
Abstract
Spinal muscular atrophy (SMA) is a severe autosomal recessive disease caused by a genetic defect in the survival motor neuron 1 (SMN1) gene, which encodes SMN, a protein widely expressed in all eukaryotic cells. Depletion of the SMN protein causes muscle weakness and progressive loss of movement in SMA patients. The field of gene therapy has made major advances over the past decade, and gene delivery to the central nervous system (CNS) by in vivo or ex vivo techniques is a rapidly emerging field in neuroscience. Despite Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis being among the most common neurodegenerative diseases in humans and attractive targets for treatment development, their multifactorial origin and complicated genetics make them less amenable to gene therapy. Monogenic disorders resulting from modifications in a single gene, such as SMA, prove more favorable and have been at the fore of this evolution of potential gene therapies, and results to date have been promising at least. With the estimated number of monogenic diseases standing in the thousands, elucidating a therapeutic target for one could have major implications for many more. Recent progress has brought about the commercialization of the first gene therapies for diseases, such as pancreatitis in the form of Glybera, with the potential for other monogenic disease therapies to follow suit. While much research has been carried out, there are many limiting factors that can halt or impede translation of therapies from the bench to the clinic. This review will look at both recent advances and encountered impediments in terms of SMA and endeavor to highlight the promising results that may be applicable to various associated diseases and also discuss the potential to overcome present limitations.
Introduction to Spinal Muscular Atrophy
A
SMA is clinically classified into four main types (I, II, III, and IV) depending on the age of onset and the severity of the symptoms. SMA type I, or Weirdnig–Hoffman disease, is characterized by severe muscular problems in infancy with death occurring before the age of 2, mainly because of respiratory insufficiency. Patients with SMA type II, or intermediate type, display their first symptoms between 6 and 18 months of age. These patients are expected to live for decades, but they face aggressive respiratory problems. In SMA type III, or Kugelberg–Welander disease, the symptoms make their first appearance after the age of 18 months. The initially unaffected patients become wheelchair bound as the disease worsens. Type IV is a rare form, with symptoms very similar to type III but with onset in adulthood (around 30 years of age).
Despite this clinical heterogeneity, 95% of cases are associated with deletions or mutations of the survival motor neuron 1 gene (SMN1) (Kolb and Kissel, 2011; Porensky et al., 2012). The SMN gene is located on chromosome 5 (5q11.2–13.3), and humans possess two almost-identical copies of the SMN gene, with similar promoters encoding the SMN protein, namely, SMN1 and SMN2. SMN1, the telomeric copy, produces the fully functional, full-length SMN protein that is necessary for normal lower motor neuron function, whereas SMN2, the centromeric copy, predominantly produces transcripts lacking exon 7, because of a translationally silent C-to-T transition, with the resultant effect of producing a truncated and unstable protein (Fig. 1). SMA has a unique genetic profile as it is autosomal recessive for the functional loss of SMN1, but its nearly identical homolog, SMN2, has been shown to act as a disease modifier in humans (Cherry et al., 2013) mainly because SMN2 does produce a low level of full-length SMN (about 10%) but it cannot adequately compensate for loss of the SMN1 gene (Lorson et al., 1999; Mitrpant et al., 2013). Above 95% of individuals with a clinical diagnosis of SMA lack exon 7 in both copies of the SMN1 gene, while the remainder are compound heterozygotes for deletion of SMN1 exon 7 in one allele and an intragenic mutation of SMN1 in the other allele (Hofmann et al., 2000; Chen et al., 2008). Therefore, mutations in SMN1 lead to SMA, while loss of SMN2 alone has no adverse effect. Since SMN2 produces small amounts of SMN, SMN2 copy number inversely correlates with clinical disease severity. It has been reported that there is a variable SMN2 gene copy number and a substantial inverse relationship between the number of SMN2 copies and SMA severity (Chen et al., 2008). The majority of type I SMA patients carry two SMN2 copies, type II carry three, and type III SMA patients carry three or four SMN2 copies (Cartegni and Krainer, 2002; Chen et al., 2008). Therefore, the SMN2 gene is considered as a major disease modifier. As activation or modulation of the SMN2 splicing pattern can impact SMN in a disease-modifying way, methods to increase full-length SMN protein levels from SMN2 have been developed as a way to aid SMA treatment (Cartegni and Krainer, 2003). SMA is thus a disease of low SMN levels and not a complete absence, which is universally cell lethal.

Genetic basis of SMA. Humans carry two almost-identical copies of the SMN gene: a centromeric copy (SMN1) and a telomeric copy (SMN2), which are located on chromosome 5 in the 5q13 region known as the SMA locus. The two genes differ by only a few bases; one of these changes, a C-to-T transition on exon 7, affects the splicing of SMN2 resulting in SMNΔ7 mRNA, which when translated produces a truncated, unstable, and easily degraded protein. However, 10% of SMN2 transcripts can undergo correct alternative splicing, including exon 7. SMA, spinal muscular atrophy; SMN, survival motor neuron. Color images available online at
Drosophila melanogaster and Caenorhabditis elegans have both been shown to carry single homologs of SMN, which are 31% (smn) and 36% (smn-1) identical to the human gene, respectively. In these models, larval lethality occurs when the protein and mRNA become depleted after homozygous deletion of the gene (Hua et al., 2008; Baughan et al., 2009). As complete loss of SMN is lethal, SMA only arises as a disease in humans owing to the presence of SMN2, which provides a sufficient residual full-length protein for cellular viability (Singh, 2007).
