 480 system. About 100 ng of high-molecular-weight genomic DNA in 2 μl was used as a template in each reaction. About 10 μl of reaction mix contained 300 nM of each primer and 5 μl Absolute Blue qPCR SYBR Green 2× Buffer (Thermo Scientific). The primers F (5′ AACTAGGGAACCCACTGCTTAAG) and R (5′ GAGTCCTGCGTCGAGAGAGC) were designed to target the late reverse transcript region of the lentivector backbone (Butler et al., 2001). In order to normalize the copy number to the cell number in each sample, β-actin qPCR was performed as above using primers F (5′ TGGCATCCACGAAACTACAT) and R (5′ TGGTACCACCAGACAGCACT). The reaction conditions were standard qRT-PCR cycling: 50°C for 2 min, 95°C for 10 min, 50× (95°C for 15 sec, 60°C for 1 min).
Linear amplification-mediated PCR
LAM-PCR was performed on high-molecular-weight DNA extracted from transduced cell populations using a protocol described previously (Schmidt et al., 2007).
Southern blotting
High-molecular-weight DNA extracted from transduced clonal populations was digested with EcoRI (IDLV-SG) or XhoI (IDLV-SGm). Southern blotting was performed as previously described (Southern, 1975). For the probe, PCR was performed to amplify a 717 bp fragment of the eGFP gene, using plasmid pRRLsc-SV40-eGFP as template, forward primer 5′ATG GTG AGC AAG GGC GAG, reverse primer 5′CTT GTA CAG CTC GTC CAT, annealing temperature of 56°C, and a total of 35 cycles. The probe was labeled with [α-32P]-dATP using random hexanucleotide priming. Hybridization and washes were done following standard procedures.
Fluorescent in situ hybridization
Cell fixation
Approximately 1–5×106 CHO cells were detached using trypsin/EDTA, and incubated in 75 mM KCl solution at room temperature for 5 min. The cells were then washed with 5% acetic acid and resuspended in 5 ml of freshly made fixative (3:1 methanol:glacial acetic acid; Sigma). Cells were washed with fixative twice more to remove any traces of medium and stored at −20°C.
Probe labeling and preparation
FISH probes were prepared by labeling the pRRLsc-SV40-eGFP transfer plasmid with dUTPs complexed either with SpectrumGreen or SpectrumOrange fluorochromes (Abbott Molecular) using the Nick Translation System (Invitrogen/Life Technologies) according to the manufacturer's instructions. Probe mixes were prepared for FISH by adding Cot-1 DNA (50-fold weight excess) to each labeled probe and precipitated by addition of two volumes of 100% ethanol. The probes were pelleted and dried using Speedy Vac apparatus and Savant Centrifuge/concentrator. The dry pellets were resuspended in Abbott LSI/WCP Hybridization buffer at a concentration of 6.67 ng/μl and stored in the dark at −20°C.
FISH run procedure
Fixed cells were dropped onto slides prewashed in fixative and allowed to air-dry, and the area containing cells was marked on the underside using a diamond pen. Slides were soaked in PBS pH 7.4 for 5 min and then rinsed twice with sterile water. They were then dehydrated by incubating for 2 min each in 70%, 90%, and 100% ethanol. The slides were air-dried, and 15 μl (50 ng) probe was applied onto a cover slip, which was then placed onto the slide and sealed with rubber solution (Weldtite Ltd.). The slides were incubated at 72°C for 6 min and then at 37°C overnight. The next day, the coverslips were removed by washing in 4× saline-sodium citrate (SSC) buffer and 0.05% Tween pH 7.0. A stringent wash was performed for 5 min at 72°C in 0.4×SSC pH 7.0. After this, the slides were incubated for 2 min at room temperature in 4× SSC/0.05% Tween pH 7.0, and then rinsed 3 times with PBS for 2 min each time. After the final PBS wash, the slides were air-dried and 15 μl of the nuclear stain DAPI (160 ng/ml; Cytocell Ltd.) was applied together with a new cover slip, which was then sealed with nail varnish.
Imaging and analysis of FISH signals
Although the cell cultures from which the FISH slides were derived were not blocked in metaphase, at least 50 spontaneous metaphases were present on all the slides. For the qualitative analysis of all cultures, images were taken of ∼20 metaphases per sample slide using 1000× magnification on a Zeiss Axioplan 2 microscope and Metasystems Isis 2 software. Four slides were designated for detailed analysis: mock transduced control, and clonal cell lines transduced with IPLV-SG, IDLV-SG, or IDLV-SGm. For each slide, 80 fields at 400× magnification were processed by counting the number of nuclei in the field, the number of signals inside nuclei, and whether signal was a singlet or doublet (the latter present on both chromatids). The average number of signals per positive nucleus was estimated as total number of “within nucleus” signals divided by total number of positive nuclei.
Stress-induced DNA duplex destabilization analysis
The computational analysis of vector sequences was performed using the WebSIDD service available online. The near-neighbor and the circular molecule settings were chosen for the analyses.
Statistical analyses
All statistical analyses were carried out using GraphPad Prism version 4.00 for Windows (GraphPad Software). For the statistical analyses of the cell cycle data, Student's t-test was used to compare the proportions of cells separately for each cell cycle phase between treatments; that is, the proportion of cells in G1 phase was compared between arrested and control. For the statistical analyses of data from the transduction experiments, the percentages of cells expressing eGFP were taken from the last measurement and one-way ANOVA was used to compare all groups. Tukey's post hoc test was used to establish whether significant differences existed between specific sets of data. For the FISH data, statistical analyses were performed on the average number of nuclear signals using one-way ANOVA.
Results
The effect of transient cell cycle arrest and S/MARs on the mitotic retention of nuclear IDLV episomes
To evaluate the impact of transient cell cycle arrest and the presence of an S/MAR element on the stability of IDLV transduction and potential establishment of stable replicating IDLV episomes, in vitro transduction experiments were conducted using two different vesicular stomatitis virus glycoprotein G-pseudotyped IDLVs. Both vectors contained the eGFP gene driven by the SV40 promoter, the difference between them being that one (IDLV-SG) lacked and the other (IDLV-SGm) contained the miniMAR element. CHO cell cultures were transduced with the IDLVs 1 day after seeding, and at day 2 (24 hr later) induced to undergo transient cell cycle arrest until day 7 in MSD medium. To ascertain the efficacy of the induced cell cycle arrest, cell cycle phase analyses were performed by propidium iodide staining and flow cytometry of CHO cells at day 2 and at day 7. For both transduced cells and mock control, MSD medium effectively increased the proportion of cells in G1 phase (from about 45% to 70%), and decreased the proportion of cells in S phase and particularly in G2 phase (Fig. 1). This confirmed the effectiveness of methionine and serum depletion for inducing cell cycle arrest in control and IDLV-transduced cells. After the cell cycle arrest period, the cultures were returned to full growth medium and allowed to proliferate. Such cultures soon required normal passaging, demonstrating the transient nature of the cell cycle arrest.
