
Abstract
Despite advances in the understanding of its molecular pathophysiology, pancreatic cancer remains largely incurable, highlighting the need for novel therapies. We developed a chimeric antigen receptor (CAR) specific for prostate stem cell antigen (PSCA), a glycoprotein that is overexpressed in pancreatic cancer starting at early stages of malignant transformation. To optimize the CAR design, we used antigen-recognition domains derived from mouse or human antibodies, and intracellular signaling domains containing one or two T cell costimulatory elements, in addition to CD3zeta. Comparing multiple constructs established that the CAR based on human monoclonal antibody Ha1-4.117 had the greatest reactivity in vitro. To further analyze this CAR, we developed a human pancreatic cancer xenograft model and adoptively transferred CAR-engineered T cells into animals with established tumors. CAR-engineered human lymphocytes induced significant antitumor activity, and unlike what has been described for other CARs, a second-generation CAR (containing CD28 cosignaling domain) induced a more potent antitumor effect than a third-generation CAR (containing CD28 and 41BB cosignaling domains). While our results provide evidence to support PSCA as a target antigen for CAR-based immunotherapy of pancreatic cancer, the expression of PSCA on selected normal tissues could be a source of limiting toxicity.
Introduction
P
The lack of curative treatment options for patients with advanced disease has prompted researchers to assay diverse experimental approaches. Several active immunotherapy clinical trials have been conducted, including vaccination with peptides derived from tumor-associated antigens or with peptide-loaded dendritic cells, but these trials failed to provide evidence of clinical responses (Koido et al., 2011). In order to expand the repertoire of molecular targets for immunotherapy of pancreatic cancer, we generated and characterized a set of chimeric antigen receptors (CARs) directed to prostate stem cell antigen (PSCA). PSCA is a 123 amino acid glycophosphatidylinositol-anchored surface glycoprotein of unknown function (Saeki et al., 2010) initially described to be highly expressed in prostate tumors, with low basal expression in prostate epithelium, urinary bladder, kidney, esophagus, stomach, and placenta (Gu et al., 2000). Later studies demonstrated that it was overexpressed in a variety of human malignancies, including pancreatic cancer, but remained undetectable in healthy pancreas (Argani et al., 2001). A recently published randomized phase II clinical trial showed that administration of a PSCA-specific antibody in combination with gemcitabine improved the 6-month survival rate over gemcitabine alone, though there was not a significant difference in progression-free or overall survival between the two groups (Wolpin et al., 2013).
Herein we report the development and characterization of potent anti-PSCA CARs entirely derived from molecules of human origin. We further demonstrated that these human antibody-based CARs had superior surface expression and increased reactivity than a mouse antibody-derived counterpart, and elicited significant in vivo antitumor activity in a humanized mouse model of pancreatic cancer.
Materials and Methods
Gene expression analysis
Commercial RNA from normal pancreas and pancreatic ductal adenocarcinoma were purchased from Origene (CR560779, CR560781, CR560929, CR56131, CR561392, CR561533, CR560122, and CR560156; Rockville, MD). Normal tissue and tumor cDNA arrays were purchased from Clontech (MTC panels I and II; Mountain View, CA) and Origene, respectively. PSCA and mesothelin (MSLN) mRNA expression was analyzed using TaqMan primer/probe sets (Applied Biosystems, Foster City, CA). Total RNA was isolated using RNeasy Mini Kit (Qiagen, Germantown, MD) and cDNA was synthesized using SuperScript III First-Strand Synthesis SuperMix for quantitative reverse transcription polymerase chain reaction (qRT-PCR; Invitrogen, Carlsbad, CA) following the manufacturer's instructions. PSCA and MSLN mRNA expression was analyzed using TaqMan primer/probe sets Hs00245879_m1 and Hs04177224_g1, respectively (Applied Biosystems). Normal tissue and tumor cDNA arrays were purchased from Clontech (MTC panels I and II) and Origene, respectively.
A custom-designed PSCA CAR-specific TaqMan primer/probe set was used for the analysis of copies of transgene in persisting splenocytes: forward primer, CACCGTGACCGTGTCCTC; reverse primer, CTCTGGGTCAGCTGGATGTC; probe, CCGCTGCCTCCACCGC. Human PSCA (Thermo Fisher Scientific, Inc., Waltham, MA) and MSLN (Clontech SC110135) cDNA clones were utilized for the generation of standard curves.