SMN is a ubiquitously expressed protein and is essential for the survival of nearly all cell types. The cause of the selective vulnerability of motor neurons to low SMN protein levels is still unclear and remains to be elucidated. The first reported and best characterized role for SMN is in the assembly of small nuclear ribonucleoproteins (snRNPs) (Pellizzoni et al., 2002). The SMN complex is comprised of at least nine constituents, including Gemins proteins 2–8 and Unr-interacting protein (UNRIP). The SMN protein has been shown to form this complex after its own self-oligomerization and subsequent interaction with these constituent features (Young et al., 2000). This formed complex then efficiently assembles a core of the Sec1/Munc family of proteins onto small nuclear ribonucleic acids (snRNAs). Through additional processing, snRNPs are synthesized before being transported into the nucleus where they are involved in the catalytic removal of introns from pre-mRNA transcripts in the process of splicing (Young et al., 2000; Cauchi, 2010). While the cardinal genetic causes of SMA are known, the same is not true of the biochemical pathways that are relevant to the disease and, as mentioned, it is still ambiguous as to why lower levels of the widely expressed multifunctional SMN protein specifically induce the loss of motor neurons. It has also been suggested that SMN plays a crucial role in the axonal transport of mRNA (Rossoll et al., 2003; Fallini et al., 2013). Therefore, two hypotheses have been proposed to explain the molecular dysfunction that gives rise to SMA. The first hypothesis states that the loss of SMN's well-known function in snRNP assembly causes an alteration in the splicing of a subset of genes essential for the survival of motor neurons. The second hypothesis proposes that SMN is crucial for axonal trafficking of mRNA-binding proteins and their target mRNAs in neurons and that disruption of this function results in SMA as it is believed that the complex of SMN with its binding partner hnRNP-R interacts with beta-actin mRNA and translocates to axons and growth cones of motor neurons (Rossoll et al., 2003; Burghes and Beattie, 2009; Fallini et al., 2012).
No effective treatments currently exist for SMA patients, but the identification of therapeutic targets and the development of suitable animal models for preclinical testing have resulted in increased drug development efforts in the last years (Van Meerbeke and Sumner, 2011; Bebee et al., 2012; Wadman et al., 2012a,b). The current gene therapy approaches primarily focus on increasing the levels of the full-length SMN protein through two main methodologies: the replacement of the SMN1 gene or regulation of SMN2 expression. Because SMA is a monogenic condition, vector-based gene replacement of the SMN1 gene is an attractive option for the treatment of the disease.
Gene Therapy
Of the approximate 25,000 genes that comprise the human genome, mutations in more than 6,000 have already been identified as causing gene-related disorders (Wei et al., 2011). Increased understanding of genotype–phenotype relationships borne from the sequencing of the human genome has allowed clinicians and researchers to gain more insight into genetic diseases and thus develop improved tools to aid in their treatment. While some genetic diseases such as hemophilia A, familial hypercholesterolemia, and sickle cell anemia (Connor and Connor, 1993; Davies and Gilmore, 2003; Srivastava, 2004) have shown some therapeutic success through metabolic manipulation and protein augmentation, it is believed that some of the major therapeutic approaches for hereditary disorders will come from the development of novel therapies that are centered on transferring genetic material to correct or compensate for an abnormal phenotype associated with a particular genotype (Hofmann et al., 2000; Chen et al., 2008). As monogenic diseases are caused by disruption in a single gene, the nature of these disorders makes them attractive for gene therapy, as stable transfer of the healthy gene could ensure long-term therapeutic benefit.
Within the past few decades, gene therapy has emerged as a powerful tool to restore defective genes that trigger some human disorders (Friedmann, 1992; Verma and Weitzman, 2005). This therapeutic strategy is broadly defined as the introduction or alteration of genetic material within a cell or organism with the intention of curing or treating a disease (Fig. 2). Different approaches are available to achieve this goal, involving either in vivo or ex vivo techniques.
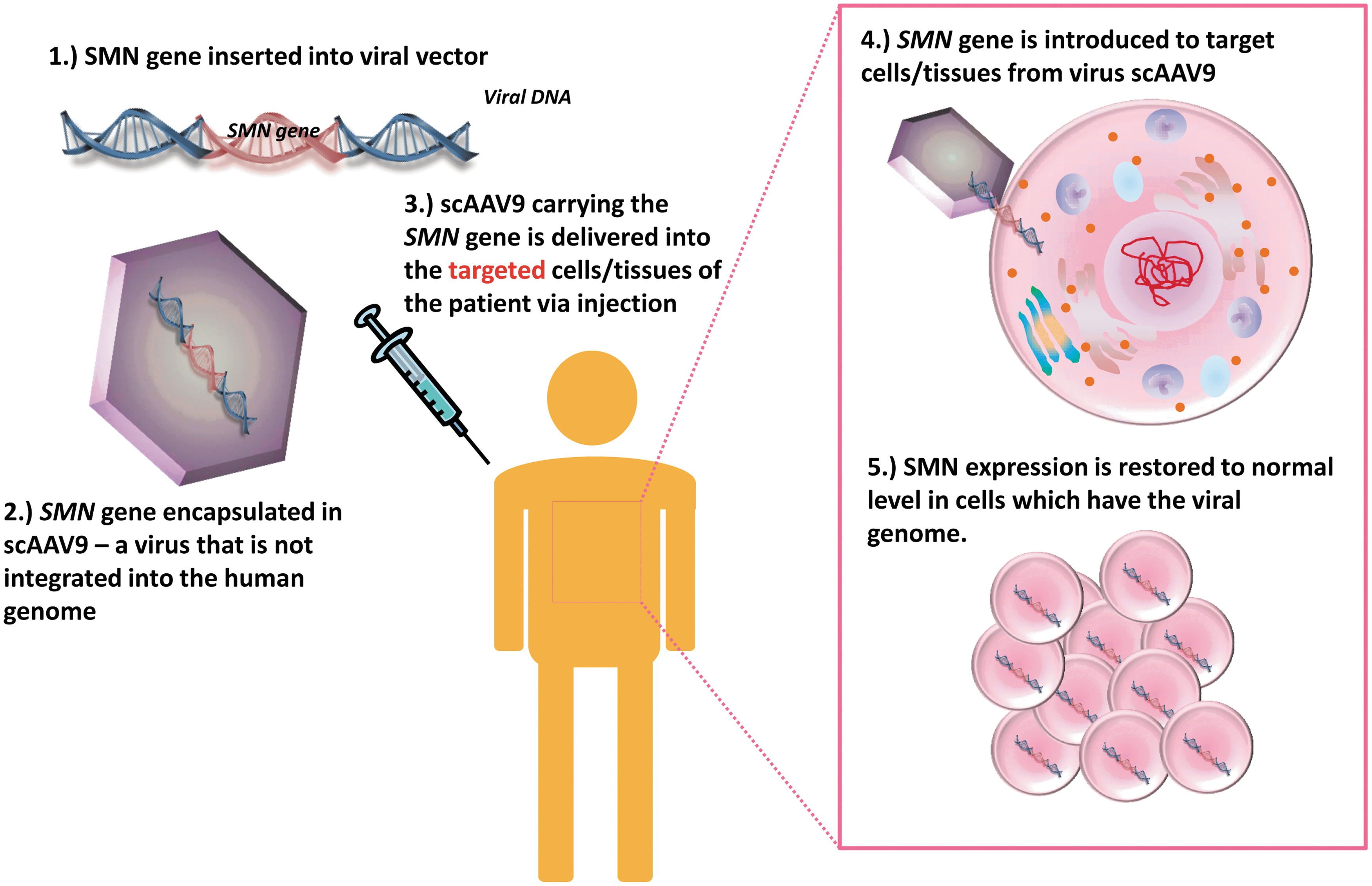
Simplified diagram of SMA gene therapy. SMA is caused by the low level of the SMN protein. Introducing a functional SMN gene to SMA patients via gene therapy can restore the SMN expression level to normality. As a first step, for example, the SMN gene is inserted into the viral vector, scAAV. The SMN-encoding AAV plasmids are then used for large-scale virus production. In clinical application, the patients would be given a dose of the virus via injection. Choices of delivery routes include intramuscular, intravenous, intracerebroventricular, and intrathecal injections. When the virus has reached the targeted cells, the SMN gene is introduced into the targeted cells for SMN protein expression, hence restoring the SMN expression to the endogenous level of a healthy individual. AAV, adeno-associated virus. Color images available online at