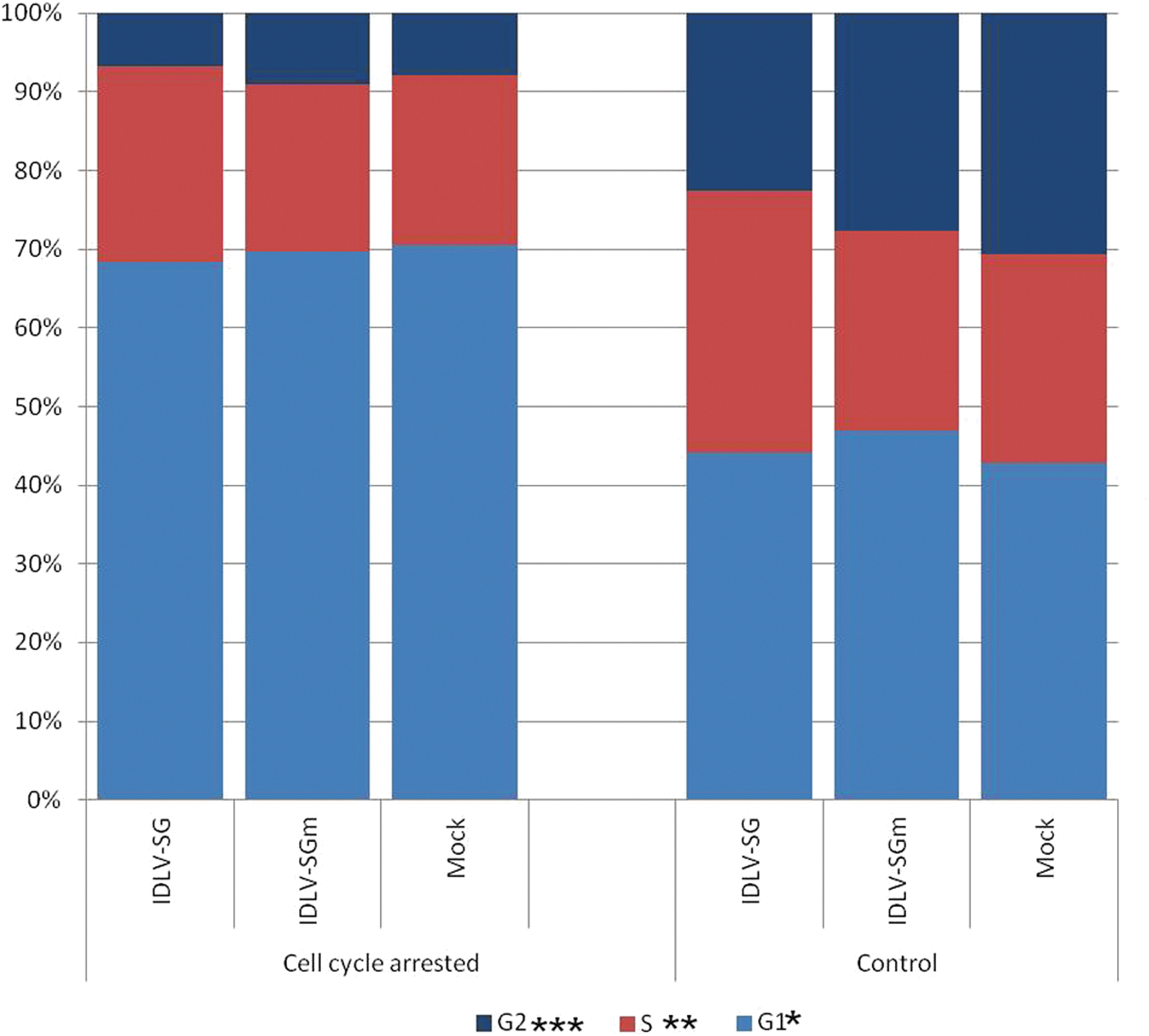
Cell cycle phase analysis of IDLV-transduced CHO cells with and without induction of cell cycle arrest by methionine and serum depletion. CHO cells were plated, transduced with IDLV vectors [IDLV-SG (IDLV-SV40-GFP); IDLV-SGm (IDLV-SV40-GFP-mMAR)] or mock control after 1 day, and then treated with MSD medium from day 2 to day 7. At day 7, cultures were returned to full growth medium and allowed to proliferate as normal with routine passaging as required. To confirm induction of cell cycle arrest, cell cycle phase analyses were performed by propidium iodide staining and flow cytometry at day 7. The nonarrested controls were analyzed at the same time point. The colored sections indicate the average percentages of cells in each cell cycle phase. The proportion of cells in different cell cycle phases that were significantly different between treatments was determined by Student's t-test (*p<0.05, **p<0.01, ***p<0.001). CHO, Chinese hamster ovary; IDLV, integration-deficient lentiviral vector; MSD medium, methionine- and serum-depleted medium.
All transductions yielded over 90% eGFP+ cells at day 5, but the eGFP expression pattern was distinctly different in cells transduced with IDLV-SG and IDLV-SGm and subjected to a transient cell cycle arrest using MSD medium, compared with those in continuous proliferative culture. Expression of eGFP after return to full growth medium declined from day 7 (>98% eGFP+) but stabilized at approximately day 18 at remarkably high levels: 22% and 17% eGFP+ cells, respectively, for cultures transduced with IDLV-SG and IDLV-SGm (Fig. 2a). After 10 weeks these substantial levels of eGFP+ cells were maintained at 25% and 15%, respectively (Fig. 2c). In the case of the IDLV-SG- and IDLV-SGm-transduced cells not treated with MSD medium and allowed to proliferate continuously, eGFP levels declined rapidly from day 3 (>98% eGFP+) to day 10 (<1% eGFP+), and by week 10 posttransduction they had dropped to 0.8% and 0.5% eGFP+ cells, respectively, with no statistically significant difference between the vectors containing and lacking the S/MAR element (one-way ANOVA, n=3) (Fig. 2b and c). These low levels in continuously grown transduced cells match what would be expected from residual integration of IDLVs (Wanisch and Yáñez-Muñoz, 2009). A control population was transduced with integrating LV-SG vector, resulting in 94% eGFP+ cells at day 4. The cells were allowed to continuously proliferate for 40 days, at which point 90.8% of the cells were still expressing eGFP (data not shown).