Cell lines and primary human lymphocytes
LNCaP, DU145, HPAC, NorP1, Panc 02.03, Panc 02.13, and T3M4 cell lines were purchased from American Type Culture Collection (Manassas, VA), and cultured as instructed. Primary lymphocytes from healthy donors were cultured in AIM-V medium (Invitrogen) as described (Zhao et al., 2009).
Retroviral constructs and analysis
Ha1-4.117-based single-chain fraction variable (scFv) cDNA was derived from a human hybridoma producing a PSCA-specific antibody (Gudas, 2012). DNA was synthesized by BlueHeron (Bothell, WA), using an optimization algorithm for codon usage in humans, and cloned into NcoI and XhoI sites of pMSGV1-28Z and pMSGV1-28-BBZ vectors. A CD28-containing second-generation CAR against MSLN was generated based on previously published sequences (Li et al., 2004; Carpenito et al., 2009) and cloned into MSGV1 retroviral vector. Evaluation of interferon-gamma (IFNg) secretion by CAR-transduced T cells was performed as described. Flow cytometry analyses were performed using a FACSCanto II flow cytometer with FACSDiva software (BD Biosciences, San Jose, CA), and analyzed using FlowJo software (Tree Star, Ashland, OR). CARs were stained with biotinylated Protein-L (GenScript, Piscataway, NJ) and phycoerythrin (PE)-conjugated streptavidin (BD Bioscience), as previously described (Zheng et al., 2012). Antihuman CD3 (FITC- or APC-H7-conjugated, clone SK7) was purchased from BD Biosciences. Unless otherwise stated, peripheral blood lymphocytes (PBL) were transduced with 1:2 dilution of viral supernatant of bm2B3-28BBZ CAR or 1:4 dilution of viral supernatant of Ha1-4.117-based CARs.
Mouse model
For the mouse adoptive cell transfer (ACT) experiments, naïve CD8+lymphocytes were isolated from cryopreserved apheresis samples using the naïve CD8+T cell isolation kit (human; Miltenyi Biotec, Auburn, CA), following manufacturer's instructions. Briefly, 5×108 peripheral blood mononuclear cells were first depleted of non-naïve cells and subsequently enriched for CD8+ cells by positive selection. Lymphocytes were stimulated with anti-CD3/-CD28 beads (Dynabeads; Invitrogen) and transduced on days 2 and 3 poststimulation. Four- to six-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) female mice (The Jackson Laboratory, Bar Harbor, ME) were used for adoptive cell transfer experiments. All procedures were approved by National Cancer Institute animal care and use committee. Briefly, each mouse received a subcutaneous injection of 2×106 HPAC cells resuspended in 100 μl of 1×HBSS with 1% Matrigel. When tumors became palpable, at approximately day 15, mice were tagged and randomized into five groups, and received the indicated treatments. Human PBLs, resuspended in 500 μl of HBSS, were injected intravenously into tumor-bearing mice. In both experiments, the number of CAR-transduced CD8 T cells administered to each mouse was equalized based on flow cytometry analysis of CAR expression. In one of the experiments, mice received three daily intraperitoneal administrations of recombinant human IL-2 (Proleukin; Prometheus, San Diego, CA) at 220,000 IU in 500 μl of phosphate buffered saline (PBS). Tumor growth was monitored three times per week by a blinded observer. At the end of the experiment, spleens were harvested and filtered through a 70 μm nylon strainer. The resulting single-cell suspension was incubated in ACK Lysing Buffer (Biosource, Rockville, MD) and resuspended in PBS.
Immunohistochemistry
Excised tumors were embedded in OCT medium (Sakura Finetek USA, Inc., Torrance, CA) and frozen. Cryosections were prepared by American Histolabs Inc. (Gaithersburg, MD) and fixed with acetone before staining. PSCA staining was performed using a peroxidase-based kit as instructed by manufacturer (EnVision+System-HRP [DAB]; DAKO, Carpinteria, CA). Monoclonal mouse antihuman PSCA antibody 1G8 was kindly provided by Dr. Robert E. Reiter, University of California–Los Angeles. For fibroblast activation protein (FAP) staining, FAP-specific antibody FAP5 (Ostermann et al., 2008) or mouse IgG2a isotype control antibodies were biotinylated (EZ-Link NHS PEG4-biotin; Pierce, Rockford, IL) and used at approximately 2 μg/ml. Sections were developed with ABC reagent (VectorLabs, Burlingame, CA) and DAB substrate (Dako). FAP5 antibody was kindly provided by Pilar Garin-Chesa (Boehringer Ingelheim, Vienna, Austria). Samples were counterstained with a 50% Mayer's hematoxylin–50% Harris hematoxylin solution (Sigma-Aldrich, St. Louis, MO). Images were acquired using an Eclipse E400 microscope (Nikon Instruments Inc, Melville, NY) coupled to a Nuance Multispectral Imaging System VIS camera (PerkinElmer, Waltham, MA).