Ex vivo gene transfer refers to a strategy in which the target cells are removed from the individual to be treated, genetically modified in the laboratory, and then re-administered to the patient (Dominguez et al., 2011; Mingozzi and High, 2011). Some of the main advantages of ex vivo gene delivery include the capability of targeting specific cell types and the safety of the manipulation (Foust et al., 2010; Mingozzi & High, 2011). As host cells are harvested, the relevant cell type(s) can be isolated and manipulated before the replacement of cells back into the host, limiting the level of off-target or transduction of irrelevant cells (Saunders et al., 2012). The genetic changes can be screened before reintroduction to assess the location and sequence of the changes. In terms of immunocompatibility, the cells are taken from the host and returned to the same host after treatment; therefore, there is no need for immunosuppression as the re-introduced cells would not be treated as foreign by the immune system (Valori et al., 2010).
There are some limitations to ex vivo gene therapies, however. Due to their inability to divide, most neuronal types cannot be transduction targets for therapies as to be maintained and modified in vitro prior to delivery they must be capable of dividing. (Lorson et al., 1999). This method of therapy is also an invasive one as the delivery and grafting of cells is a much more intensive process than injection of in vivo gene therapy vectors. Finally, the delivery of dividing cells potentially bears the risk of tumor formation (Lorson et al., 1999; Tuszynski et al., 2002).
Ex vivo strategies are best suited to applications in which the corrected cells can be easily obtained, such as bone marrow, or where the corrected cells have a selective advantage when they are returned to the patient (Capowski et al., 2007; Hacein-Bey-Abina et al., 2010; Kashiwakura et al., 2012; Biffi et al., 2013). The evolution of ex vivo gene therapies for SMA has to date been very limited because of the postmitotic nature of the CNS cells affected during disease progression. By far and away, however, the majority of the gene therapy research that has been carried out in SMA to date has been done utilizing direct in vivo techniques (Bouard et al., 2009; Kal Van Tam et al., 2014).
In vivo gene therapy approaches introduce genetic material directly into the host cells (Maguire et al., 2008; Valori et al., 2010). The delivery of the therapeutic gene into the target cells frequently requires the use of a carrier vector, in which the gene is encapsulated and produced in large amounts (Kay et al., 2001; Pezzoli et al., 2012; Chuah et al., 2013). The nature of this vector may be an attenuated and modified virus, which has lost some structural genes to avoid nondesirable infections and incorporate specific promoters to facilitate the achievement of strong transgene expression. RNA viruses such as lentivirus and retrovirus, or DNA viruses such as adenovirus and adeno-associated virus (AAV) are frequently used in gene therapy and have been broadly reviewed in many articles (Graham and Prevec, 1995; Coura Rdos and Nardi, 2007; Valori et al., 2008; Kay, 2011; Vannucci et al., 2013). However, there are other approaches that do not involve the use of viral particles to release the healthy gene, for instance, the direct injection of naked DNA, or the use of nonviral vectors such as cationic lipids and cationic polymers, which establish electrostatic interactions with negatively charged DNA to form complexes (Al-Dosari and Gao, 2009; Wang et al., 2013). While nonviral techniques are becoming more common, according to databases of gene therapy clinical trials, more than 60% of the gene therapy human trials approved worldwide so far involve the use of viral vectors (Ginn et al., 2013).
Gene Therapies in SMA
Gene targeting methods should fulfill a commitment in terms of specificity, efficiency, and safety to achieve successful gene therapy. These approaches, therefore, must be specific to select only the target cells, displaying highly specific gene delivery efficiency and also long-term gene expression (Wilson, 1996; Campbell and Hope, 2005). SMA is a monogenetic disease that has been the focus of extensive efforts by several research groups with the aim of developing efficient gene therapy strategies for restoring SMN protein levels in motor neurons. Encouraging results are emerging from several gene therapy approaches and are ready for human clinical applications (Azzouz et al., 2004b; Passini et al., 2010; Valori et al., 2010; Dominguez et al., 2011; Glascock et al., 2012a; Benkhelifa-Ziyyat et al., 2013). At present, the main targets for therapeutics in SMA involve stimulating SMN2 function in order to increase SMN protein levels or using viral therapies to restore SMN levels. (Arnold and Burghes, 2013). Currently, preclinical studies show that therapies targeting SMN protein restoration levels are most promising approaches to disease management and treatment.