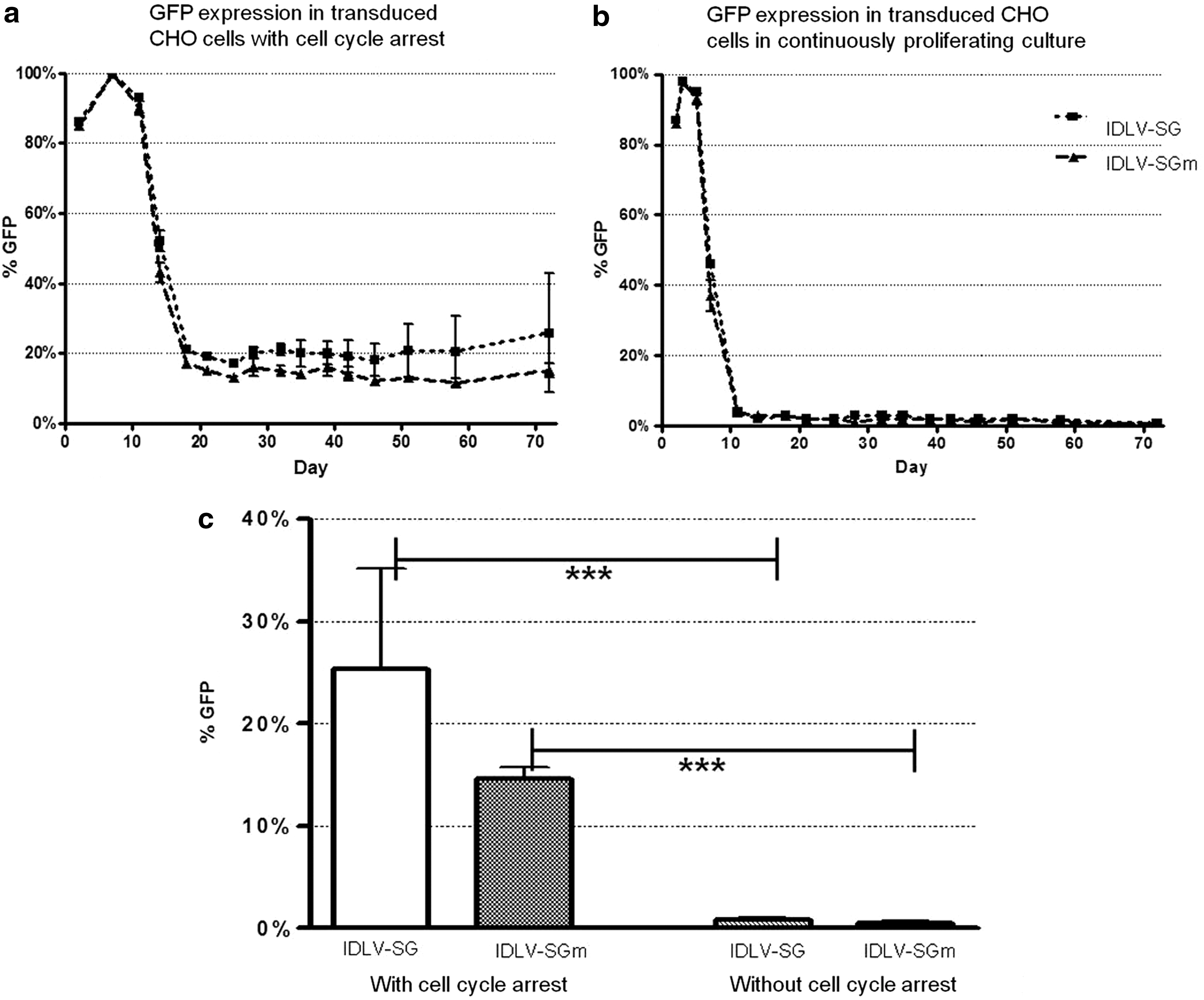
Transient induction of cell cycle arrest leads to substantial levels of stable eGFP expression in IDLV-transduced CHO cells. CHO cells were seeded and transduced at MOI 0.5 with vectors IDLV-SG or IDLV-SGm.
Much higher levels of eGFP+ cells were observed in IDLV-transduced cultures initially subjected to the MSD medium treatment compared with untreated cultures, and this is a highly statistically significant effect for cells transduced with both vectors (p<0.001; one-way ANOVA with Tukey's post hoc test, n=3). Previously reported levels of stably expressing cells achieved with S/MAR-element-containing episomal vectors are up to 10-fold lower at 3–4.5% in the absence of selection pressure (Broll et al., 2010). Here, with MSD medium treatment the IDLV-SG vector actually yielded significantly higher levels of stable transduction compared with IDLV-SGm (22% and 17%, respectively, p<0.01, by one-way ANOVA followed by Tukey's post-test).
Lack of effect of transient cell cycle arrest and S/MARs on the mitotic retention of AAV, plasmid, and minicircle vector genomes
Next, we set forth to evaluate the impact of the episome background sequence and method of introduction by combining the miniMAR element and transient cell cycle arrest with a range of different vectors. We constructed four different AAV vectors: a single-stranded eGFP vector with and without miniMAR (ssAAV-CG and ssAAV-CGm), and two corresponding self-complementary vectors (scAAV-CG and scAAV-CGm). The ssAAV vectors comprised the eGFP gene driven by the CMV promoter, and the scAAV vectors in addition contained the chicken β-actin enhancer. In vitro transduction of CHO cells was performed 24 hr after seeding using an MOI of 105, and a transient cell cycle arrest using MSD media was induced 24 hr posttransduction and maintained until day 7.
The in vitro transduction capacity of AAV vectors is generally lower than that of IDLV vectors (Ellis et al., 2013), and here the single-stranded vectors yielded a reasonable 15% and 16% eGFP+ cells 5 days after transduction for ssAAV-CG and ssAAV-CGm, respectively (Fig. 3a). The scAAV vector without miniMAR achieved the highest initial transduction level at 72% eGFP+ cells. Interestingly, the corresponding miniMAR-containing scAAV vector only achieved 5% eGFP+ cells by 5 days posttransduction (Fig. 3b).