Statistical analysis
Comparisons between groups were performed using Mann–Whitney's U-test. p-Values were calculated using Prism 5.04 (GraphPad Software). Tumor growth statistics were calculated using the Wilcoxon rank-sum test, based on linear slopes of the tumor growth curves at each data point. p-Values of 0.05 or lower were considered statistically significant.
Results
PSCA mRNA expression in tumors, pancreatic cancer cell lines, and normal tissue
To evaluate the pattern of expression of PSCA in normal and malignant tissues, we characterized the levels of mRNA expression using qRT-PCR. First, we evaluated PSCA expression in a cDNA array derived from 437 tumor samples of diverse histologies. With a threshold of 1000 copies of PSCA per 106 copies of beta-actin (ACTB) mRNA defined as the criterion for positivity, qPCR analysis showed that the majority of specimens of pancreatic, prostatic, and urinary bladder tumors were positive for PSCA, similar to previous reports (Gu et al., 2000; Argani et al., 2001; Elsamman et al., 2006) (Fig. 1a). Interestingly, unlike a previous report (Bahrenberg et al., 2000), our results indicate that esophageal and gastric tumors were also positive for PSCA, with median expression levels comparable to those of prostate and pancreatic cancer (Fig. 1a). Moreover, positive expression of PSCA was observed in cervical tumors (Fig. 1a).

PSCA expression in tumor and normal tissues.
We next analyzed RNA samples from 40 cell lines derived from pancreatic adenocarcinomas. As shown in Fig. 1b, 18 out of 40 cell lines demonstrated PSCA mRNA levels greater than 1000 copies per million copies of ACTB, with SKPC-1, HPAC, Panc 02.03, and Panc 8.13 having greater than 10,000 copies of PSCA per million copies of ACTB.
When we analyzed a panel of cDNA from 15 normal tissues, we found that normal prostate and placenta tissues expressed high levels of PSCA mRNA, while normal lung, kidney, and pancreas expressed a much lower amount of the transcript. PSCA expression in other normal tissues remained below 1000 copies per million copies of ACTB (Fig. 1c). These results suggest that PSCA has restricted expression in normal tissues and is highly overexpressed in a variety of human malignancies, including pancreatic cancer.
Superior antitumor activity of an anti-PSCA CAR derived from human antibody Ha1-4.117
We sought to develop a fully human anti-PSCA CAR in order to circumvent potential antimouse immune responses shown to arise in trials using mouse antibody-based CARs (Kershaw et al., 2006; Lamers et al., 2006). We designed a CAR containing the antigen-recognition domain from Ha1-4.117, a human antibody described to be highly specific in its recognition of PSCA (Gudas, 2012). Two versions of this CAR were generated; a second-generation CAR containing costimulatory molecule CD28, linked to the zeta-chain of CD3, and a third-generation design with CD28 and 4-1BB costimulatory domains linked to CD3-zeta (Fig. 2a). In addition, we generated an anti-PSCA CAR derived from humanized mouse antibody bm2b3 (Fig. 2a) (Leyton et al., 2008, 2009). These CAR sequences were inserted into retroviral vectors and used to transduce human PBL. As shown in Fig. 2b, Ha1-4.117-derived CARs were more efficiently expressed in human T cells than the bm2b3-derived CAR, and more abundantly than a control anti-Her2/neu CAR (Fig. 2b).