Lentiviral Vectors
Lentiviral vectors are inactivated retroviruses commonly used for in vivo applications for neurodegenerative diseases. Typically, the most widely used backbone of lentiviral vectors is based on the human immunodeficiency virus 1 (HIV-1), using a three-plasmid system composing of a packaging plasmid, vector plasmid, and envelope plasmid (Naldini et al., 1996). Lentiviruses can intergrate into the host genome, offering long-lasting and efficient transgene expression without eliciting an immune response in patients (Azzouz et al., 2004a), and are able to transduce nondividing cells, which is essential in the treatment of neurodegenerative diseases, where the target cells are predominantly postmitotic neurons.
Although random integration of lentiviral vector genetic information into the host cell genome remains of paramount concern, one ongoing phase I/II trial using lentiviral vectors (ProSavin Oxford BioMedica) for treating Parkinson's disease shows a long-term safety profile, and all treated patients demonstrated improvements in motor activities (Nakata et al., 2012; Palfi et al., 2014).
Earlier reports of gene therapy in the SMA mouse model demonstrated successful transduction of SMN to the motor neurons in the lumbar spinal cord after delivery to multiple muscles and retrograde delivery of the rabies G pseudotyped virus to the motor neurons (Azzouz et al., 2004b). Azzouz and colleagues employed lentiviral vectors expressing SMN cDNA to restore SMN protein levels in cell culture (Azzouz et al., 2004b). Subsequent animal experiments reported that the vector restored the amount of SMN in various muscles of SMA mice, reduced motor neuron death, and extended the life span of SMA mice by an average of 20–38% from multiple single injections of the vector (Azzouz et al., 2004b). Nevertheless, the efficiency of the latest proof-of-concept studies was shown to be marginal and thus, cannot form a strong basis for clinical applications in humans (Arnold & Burghes, 2013; Azzouz et al., 2004). Another major obstacle using the latest strategy is the production of the large amount of virus required for muscle delivery in human therapeutics (Aiuti et al., 2003; Ott et al., 2006; Arnold & Burghes, 2013).
Adeno-Associated Virus Vectors
An alternative approach to lentiviral vectors for SMA is the use of the AAV-based gene therapy vectors. AAV is a member of the parvoviridae family and a dependovirus. It is ideal for gene therapy because of its long-term gene expression, the inability to autonomously replicate without a helper virus, transduction of nondividing cells, and the lack of pathogenicity from wild-type infections. AAV gene therapy has attracted considerable interest from researchers; as of 2010, there were already 18 active trials in different stages of the development pipeline (Bevan et al., 2010). The family of AAV contains more than 100 serotypes, and AAV9 stands out for the treatment of neurological diseases such as SMA because of its ability to cross the blood–brain barrier through the vascular system (Dayton et al., 2012). Additionally, early studies showed that all AAV vectors do not integrate into the genome of host cells and can transduce quiescent cells (Mingozzi & High, 2011), such as spinal motor neurons and astrocytes, after systemic delivery in rodents (Foust et al., 2010; Valori et al., 2010; Dominguez et al., 2011). These features make AAV an attractive candidate for clinical development in SMA.
In 2010–2011, successful rescue of SMA mice was reported by four groups using gene therapies to replace SMN with an AAV-based vector (Fig. 3) (Foust et al., 2010; Passini et al., 2010; Valori et al., 2010). Several groups have engineered a self-complementary AAV9 (scAAV9) vectors designed to overexpress a sufficient amount of the human SMN protein in a mouse model with severe SMA (Foust et al., 2009, 2010; Valori et al., 2010; Dominguez et al., 2011). Compared with canonical AAV vectors, the advancement of the scAAV vectors offers huge advantages in transduction efficiency and transgene expression for relatively small genes (McCarty, 2008). In these studies, mice with SMA were treated in vivo with a single intramuscular or intravenous injection of SMN-expressing scAAV9 vector. Encouragingly, this intervention resulted in extending the lifespan of SMA mice and improved some clinical parameters, such as weight gain and restoration of motor functions. Akin to scAAV9, similar positive results have been demonstrated by other groups using scAAV8 vectors and different strains of SMA mouse models (Passini et al., 2010; Bevan et al., 2010, 2011; Benkhelifa-Ziyyat et al., 2013). These compelling examples lay a foundation for human clinical studies using the AAV vector.