The establishment of stable episomes is specific to IDLV-transduced cells. CHO cells were plated into culture, and after 1 day transduced at an MOI of 105 with single-stranded AAV vectors
The transduced cells were successfully induced to undergo a transient cell cycle arrest for 5 days, and allowed to proliferate freely afterward. However, the proportion of eGFP+ cells in all transduced populations continued to decline until, by 42 days after transduction, it had reached a level of 0.2–1.1% above background (Fig. 3d). None of the AAV vectors showed a significant increase in retention of transgene expression.
As the inclusion of the miniMAR element did not increase the proportion of stable episomes for either the IDLV or the AAV vectors, we hypothesized that the cell cycle arrest alone may be enough to enable the establishment of the nuclear episomes provided that they are free of prokaryotic sequences. We therefore combined the transient cell cycle arrest protocol with a minicircle and control plasmid vector transfection. CHO cells were transfected at 2 μg per 105 cells 24 hr after seeding, and subjected to an MSD medium-induced cell cycle arrest as above. The proportion of eGFP+ cells reached 86% for the minicircle and 81% for the control plasmid, but dropped quickly after the cells were allowed to proliferate and reached levels of 0–0.7% above background by 30 days posttransduction (Fig. 3c and d). Therefore, under these conditions neither the minicircle nor the plasmid vector supported significant frequencies of episome establishment.
Clonal stability of IDLV episomes
To evaluate the stability of eGFP expression in CHO cells after IDLV transduction, a number of clonal cell populations were derived by dilution cloning. The IDLV-transduced parent populations subjected to an initial period of cell cycle arrest were allowed to proliferate freely with routine passaging for 100 days without any form of selection (Fig. 4a and c). The populations were then subjected to the dilution cloning procedure, and clones arising after a further 14-day culture period were scored as either eGFP+ or eGFP− by fluorescence microscopy. Three eGFP+ clones from each parent population were maintained in culture for up to a further 60 days with routine passaging. The clonal populations were examined every 10 days by flow cytometry to ascertain the stability of the mean fluorescence intensity (MFI) and the proportion of eGFP+ cells. For clones transduced with IDLV-SG, all clones exhibited consistent eGFP+ cell numbers and MFI levels over 50 days in culture (Fig. 4b). For the IDLV-SGm vector, all three clones also exhibited consistent eGFP+ cell numbers over 50 days in culture, and continued to retain a Gaussian although somewhat variable MFI distribution (Fig. 4d).

Clonal stability of eGFP expression during long-term proliferation of CHO cell lines derived by IDLV transduction with transient cell cycle arrest. CHO cell lines derived by dilution cloning from parent populations transduced with IDLV-SG and IDLV-SGm and subjected to transient cell cycle arrest were maintained in continuous proliferating culture for up to 60 days with routine passaging approximately 3 times weekly at a 1:5 split.
The episomal copy number was analyzed in each of the clonal populations by performing quantitative real-time PCR with primers targeting the vector backbone, and the CHO β-actin gene to estimate the number of cell equivalents per sample. The clonal populations transduced with IDLV-SG averaged 4.1±0.2 (clone 4), 0.9±0.05 (clone 7), and 1.6±0.3 (clone 10) episome copies per cell. For cells transduced with IDLV-SGm, the average backbone copy numbers were 7.0±4.0 (clone 8) and 8.8±4.6 (clone 11) per cell. No signal was obtained for IDLV-SGm clone 5. Three controls were included in the experiment: DNA extracted from CHO cells transduced with IDLV-SG and integrating LV-SG 24 hr after transduction, and a clone derived from cells transduced with integrating LV-SG, all with MOI of 2. All three yielded high amounts of vector signal, with vector genome copy numbers estimated at 35, 37, and 45 for IDLV, ICLV, and ICLV clone, respectively.
Integration status analysis of IDLV episomes
To overcome limitations associated with any one technique, we employed an array of experiments to analyze the possible integration of the IDLV episomes in the clonal, eGFP+ cell populations, and assessed the combined results. LAM-PCR was used to amplify regions adjacent to the vector long terminal repeats (LTRs), followed by next-generation sequencing and bioinformatic analysis to distinguish nonintegrated vector genomes from those associated with host DNA. Southern blotting was used to assess vector genome restriction enzyme digestion patterns for evidence of integration. Finally, vectors in transduced nuclei were visualized using FISH, seeking the presence of symmetrical signals in sister chromatids of metaphase chromosomes as evidence of integration.
LAM-PCR and deep sequencing of vector–genome junctions
LAM-PCR (Schmidt et al., 2007) was used to analyze the six selected eGFP+ clonal populations of CHO cells derived from transduction with IDLV-SG and IDLV-SGm and subjected to transient cell cycle arrest. In the analysis, different products result from the various vector genome configurations: (1) amplification from the 5′-LTR into the vector backbone yields a 283 bp fragment (210 bp vector fragment plus LAM-PCR linkers), which is produced by all vector genome conformations: linear, circular, and integrated (Fig. 5a); (2) a two-LTR circle will produce a 174 bp band; and (3) a variety of fragments with a minimum size of 163 bp may result by amplification from the 3′-LTR into the host DNA where integrated vector genomes are present.