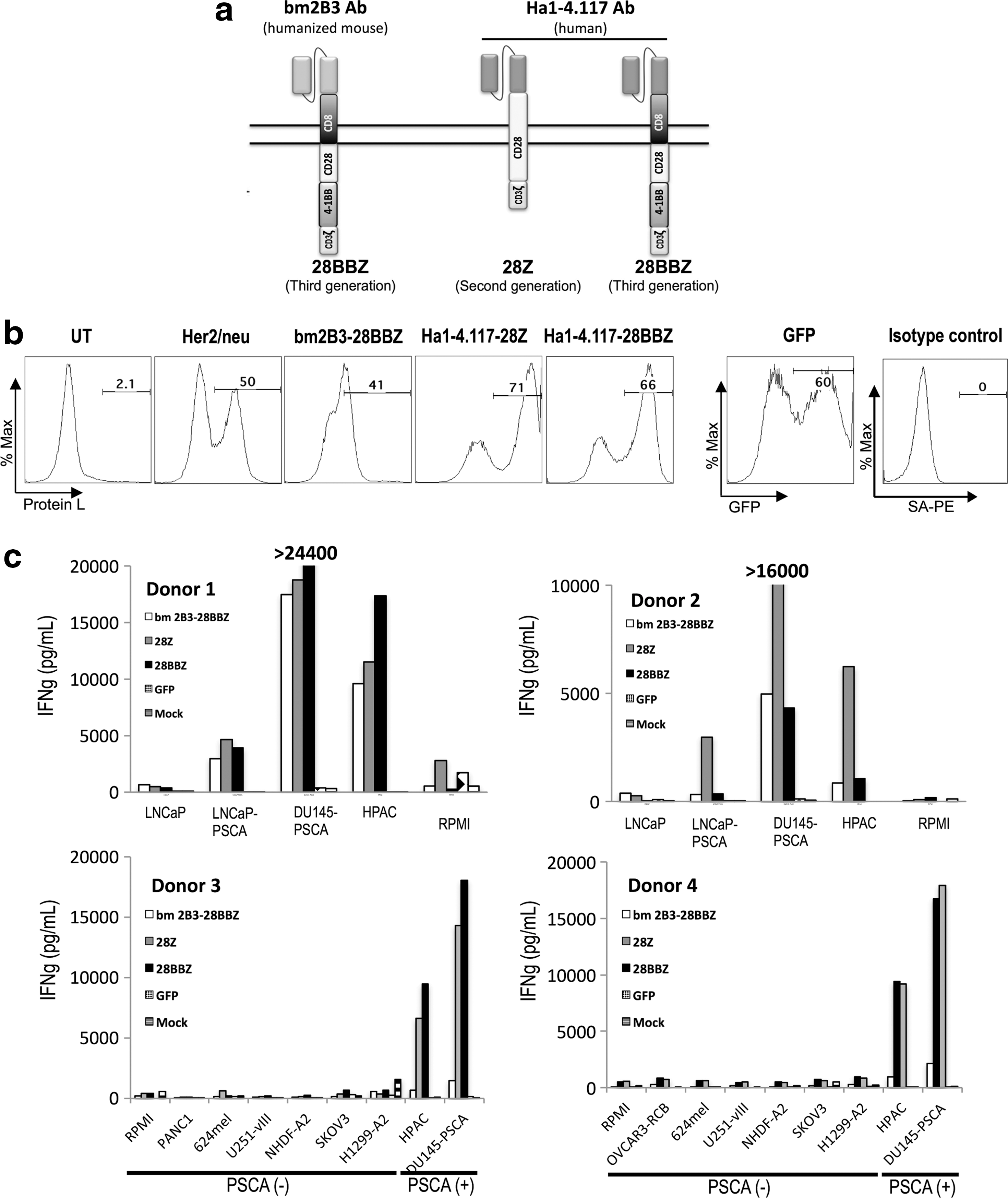
Human monoclonal antibody-based anti-PSCA CARs demonstrate superior expression and reactivity.
Specific recognition of PSCA-expressing target cells was tested in coculture experiments (Fig. 2c). Antigen-specific IFNg release was observed for PSCA-expressing prostate cancer lines LNCaP-PSCA and DU145-PSCA, but not PSCA-negative LNCaP. Similarly, strong recognition of PSCA-expressing pancreatic cancer line HPAC was observed, whereas PSCA-negative lines PANC-1, 624mel, U251-vIII, NHDF-A2, SKOV3, and H1299-A2 were not recognized by PSCA CAR-transduced lymphocytes. These results demonstrated that the potent reactivity conferred by these CARs is highly antigen-specific (Fig. 2c). In four of four donors evaluated, the Ha1-4.117-based CARs induced stronger release of IFNg than the murine bm2b3-derived counterpart.
PSCA CARs induce greater reactivity against pancreatic cancer cell lines than MSLN CARs
MSLN is overexpressed in pancreatic cancer and mesothelioma cells and is currently being tested as a target in a CAR-based immunotherapy trial for pancreatic cancer (NCT01583686;

PSCA and MSLN as targets for pancreatic cancer immunotherapy.