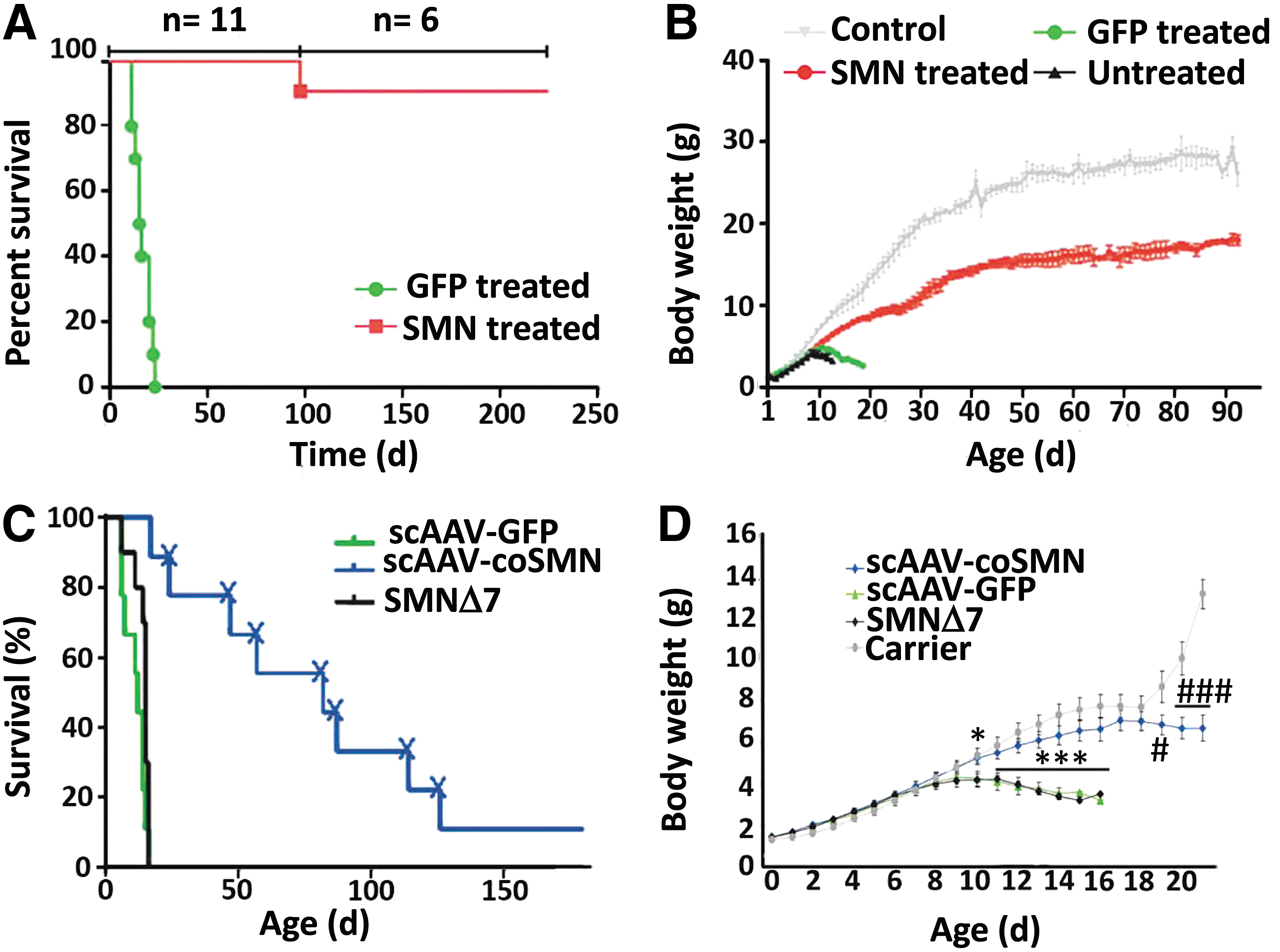
Successful phenotypic correction by using gene therapy on P1 SMA mice.
Various routes of delivery of scAAV9-SMN1, including intravenous, intracerebroventricular, intramuscular, and combined routes, have been investigated for SMA therapy (Foust et al., 2009; Dominguez et al., 2011; Glascock et al., 2012b; Benkhelifa-Ziyyat et al., 2013). Treatment of SMNΔ7 mice with intramuscular doses of scAAV9-SMN1 caused widespread transduction of the lumbar, thoracic, and cervical regions (via GFP detection) or the spinal cord 4 weeks after administration and led to a significant increase in survival as compared with noninjected littermates. These animals displayed an increased lifespan of over 160 days, while the mean survival for untreated animals was typically around 12 days (Benkhelifa-Ziyyat et al., 2013). Promising results have been achieved in larger animals, including primates and pig after AAV administration. When scAAV9 was introduced into the vasculature, it crossed the blood–brain barrier successfully and achieved efficient transduction of motor neurons in various regions of the spinal cord (Duque et al., 2009; Foust et al., 2009, 2010; Valori et al., 2010; Bevan et al., 2011; Dominguez et al., 2011). This translation into larger animals is encouraging for the transition of SMA therapy into humans.
Preclinical toxicology testing in primates and mice showed good safety profile for scAAV9-SMN, and an initial human clinical trial has been approved by the U.S. Food and Drug Administration (FDA) in a fast-track process. In larger animals, intrathecal delivery also shows efficient transduction of motor neurons and allows for a reduced viral dose to be used (Bevan et al., 2011; Federici et al., 2012; Gray et al., 2013). Autoimmunity against restored SMN, as seen in other gene therapy trials, is not predicted to occur in SMA because of the presence of endogenous SMN levels produced by the SMN2 gene. scAAV9-SMN offers the potential of one-time dosing without the requirement of repeated treatment. Studies are underway to fully optimize this route of delivery and to obtain the required toxicology studies to move this treatment to clinical trials (Arnold & Burghes, 2013). Brian Kaspar and colleagues have recently been given approval by the FDA of an Investigational New Drug application to begin a phase I clinical trial of a systemic AAV9-delivered human SMN gene to SMA type I infants, which is expected to start in early 2014.
Antisense Oligonucleotides
Apart from the vector-mediated gene delivery approaches, antisense oligonucleotides (ASOs) can increase SMN expression by abolishing the exon 7 skipping mechanism of the SMN2 gene. ASOs are therapeutic RNA molecules that block cis-acting splice modifiers such as exon splice enhancers or intron splice silencers that can either enhance or disrupt the targeted splicing events. ASO-10-27, a 2′-O-methyl–modified oligonucleotide (5′-TCACTTTCATAATGCTGG-3′), is the most commonly used one. In SMA, ASO-10-27 hybridizes to the intronic splicing silencer N1 (ISS-N1) in SMN2 intron 7 and has been demonstrated to promote intron 7 inclusion in targeted transcripts and a corresponding increase in the level of the SMN protein in vitro (Singh et al., 2004).
To apply ASO-10-27 for in vivo clinical application, several research groups have shown promising results in delivering this oligo into the CNS (Hua et al., 2011). Passini and colleagues administered the ASO into the cerebral lateral ventricles of mice with a severe form of SMA by direct injection, and this resulted in a significant increase of splice-mediated SMN proteins and in the number of motor neurons in the spinal cord (Passini et al., 2011). These molecular and cellular changes improved the survival benefits of mice, such as the muscle physiology, motor functions, and survival. Similarly, the same group tested the ASO-10-27 in cynomolgus monkeys by intrathecal infusion, which refers to the direct injection of the ASO into the cerebrospinal fluid surrounding the brain and the spinal cord tissue. The administration of the drug achieved putative therapeutic levels of the ASO in various areas of the brain and the spinal cord in vivo (Federici et al., 2012). These preclinical data set a benchmark in using the ASO-10-27 in later human trials.