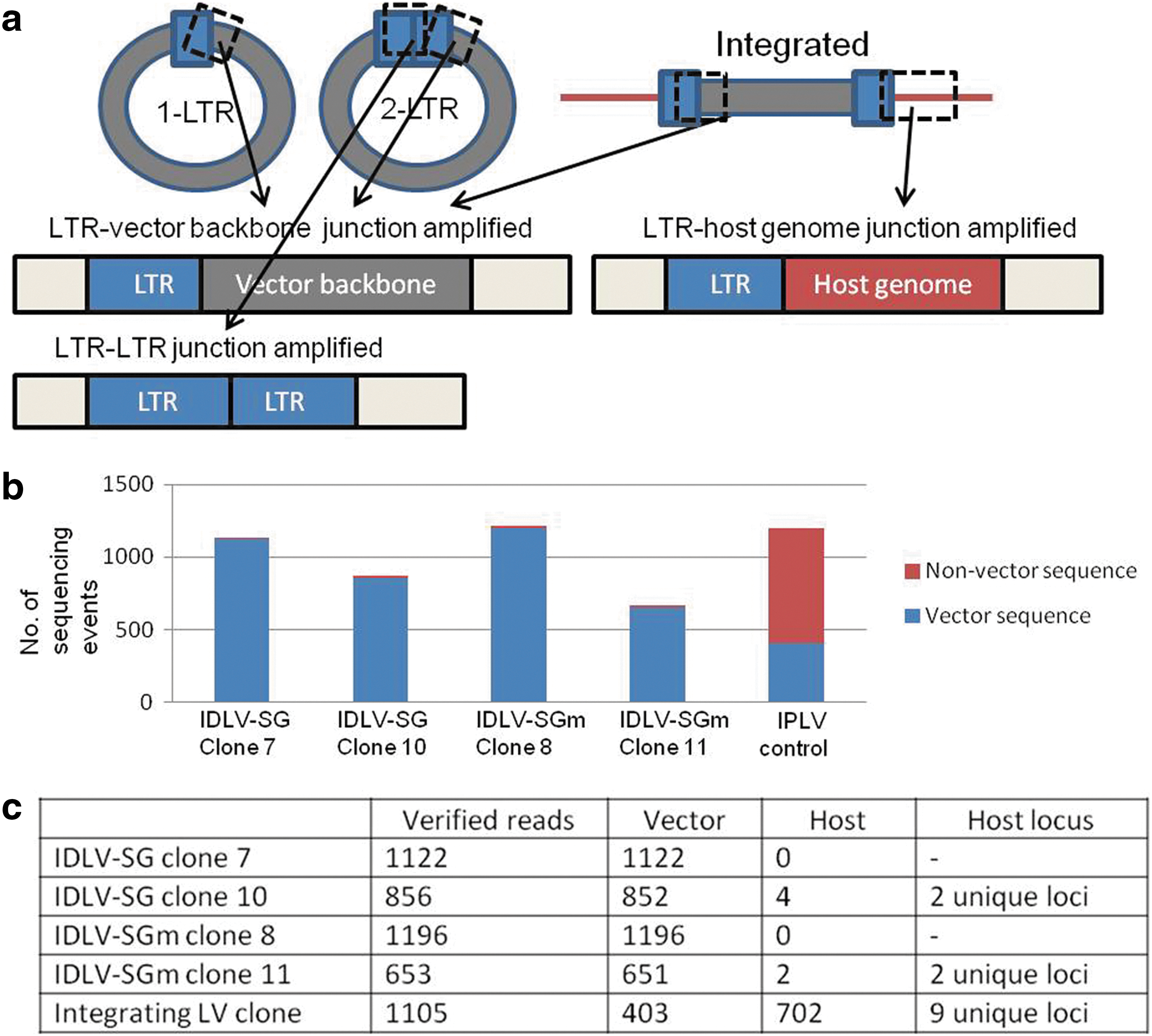
Episomal status of vector sequences in CHO cell clones derived by IDLV transduction and transient cell cycle arrest is confirmed by LAM-PCR analyses and high-throughput deep sequencing. After 60 days of continuous proliferation, genomic DNA was prepared from the stable GFP-expressing CHO clones derived from IDLV transduction and transient cell cycle arrest. DNAs were subject to LAM-PCR, and the amplicons were analyzed by high-throughput deep sequencing.
The PCR products from LAM-PCR were subjected to high-throughput sequencing, resulting in an average of 1100 reads per sample for successful cases. Two samples resulted in less than 50 sequence reads in total (IDLV-SG clone 4 and IDLV-SGm clone 5), and were discounted from further analysis. The sequencing results were divided into two categories, the first containing only vector DNA, and the second nonvector DNA. The vast majority of LAM-PCR sequencing reads for the CHO clones transduced with IDLV-SG and IDLV-SGm yielded only vector sequences (Fig. 5b). To investigate the sequence reads classed as nonvector, they were aligned against the Chinese hamster genomic sequence by applying the QuickMap parameters (Appelt et al., 2009). After analysis, six events were accepted as probably arising from true genomic integrations: two lone events (0.2% of the total number of reads) from IDLV-SGm clone 11 corresponding to two different loci, and four events (0.4% of the total number of reads) from IDLV-SG clone 10 with three reads corresponding to one genomic locus and the remaining single read to a different one. All of these events consisted of a truncated LTR sequence followed by a short sequence corresponding to a defined region in the hamster genome; this is the typical pattern of the rare integration events from IDLVs (Wanisch and Yáñez-Muñoz, 2009; Mátrai et al., 2011). In contrast, the control cell line transduced with IPLV-SG lentivector produced 702 separate events (63.5% of the total number of reads) containing full LTRs and corresponding to 9 unique sequences in the hamster genome (Fig. 5b and c).
Integration analysis by Southern blotting
High-molecular-weight DNA extracted from transduced cells was subjected to a restriction enzyme digest that cuts once within the vector genome, and analyzed by Southern blotting. If episomal, the resulting bands should correspond to the expected size of the 1-LTR circle (3.0 or 3.7 kb for SG and SGm, respectively) or 2-LTR circle (3.3 or 3.9 kb, respectively). If integrated, each integration event should produce a band not likely to correspond to the expected episome sizes. The minimum size for the fragments detectable by the GFP probe (and therefore capable of expressing GFP) is 2967 for IDLV-SG and 2169 bp for IDLV-SGm (Fig. 6a).