We then compared the transduction efficiency of the second- and third-generation Ha1-4.117-derived anti-PSCA CARs to the anti-MSLN CAR currently used in clinical trial (NCT01583686;
PSCA CAR-transduced human CD8+T cells induce potent antitumor effects in vivo
Finally, we sought to evaluate whether human lymphocytes expressing PSCA-specific CARs could mediate an antitumor response in vivo. To that end, we established a murine model of adoptive cell transfer consisting of immunodeficient NSG mice engrafted with established HPAC pancreatic cancer cells followed by intravenous infusion of CAR-engineered human CD8+ cells.
Two experiments were performed following the design shown in Supplementary Fig. S1 (Supplementary Data are available online at

PSCA CARs effectively treat established tumors in NSG mice.
At the end of the experiments, splenocytes were isolated and screened for the presence of human CD3+ cells by flow cytometry. Human lymphocytes were observed in all groups except for mice treated with vehicle alone (Supplementary Fig. S2). Persistence of gene-modified lymphocytes was analyzed by PCR for the integrated proviral DNA in splenocytes. In both experiments, the groups treated with the third-generation CAR had a significantly higher number of CAR-transgene DNA than the groups treated with the second-generation CAR. The mean numbers of integrated copies per 106 copies of ACTB in the PSCA-28BBZ and PSCA-28Z groups were 2379 versus 198 (p=0.029) in the first experiment, and 23,104 versus 7307 (p=0.0079) in the second experiment (Fig. 4b). A similar trend was observed for the number of integrated CAR cDNA in lymph node-derived T cells (Supplementary Fig. S3). These results suggest that, in this model, a higher persistence of CAR-transduced cells does not correlate with better antitumor response.
Residual tumor masses from experiment 1 were subject to immunohistochemistry. A reduction of PSCA expression was observed in the CAR-transduced T cell-treated groups, but not in control groups, supporting the notion of a target-specific antitumor effect (Fig. 5a). We also stained residual tumor masses for FAP, a protein expressed in tumor-associated stromal cells (Ostermann et al., 2008). PSCA staining and FAP staining were mutually exclusive (Supplementary Fig. S4), and while FAP expression was restricted to stromal bands in untreated tumors, the vast majority of cells remaining in treated tumor masses stained positive for FAP (Fig. 5b), suggesting that the treated residual masses were comprised predominantly of stromal cells.

Immunohistochemistry staining of PSCA CAR-treated tumors.
Discussion
Recent advances in the understanding of the molecular events underlying pancreatic cancer pathophysiology have permitted the identification of genetic alterations associated with specific stages of malignant transformation. In a proposed linear progression model, PSCA upregulation is an early event, detected in premalignant pancreatic intraepithelial neoplasias (PanIN), while MSLN upregulation occurs later and is present only in fully transformed cells that have acquired metastatic potential (Feldmann et al., 2007). Thus, elimination of MSLN-expressing cells may result in reduction of tumor burden but might fail to eliminate PanINs, whereas targeting PSCA-expressing cells might also eliminate premalignant neoplastic cells, thus reducing the likelihood of relapse. Furthermore, we demonstrated that anti-PSCA CARs have superior surface expression and induce higher reactivity than an anti-MSLN CAR currently being tested in a clinical trial, suggesting that PSCA-targeted adoptive cell transfer therapies may be more efficient than MSLN-targeted adoptive cell transfer therapies in inducing antitumor responses in pancreatic cancer patients.
Several CARs have been tested in vitro and/or in animal models as potential immunotherapy agents for pancreatic cancer, targeting CEA (Chmielewski et al., 2012), EGFR (Zhou et al., 2013), CD24, or Her2/neu (Maliar et al., 2012). A report by Morgenroth et al. (2007) described a first-generation CAR against PSCA based on mouse monoclonal antibody 7F5. This design was shown to induce in vitro cytotoxicity in a mouse CTL cell line when exposed to PSCA-expressing targets as a model of prostate cancer, but no data on human primary T cells was reported (Morgenroth et al., 2007). More recently, by using a 2b3-based first-generation CAR, Katari et al. (2011) showed that CAR-transduced human T cells were able to recognize and lyse PSCA-expressing cells, but did not show any evidence of in vivo antitumor effect. The same group showed later, using a CAPAN1 intraperitoneal xenograft model in SCID mice, that intraperitoneal administration of T cells expressing the 2b3-derived CAR resulted in selection of PSCA tumor cells in vivo but failed to control tumor growth (Anurathapan et al., 2014).