In 2012, ISIS pharmaceuticals renamed ASO-10-27 to ISIS-SMNRX using their proprietary oligonucleotide modification technology and partnered with Biogen Idec to launch the first-in-human phase I clinical trial. In this single-dose, open-label trial, a total of 28 SMA children were enrolled, including 15 with type II SMA and 13 with type III SMA. The patient received SMNRX as a single-dose treatment of 1, 3, 6, or 9 mg administered intrathecally. The drug was reported to be safe and well-tolerated in children with SMA. Although ISIS-SMNRX is an oligonucleotide-based drug that is subject to nuclease degradation in the body, the drug was heavily modified and showed very long half-life in cerebrospinal fluid, and dosing once every 6–9 months is feasible. These data are consistent with drug dosage estimated from preclinical animal investigations. Moreover, the primary goal of this phase I trial was designed to evaluate the safety and pharmacokinetics of the drug in humans. It is encouraging to note that ISIS-SMNRX significantly improved the patient's muscle function, in terms of the hammersmith functional motor scale expanded, in a dose-dependent manner. The drug is currently being evaluated in a phase Ib/IIa multiple-dose dose-escalation study in patients with SMA (ClinicalTrials.gov Identifier: NCT01839656).
Limitations of Gene Therapy in SMA
Despite having several promising preclinical studies with different vector approaches to treating SMA, there are limitations of the disease that are hindering translation into clinical trials. Multiple reports have shown that there is a therapeutic window for the treatment of SMA during which increased SMN levels have led to survival of motor neurons and improvement in phenotype (Bevan et al., 2011; Lutz et al., 2011; Arnold & Burghes, 2013). The comparatively high SMN protein levels in the spinal cord during development and the apparent onset of SMA during a restricted perinatal period suggest that the requirement for SMN is temporally regulated in the nervous system (Sleigh et al., 2011). Furthermore, SMA mice have a critical period when a sufficient amount of the SMN protein is required during motor neuron development (Nurputra et al., 2013). The delivery of SMN using scAAV9 at different postnatal stages in SMNΔ7 mice demonstrated that, while P10 administration was not effective and P5 administration showed marginal survival extension, P1 administration significantly extended the lifespan of the transgenic mice to over 250 days (Fig. 4) (Foust et al., 2010). Another study showed that, by delivering compounds at different developmental stages (Narver et al., 2008; Butchbach et al., 2010), early but not late embryonic restoration of SMN by two inducible SMN alleles rescued lethality (Hammond et al., 2010; Sleigh et al., 2011). In addition, mouse models of differing severities show similar degrees of motor neuron loss, which indicates that the developmental stage at which degeneration occurs is a key determinant of disease progression (Sleigh et al., 2011). To apply the treatment to patients, it is hence crucial to administer the therapy for SMN replacement at the time during which SMN is required for motor neuron development.

Therapeutic effect after treating SMA mice with scAAV9-SMN at different time points during postnatal development.
As with many neurodegenerative diseases, unless there is a family history and prenatal screening for SMA, the disease will not be diagnosed until a significant amount of neuronal loss and muscle atrophy has occurred. Gene therapy can manipulate the genes but may only halt progression of the disease rather than reverse damage. In this case, the ideal approach would be prenatal gene therapy, to introduce the SMN protein in utero. This brings a whole host of concerns, including worries of germline transmission and developmental aberrations. Prenatal gene therapy remains in its infancy and more research is needed to understand the safety and limitations of this technique before it can be employed for clinical trials. Alternatively, administration of vectors carrying the transgene immediately after birth in cases where prenatal screening has been performed would also be beneficial. The blood–brain barrier is expected to be relatively permeable or “leaky” shortly after birth (Saunders et al., 2012), so the transduction of motor neurons would be efficient.
Since the application of viral vectors to large-scale human therapy in SMA requires the production of large quantities of vector, this leads to the “scale up” problem of therapies that are using vector systems like AAV and lentivirus (Carmo et al., 2008, 2009). However, herpes simplex virus vectors, for example, have been adopted in various clinical trials and have proven relatively easy to produce in large quantities (Federici and Boulis, 2006), and the next challenge for promising vector candidates like AAV and lentivirus would be developing low-cost high-quantity production methods.
Discussion
Even with the tremendous effort that has been carried out to date, we are still at a point where direct correction of disease-causing mutations is not a practical option in humans. All currently available genetic medicine strategies to date are based on halting or slowing the progression of the disease and treating it in a symptomatic fashion only. Alteration of the ultimate phenotype is not yet a reality. There are large societal hurdles, which result in regulatory hurdles that can be overcome, but ultimately slow the development process. However, it may also be that genetic medicines have not yet developed to the extent that the technology is sufficiently robust to “cure” a human monogenic disorder. In addition to developing the technology of genetic medicines and applying them to treating experimental models and humans with hereditary disorders, some regulatory, economic, and sociopolitical issues must also be overcome before genetic medicines can become a reality. There are many social and ethical considerations surrounding gene therapy, especially concerning safety and the permanence of the changes made to a person's genetic code. Many people are concerned at where the line between a disease and an undesirable trait is drawn and whether having the ability to change a trait makes us more intolerant of differences. SMA is no doubt a life-limiting condition that is an ideal target for gene therapy. It would be agreed by the majority that diseases such as this are worthy of the intense intellectual and economic commitment necessary to make treatment a reality, but certain groups may still not be convinced. Time to become more familiar with and educated about the idea of genetic therapeutic strategies may be all that is required to show the true potential of these treatments to the wider public, in a similar fashion to the initial days of in vitro fertilization and organ transplant.