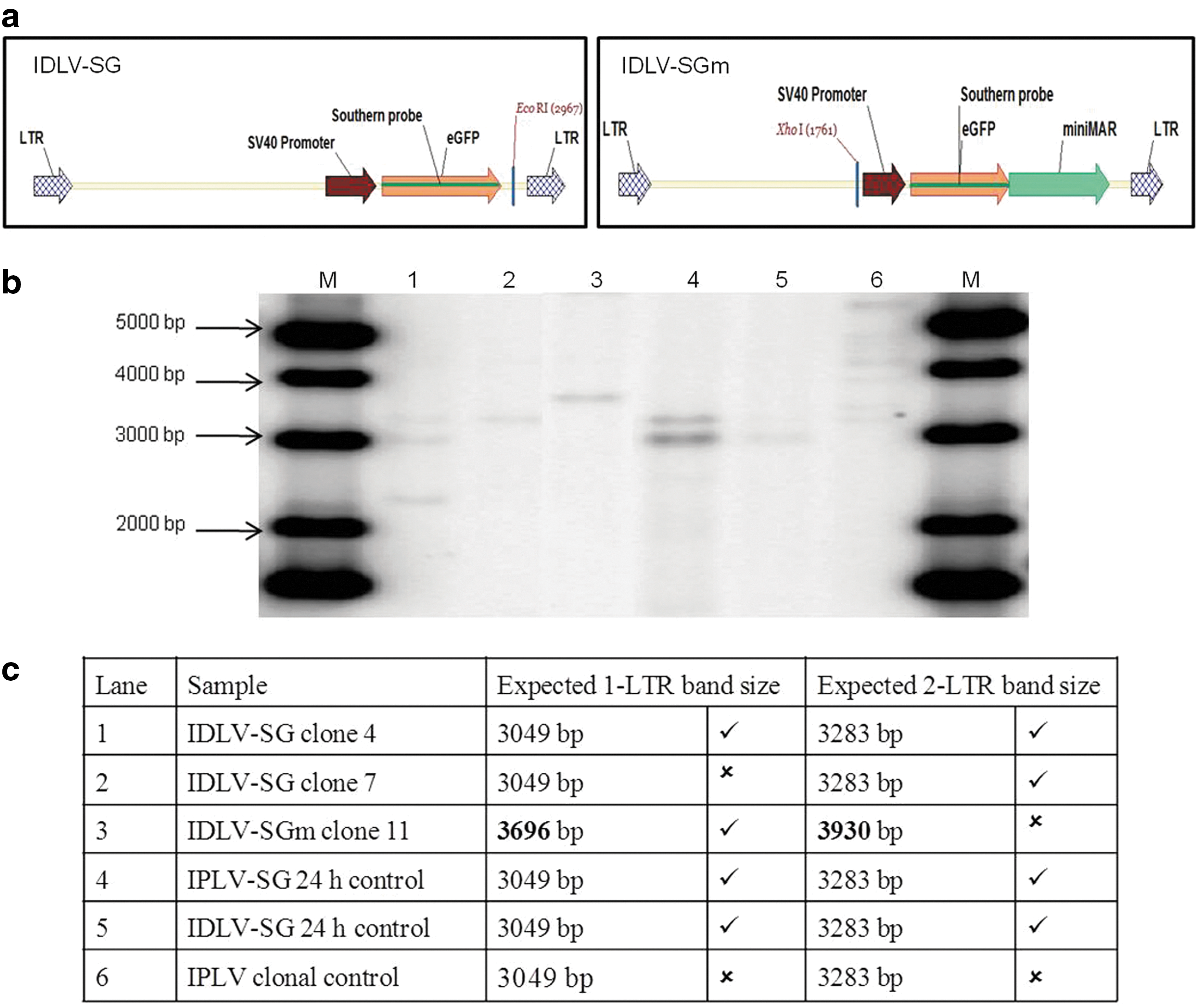
Evaluation by Southern blot hybridization of the episomal status of vector sequences in CHO cell clones derived by IDLV transduction and transient cell cycle arrest. Genomic DNA was prepared after 60 days of continuous proliferation from the stable GFP-expressing CHO clones derived from IDLV transduction and transient cell cycle arrest, as well as 24 hr after transduction for IDLV and IPLV control populations. Genomic DNAs were digested with EcoRI (IDLV-SG and IPLV-SG) or XhoI (IDLV-SGm) and 10 μg subjected to agarose gel electrophoresis, Southern blot transfer, and hybridization with a 32P-labeled probe corresponding to GFP transgene sequences. A diagrammatic linear representation of the expected episomes indicating the location of the probe and the restriction enzyme sites is shown in
Lanes 1 and 2 in Fig. 6b contain DNA from clonal populations transduced with IDLV-SG; clone 4 shows bands corresponding to both 1-LTR and 2-LTR episomal forms and clone 7 shows a 2-LTR sized band. In lane 3, clone 11 from cells transduced with IDLV-SGm shows a band corresponding to the 1-LTR episome. In contrast, the multiple integration events present in the clonal population transduced with the integrating IPLV-SG vector can be seen as discrete bands of varying sizes above 3 kb (Fig. 6b, lane 6), as expected from this type of vector. Lanes 4 and 5 contain total DNA harvested 24 hr after transduction with IDLV-SG and IPLV-SG, respectively; both show bands corresponding to 1-LTR and 2-LTR circles as expected at this early time point in the transduction process. A summary of the Southern blot results is provided in Fig. 6c.
Vector genome visualization using FISH
FISH is commonly used to scan for relatively large chromosomal abnormalities, and successes have also been reported with the detection of smaller single-copy targets (Rupprecht and Lipps, 2009). To examine the cytogenetic distribution of the vector genomes in the CHO cell clones stably transduced with IDLVs, FISH analyses were performed on 15 cell populations: IDLV-SG-transduced polyclonal population and clones 4, 7, and 10; IDLV-SGm-transduced polyclonal population and clones 5, 8, and 11; two control polyclonal populations transduced with IDLV-SG and IPLV-SG and harvested 24 hr after transduction; a control polyclonal population transduced with IPLV-SG and three derived clones; and an untransduced control population. The samples were analyzed in two different ways: first, a qualitative analysis of signal quality on FISH metaphase images was performed in all samples; second, selected clones (IDLV-SG clone 4, IDLV-SGm clone 11, IPLV-SG clone 6) and the untransduced negative control were subjected to detailed quantitative analyses.
The FISH probe was separately labeled with green and red fluorochromes, and both labeled probes were used in combination; only signals positive for both colors were accepted as true signals (Fig. 7a–l). In the slide containing untransduced cells, some signals containing only one color were observed (Fig. 7b). In contrast, the majority of signals observed in transduced cells are yellow, which results from overlapping red and green (arrows in Fig. 7d–l). A statistical analysis was performed on the selected samples. The number of specific signals inside nuclei was 5–7-fold higher in the transduced clonal samples than in the negative control (Fig. 7m; n=20, p<0.0001, one-tailed Student's t-test). This strongly supports the conclusion that the signals arise from the target sequence rather than background. An integrated transgene is expected to produce a doublet signal where the signal is present at the same location on both sister chromatids. Only 1.9% of metaphase images taken of cell populations transduced with IDLVs contained a doublet signal. In contrast, 22.9% of metaphase images taken of clonal samples transduced with IPLVs showed clear doublets (Fig. 7n). The metaphase images therefore provide significant supportive evidence for the presence of episomes in the cells transduced with nonintegrating vectors.