In the present study, we describe the development of anti-PSCA CARs based on the fully human antibody Ha1-4.117. We demonstrate that Ha1-4.117-based CARs presented superior surface expression in human T cells and conferred higher target recognition capabilities than a CAR based on bm2b3 (a variant of 2b3 with enhanced affinity) (Leyton et al., 2009), without compromising antigen specificity. This design is expected to prevent the development of an immune response directed to the remaining murine immunoglobulin regions present in 2b3-based CARs. The majority of CARs utilized in current clinical trials contain scFv fragments derived from murine monoclonal antibodies, which may lead to the induction of humoral immune responses in the host as a reaction to the xenogeneic proteins. For instance, the development of anti-CAR immune responses in metastatic renal cell cancer patients treated with a murine anti-CAIX CAR was analyzed in detail, and it was postulated that T cell function and persistence may have been negatively impacted by this effect (Lamers et al., 2011). Most concerning, it was recently reported that repeated administration of murine CAR-engineered T cells led to development of antimouse antibody responses, associated with anaphylaxis and cardiac arrest in one subject (Maus et al., 2013). The use of a human antibody such as Ha1-4.117 instead of mouse antibodies such as 2b3 would prevent this type of reactions.
To our knowledge, this is the first report of an in vivo antitumor response using PSCA-targeted intravenous ACT approaches against established pancreatic tumors. This antitumor effect was not limited to a delay in tumor growth, but involved tumor shrinkage with replacement by stromal cells or even complete regression of two tumors when IL-2 was administered following ACT. After the submission of this article, Hillerdal et al. (2014) reported the use of a 1G8-based CAR against xenografts derived from melanoma cells engineered to express PSCA, as a model for prostate cancer. In their experimental setting, mice received three intravenous infusions of CAR-transduced cells, starting 1 day after subcutaneous injection of the tumor cells. The other two doses were administered 7 and 14 days later. This treatment resulted in a slight delay in tumor implantation in the CAR-treated group, but all tumors grew exponentially after the last infusion of CAR-T cells (Hillerdal et al., 2014). In contrast, our results showed that a single administration of human T cells transduced with a second-generation Ha1-4.117-based CAR could mediate growth delay and regression of established tumors.
Our results indicate that third-generation CARs containing a 4-1BB signaling domain in addition to a CD28 moiety may endow human T cells with a survival advantage over those expressing second-generation CARs containing only the CD28 costimulatory domain. This advantage, however, did not correlate with an enhanced antitumor effect. A similar phenomenon was described by Hombach et al. (2013), who described that cytokine-induced killer (CIK) cells expressing a second-generation CAR containing CD28-CD3zeta domains presented superior antitumor efficacy than CIK expressing a third-generation CD28-Ox40-CD3zeta CAR. In that setting, the detrimental effect of the additional costimulatory domain was attributed to increased activation-induced cell death in CIK cells expressing the third-generation CAR (Hombach et al., 2013). On the other hand, other authors postulate that third-generation CARs containing both CD28 and 4-1BB outperform second-generation CARs containing only CD28, as a general rule (Tammana et al., 2010; Zhong et al., 2010). The discrepancy between our results and previously published reports imply that the optimal CAR design might need to be empirically determined for each individual target. Interestingly, in our current clinical trials, we have not observed any correlation between transgene persistence in blood and clinical response (Robbins et al., 2011; Morgan et al., 2013).
Our results on PSCA mRNA expression in some normal tissues, together with a recent report showing PSCA expression in normal pancreas islet cells (Ono et al., 2012), are a concern for potential on-target/off-tumor toxicity, as has been observed in other CAR trials. On-target/off-tumor toxicity has been documented in patients treated for renal cell carcinoma (targeting CAIX) (Lamers et al., 2006), colorectal cancer (targeting Her2/neu) (Morgan et al., 2010), and B cell malignancies (targeting CD19) (Brentjens et al., 2010; Kochenderfer et al., 2012). Nevertheless, given the paucity of truly tumor-specific antigens to target, the outstanding target-specific reactivity conferred by our CARs suggests that PSCA deserves further consideration as a clinically relevant antitumor target in pancreatic cancer patients with otherwise no treatment options. Strategies involving the use of complementary CARs (Kloss et al., 2013) or suicide gene safety switches (Di Stasi et al., 2011) may be devised to capitalize on the potent target recognition of this CAR, avoiding the toxicity related to recognition of normal tissues.
Footnotes
Acknowledgments
We would like to thank Arnold Mixon, Shawn Farid, and Dave Jones for technical support. This project was supported in part by a generous contribution from the Milstein Family Foundation and by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, Bethesda, MD.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.