As discussed above, viruses have developed specialized mechanisms of getting DNA into the cell, increasing efficiency, and targeting specific cell types, making them an ideal method for gene delivery. However, there are safety concerns over the use of viral vectors. Some viral vectors, such as retroviruses, have the ability to integrate into the host's genome. This integration happens randomly with the potential to occur in an undesirable location, such as in the middle of a vital gene. As mentioned earlier, in the first gene therapy trials to overcome severe combined immune deficiency (SCID) using gene therapy, 2 of the 10 children later developed leukemia because of the insertion location of the gene (Kohn et al., 2003). Many of these viruses also illicit an immune response, which may cause deleterious side effects through inflammation. To overcome some of these safety concerns, the AAV is being developed as a vector. This virus does not naturally cause disease, elicits a very mild immune response, and does not integrate into the host genome. The target in SMA is the neuron, a nondividing cell; therefore, the lack of integration into the genome would not limit the lifespan of the treatment. Current research looks to be driven toward the further development of AAV vectors for in vivo studies. Human trials to date have shown that with gene transfer to humans it is possible to achieve persistent expression, and to induce phenotypic modifications, at least at the gene expression level. One of the foremost remaining issues is how to achieve expression that is sufficient to correct the clinical phenotype without inducing host defenses that compromise safety, and, for the integrating vectors, how to minimize the risk of insertional mutagenesis, particularly if the corrected cells have a subsequent selective advantage and are continuing to proliferate. As the vectors are further developed, particularly with identification of serotypes and modifications of coat proteins that enhance gene transfer, the doses that are required to gain adequate expression will need to be reduced, with consequent enhancement of safety and efficacy. The use of the very current hybrid vector systems may come to the fore in this respect.
Final Considerations and Conclusions
While there is as yet no cure for SMA, gene therapy is emerging as a very promising potential approach to treating this disease. Over the past two decades, gene therapy has moved from preclinical to clinical studies for many inherited diseases such as cystic fibrosis, acquired disorders such as AIDS, and cancers. To date, 1996 gene therapy clinical trials have been completed, are ongoing, or have been approved in 34 countries worldwide as reported on The Journal of Gene Medicine Gene Therapy Clinical Trials Worldwide website. The first successful clinical trial that brought gene therapy to the forefront of medical research was an attempt to treat a form of SCID, which, like SMA, is a single-gene disorder. This success was subsequently followed by the death of Jesse Gelsinger, who died in a gene therapy clinical trial for ornithine transcarbamylase deficiency in 1999. This tragedy held gene therapy back for many years. Novel developments, though, of both viral and nonviral vectors aiming at high safety and efficiency have renewed the optimism that gene therapy can be used to treat many diseases. A major step forward in making gene therapies available was taken in 2012 when Glybera formally became the first gene therapy to be approved in a regulated market. It has been approved by the European Commission for sale in Europe with hopes that North America will follow in the near future. Glybera is a treatment for patients with lipoprotein lipase deficiency, leading to pancreatitis. This advancement has brought great excitement and renewed hope both to those in the scientific community and to the wider population in general.
SMA is an ideal candidate for gene therapy as it is caused by just one gene, SMN1. There are three main gene therapy approaches to treat SMA: first, by replacing the faulty gene SMN1; second, by introducing oligonucleotides into the cells that will alter the SMN2 gene, resulting in a functional SMN protein product; and, finally, by delivering neuroprotective proteins to degenerating motor neurons. Even though the results of preclinical trials are rather exciting and very promising, we are currently facing several major challenges that will need to be addressed in order to optimize and streamline gene therapy as a viable and practical approach to treating SMA.
Even in terms of monogenic diseases such as SMA, the question of which type of the disease should be focused on to gain the best possibility of a positive therapeutic outcome needs be addressed. Of the four main types of SMA, type I, as mentioned earlier, is the most severe and the most life-limiting. It would seem that type I would be the most appropriate initial target for gene therapy intervention because of this fact and also, as progression of the disease is so rapid, the efficiency of the treatment could be assessed more quickly. Adding weight to this argument is the evidence that type I accounts for between 50% and 70% of all cases of childhood onset SMA (Meldrum et al., 2007). A focus on this type would seem to be the most appropriate in terms of outcome measures. Much of the successful preclinical research that has led to promising results has been carried out in postnatal mice. The administration of therapeutics at the earliest time points has shown to be the most beneficial in terms of long-term improvements. This immediate intervention may be of great importance when treating patients, especially those with type I SMA. This administration regime does, however, open up clinical trials and eventual therapies to a new set of issues. It has been shown that mice injected with AAV at postnatal day 1 have a much higher survival rate than those injected at postnatal day 5. The question then arises, when would a therapeutic strategy need to be initiated to show the most benefit in human? Type I SMA features can be evident from birth to a few months of age. Genotyping for the disease currently takes about 2 weeks, so this may be seen as a rate-limiting factor in the initiation of treatment. For those patients who do not display symptoms for the first few months of life, it is currently unknown what the long-term effects of this delayed diagnosis might be. In cases where both parents know that they are carriers (the chances of which being relatively high as approximately 1/40 people is a carrier), there are prenatal options available. Chorionic villus sampling or an amniocentesis can be carried out from week 11 to 14 and 15 to 18 of gestation, respectively, and can indicate from an early time point if there are any genetic abnormalities, including SMA, present. For these children displaying a positive SMA genotype, the question of when to treat then arises. Would the best outcomes and lowest risk come from in utero treatment where the condition is known early on, or should treatment be used solely after the mother has given birth? At the moment, there is not enough evidence to back either side conclusively, and answers may only reliably come from phase I clinical trials. As a means of achieving the best possible results, the issue of timing is one that will have to be looked at from all levels, and flexibility will be paramount. Early testing and disease confirmation will need to be carried out by the medical staff on the ground, who will then need to relay the information to specialist consultants and research centers carrying out the trials without delay. Good practices in trial recruitment and coordination will go a long way to ensuring that treatment can begin at the earliest possible point so as to maximize the therapeutic benefit and research data that can be obtained.