Cytogenetic FISH analysis of the episomal status of vector sequences in CHO cell clones derived by IDLV transduction and transient cell cycle arrest. Stable GFP-expressing CHO clones derived from IDLV transduction and transient cell cycle arrest [IDLV-SG (clone 4) and IDLV-SGm (clone 11)], and clonal CHO cells stably transduced with an integrating lentivector (IPLV-SG), along with nontransduced cells were prepared for FISH and hybridized simultaneously to a mixture of two probes labeled with either SpectrumOrange or SpectrumGreen fluorochromes. Slides were counterstained with DAPI, and at least 20 metaphases were located on each slide and examined by epifluorescence microscopy, here shown at ×100 magnification
Putative origins of replication
To investigate potential lentivector sequence elements that could be functioning as origins of replication, a stress-induced DNA duplex destabilization (SIDD) analysis was conducted on the lentiviral episome sequences. SIDD refers to the tendency of the DNA helix to undergo strand separation when placed under negative superhelicity, and has been implicated in the mechanisms of activity of numerous biological processes, most notably the initiation of transcription and replication (Benham, 2001). Research has indicated that chromosomal sites that are susceptible to superhelical strand separation are required in both prokaryotes and eukaryotes for the initiation of DNA replication. In some instances an SIDD region is sufficient to initiate these processes without the help of DNA-binding proteins (Benham and Bi, 2004; Bode et al., 2006). Usually, the least stable regions tend to be A-T rich, but whether a transition occurs at a given site depends not just on the local sequence properties but also on how the site competes with all the others in the domain (Benham and Bi, 2004).
The WebSIDD algorithm was used to calculate the transition probability and destabilization energy of each base pair in the predicted episome sequences (Bi and Benham, 2004) (Fig. 8). The clearest troughs are seen in regions involved in transcription initiation and termination, which are known to exhibit duplex destabilization (Benham, 2001). The central destabilized region (1350–1500 bp) contains the dysfunctional Env coding sequence, showing a typical incremental destabilization profile for nontranscribed regions. The S/MAR element (Fig. 8b) is characterized by a series of destabilization troughs. In addition to these, the central polypurine tract (cPPT) can be seen to be highly and sharply destabilized (Fig. 8a and b, red arrows).
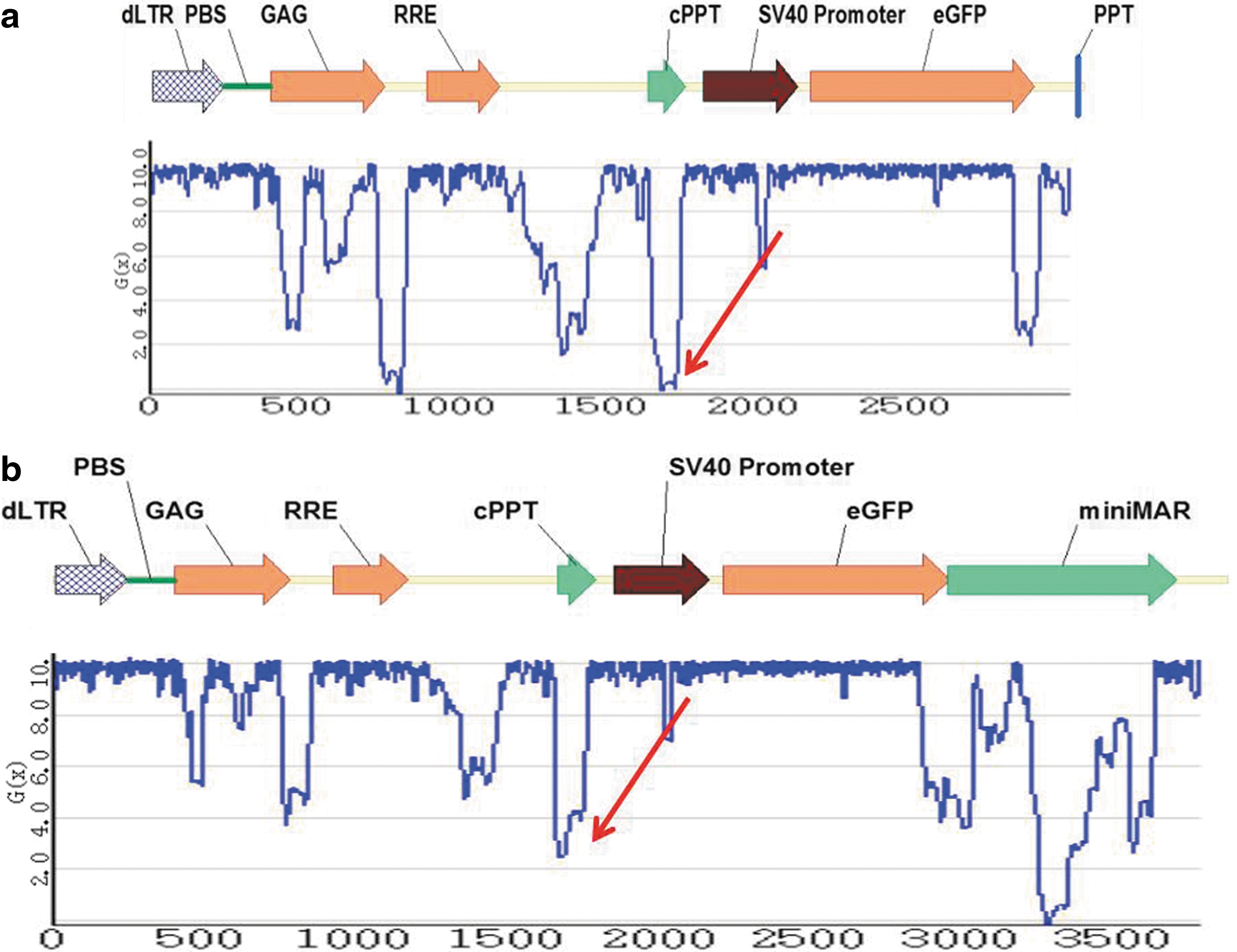
Potential origins of replication in circular episomes. Schematic representations of 1-LTR episomes expected to arise during cell transduction with
Discussion
In this study, we have established replicating episomes by using a combination of IDLV transduction and transient cell cycle arrest. The presence of the miniMAR element did not lead to higher retention levels, and may even have been slightly detrimental, which suggests that in this case the replication potential is related to a feature present elsewhere in all lentiviral genomes. The unprecedentedly high retention of the transgenes in the transduced cells initially held in cell cycle arrest can be attributed to two mechanisms: either the viral episomes undergo an epigenetic event that activates nuclear retention and DNA replication during cell cycle arrest and thus function as mitotically stable entities, or the cell cycle arrest induced an unexpectedly high level of integration.
The Gaussian distribution of the MFI in the clonal populations suggests variability in the vector copy number. The number of nonintegrated vector genomes may vary according to cell cycle status as well as other as yet unknown factors.
All the IDLVs used here carried a specific inactivating D64V mutation in the IN enzyme gene that normally mediates lentiviral integration. Estimates of integration frequency in IDLVs with this mutation set it approximately 2–3 logs lower than wild-type vector, so it is unlikely that a high integration frequency in our study may have been IN mediated (Gaur and Leavitt, 1998; Wanisch and Yáñez-Muñoz, 2009).
However, we conducted a thorough analysis of the retained IDLV genomes by LAM-PCR sequencing, Southern blot, and FISH to assess the relative levels of episomal versus integrated IDLV genomes in clonal populations of transduced cells. LAM-PCR and subsequent deep sequencing of the PCR products is considered a reliable method for detecting even single clones with integration events present in a cell population. In cells transduced with standard integrating IPLVs, the analysis yielded the expected ∼50% level of detected chromosome integration junctions (64%: 702 out of 1105 sequence reads for the IPLV-SG clone). However, in contrast, in four clonal populations derived from the IDLV transduced samples that had undergone over 100 population doublings, ≤0.2% integration junctions were detected (6 out of 3827 sequencing reads for clones from IDLV-SG and IDLV-SGm). Indeed in two clones no integration junction whatsoever could be detected by LAM-PCR deep sequencing analysis. The very small number of reads indicating potential integration events in two out of the four clonal IDLV samples analyzed (2/653 for IDLV-SGm clone 11 and 4/856 for IDLV-SG clone 10) may reflect sporadic events arisen in a very small proportion of cells during extensive growth, freeze–thawing, and passage in culture.
Southern blotting was also used to evaluate the conformation of the vector genomes in three IDLV clones and yielded strong evidence supporting the episome hypothesis: IDLV-SG clones 4 and 7 and IDLV-SGm clone 11 produced bands consistent with the expected size for episomal vector genomes (Fig. 6b). The unidentified smaller ∼2200 bp fragment present in IDLV-SG clone 4 must be the result of an internal rearrangement of the episome, since based on the configuration of restriction enzyme digest site and probe location, the size of predicted vector–genome integration fragments must be >2967 bp (Fig. 6a and b).
Finally, the FISH evaluation was performed on two stable IDLV-transduced clones. The results from extensive FISH analyses of IDLV-SG clone 4 and IDLV-SGm clone 11 indicated that the IDLV vector genomes produced nuclear signals, which were uniquely and predominantly singlet; this is consistent with their existence as nonintegrated episomal elements.
In conclusion, these combined data of LAM-PCR sequencing, Southern blot, and FISH strongly suggest that the vast majority of IDLV genomes in the CHO clones derived from transduction combined with cell cycle arrest are not integrated, and support the hypothesis that they exist as autonomous replication episomes. While the existence of episomal concatenated structures cannot be discounted, the presence of integrated vector concatemers would have generated at least one vector–host junction signal for each concatemer during LAM-PCR. As less than 0.2% of sequencing reads produced such a signal, the integrated episome hypothesis can be discounted in the majority of cells.
The episomal configuration was established in cells transduced with IDLVs both containing and lacking miniMAR, and it is apparent that while the S/MAR may act to enhance replication, they are dispensable for IDLV episome retention in our protocol. Further evidence against the involvement of the miniMAR element in the formation of the nuclear episomes is provided by the failure of the miniMAR-containing AAV vectors, plasmids, and minicircles to support long-term transgene expression. The minicircle vector also did not result in the establishment of replicating episomes at significant levels. Our observations thus suggest that a functional origin of replication (ori) yet to be identified must be present in the retained circular IDLV episomes. This could be a cryptic function, perhaps irrelevant in integrated proviruses but surfacing in episomal molecules under adequate conditions such as those reported here.
A search for the putative replication origin was carried out by scanning the episome sequences for DNA duplex instability. In addition to the expected destabilized regions, the cPPT was found to be highly destabilized. The cPPT is known to be involved in nuclear import and plus-strand synthesis, and has been shown to increase the transduction efficiency of lentivectors (Follenzi et al., 2000; Zennou et al., 2000; Manganini et al., 2002). However, the destabilization profile suggests the possibility for additional cPPT functions associated with high destabilization, such as replication initiation, and the testing of this theory would provide for some interesting further experiments.
The experiments here were set out to investigate the possibility for developing stable, episomal gene transfer for therapeutic and biotechnological purposes. By combining IDLV transduction with an induced cell cycle arrest, this aim was achieved. While CHO cells are a suitable biotechnology platform, clinical application of this system will require translation to relevant stem cell populations. If this can be achieved, the ability to generate mitotically stable gene expression from nonintegrating episomes would significantly advance the safety aspect of gene addition therapy.
Footnotes
Acknowledgments
This work was supported by the Sixth EU Framework Programme (Clinigene, Grant Agreement No. 18933) and a Reid Scholarship from Royal Holloway-University of London to H.K. Thanks to Cecilia Good for molecular cloning and to Christina Lehrer for help with LAM-PCR.
Author Contributions
G.D. and R.J.Y.-M. designed and supervised the study. H.K. also designed the study and was involved in the conduct of all experiments. M.S., F.A.G., J.U.A., and S.L. assisted with the design, conduct, meta-analysis, and bioinformatic analyses of LAM-PCR and deep sequencing experiments. C.M.O. and A.F.D. assisted with the design and interpretation of FISH experiments. S.G.A. supervised the molecular cloning. J.B. provided the truncated S/MAR element. H.K. drafted the article, which was then edited by G.D. and R.J.Y.-M., with contributions from all other authors.
Author Disclosure Statement
The authors declare the following personal financial interest: G.D., H.K., and R.J.Y.-M. are named inventors on a patent application for the stable episome gene transfer system described in this article. No competing financial interests exist for the other authors.