Considering the route of delivery raises the concern of which site(s) of the patient should be administered with the therapeutic virus. Preclinical testing has shown that intramuscular (Azzouz et al., 2004b; Benkhelifa-Ziyyat et al., 2013), intravenous (Foust et al., 2009; Valori et al., 2010; Dominguez et al., 2011), intracerebroventricular (Coady and Lorson, 2010; Shababi et al., 2011), and intrathecal (Passini et al., 2010) injections are efficient delivery methods that can lead to survival benefits at the correctly administered dosage. However, choosing the appropriate delivery route based solely on the preclinical data is insufficient. While intramuscular injection is comparatively noninvasive, it is not a very practical approach to deliver the viral vectors since all affected skeletal muscles would be necessary to be injected to achieve global motor neuron transduction. This will be especially true should in utero administration prove to be a viable option. Aside from that, as the disease progresses, the reduction of neuromuscular junctions would significantly reduce the uptake of viral particles since the uptake event is only possible with innervated muscles (Goulet et al., 2013; Hamilton and Gillingwater, 2013). Intravenous injection may be a good alternative to intramuscular injection for its noninvasive nature, since the viral vectors could reach every single motor neuron via the systemic route. The drawback is that the potentially low efficiency for the viral vectors to cross the blood–brain barrier would result in the necessity to inject a relatively higher dose of virus to the patient. This could lead to the problem of capsid-specific T cell activation in the liver after vector infusion as it is dose dependent (Mingozzi & High, 2011). Moreover, when the vector is delivered through the bloodstream, it could trigger humoral immunity in the patient, whereby the neutralizing antibodies would destroy the transduced cells via cytolytic granules and cytokines (Mingozzi & High, 2011). Intracerebroventricular or intrathecal infusion would overcome the issue of crossing the blood–brain barrier, but the higher surgical risks and ethics concerns would be the major issues brought along by these routes of administration. Since the virus is directly delivered to the CNS of the patient, a lower dosage of viral vectors would be required when compared with the other injection routes. This leads us to the next question pertinent to the manufacturing of virus for clinical applications. Reducing the dosage required for each patient is desirable since producing a virus for clinical purposes has high demand on the purity of the virus. Producing a virus at good manufacturing practice level is very time-consuming and comes at a high financial cost. Moreover, scaling up the production line, the shelf life of the virus, its stability over the required storage period, and toxicity and/or immune response validation are crucial factors for consideration before each gene therapy could reach the patients. In terms of scaling up virus production, the major issues may not arise in phase I and II clinical trials but rather when a therapy is being marketed on a much larger scale. Practical production bottlenecks will need to be assessed to allow the treatment to be rolled out more easily without compromising the production process in place. Hence, careful considerations must be taken into account in choosing the route of administration and virus manufacturing, as they depend on the type of SMA patients, their age, and health conditions at the time of treatment. AAV immune response is another issue to be considered in gene therapy clinical trials. The modified virus contains a viral capsid, which envelops the transgene to be delivered, and minimizes the viral capsid for the same wild-type virus. The natural exposure to wild-type AAV can potentially trigger humoral immunity against the capsid as early as 2 years of age (Erles et al., 1999; Calcedo et al., 2011; Li et al., 2012; Mingozzi and High, 2013). Therefore, this previous exposure can prompt a specific immune response against AAV, which could compromise the treatment for some patients. While this may not be an issue for early SMA treatment, it will need to be bore in mind for other late-onset diseases in order to elucidate a strategy that will allow the potential AAV humoral activity to be overcome to increase success in the clinical trial setting. Mindful of the fact that almost 70% of population present antibodies against AAV, the best solution would not seem to be to exclude these patients in the treatments, but rather to switch viral serotypes (such as to AAV9) to reduce the propensity for an immune response in patients (Mingozzi et al., 2013). However, very recently reported, the successful treatment with Glybera after direct intramuscular injection is a strong proof of concept indicating that it is possible to bypass systemic and local immune responses while also allowing for successful disease treatment (Ferreira et al., 2014).
The number of gene therapy clinical trials approved worldwide has been increasing dramatically since the last decade of the past century, with U.S.- and European-based trials leading the way. Human research ethics is regulated under the Declaration of Helsinki (1964) and the proceeding enacted reviews. While the majority of countries have signed up to this declaration, each country legislates with specific laws governing their own gene therapy clinical trials. In the European Union, clinical trials involving genetically modified viral vectors are mainly regulated under the Directive 2001/20/EC, which ensures the safety and responsibility for the humans involved in clinical trials, and also the quality and the potential risks for the environment and human health. Glybera, as mentioned, has been the first gene product approved by the European Medicine Agency in 2012 (Yla-Herttuala, 2012). However, this drug is not available yet in the United States. In fact, according to the American Society for Gene and Cell Therapy, there are no gene therapy products currently permitted by the FDA, the agency overseeing the Health and Human Services in the United States. Here, federal laws regulate gene therapy clinical trials helped by a committee of independent experts (Institutional Review Board), who also review and must approve each clinical trial. Along with robust legislation regarding safety and ethics in gene therapy clinical trials, the existence of differences in the regulatory framework between different countries is one of the main issues to be solved in this field. A stronger coordination among different institutions and agencies will be key to allowing the smooth running of gene therapy clinical trials and ultimate therapies between different countries. This increased cooperation will be especially needed for SMA and other rare diseases where patient recruitment will already be a limiting factor.
It is clear that these are exciting and hopeful times in the field of gene therapy. Major recent advances have paved the way for therapeutic strategies to be translated from the bench to the patient, with the increasing number of clinical trials being a strong testament to this. The field of SMA research is in a strong position to learn from currently available clinical trial data and move forward with prospective treatments in the near future. The monogenic nature of the disease makes SMA a good candidate for gene-based therapeutics, and while practical and regulatory issues may temporarily impede clinical success, there is still much to be optimistic about.
Footnotes
Author Disclosure Statement
No competing financial interests exist.