
Abstract
The interaction of human adenovirus (HAdV)-C5 and many other adenoviruses with blood coagulation factors (e.g., human factor X, FX) involves the binding of their GLA domain to the hexon capsomers, resulting in high levels of hepatotropism and potential hepatotoxicity. In this study, we tested the possibility of preventing these undesirable effects by using a GLA-mimicking peptide as a competitor. An FX GLA domain-derived, 40-mer polypeptide carrying 12 carboxyglutamate residues was synthesized (GLAmim). Surface plasmon resistance (SPR) analysis showed that GLAmim reacted with free and capsid-embedded hexon with a nanomolar affinity. Unexpectedly, GLAmim failed to compete with FX for hexon binding, and instead significantly increased the formation of FX–hexon or FX–adenovirion complexes. This observation was confirmed by in vitro cell transduction experiments using HAdV-C5-Luciferase vector (HAdV5-Luc), as preincubation of HAdV5-Luc with GLAmim before FX addition resulted in a higher transgene expression compared with FX alone. HAdV-C5 virions complexed with GLAmim were analyzed by cryoelectron microscopy. Image reconstruction demonstrated the bona fide hexon–GLAmim interaction, as for the full-length FX, although with considerable differences in stoichiometry and relative location on the hexon capsomer. Three extra densities were found at the periphery of each hexon, whereas one single FX molecule occupied the central cavity of the hexon trimeric capsomer. A refined analysis indicated that each extra density is found at the expected location of one highly variable loop 1 of the hexon, involved in scavenger receptor recognition. HAdV5-Luc complexed with a bifunctional GLAmimRGD peptide showed a lesser hepatotropism, compared with control HAdV5-Luc alone, and efficiently targeted αβ-integrin-overexpressing tumor cells in an in vivo mouse tumor model. Collectively, our findings open new perspectives in the design of adenoviral vectors for biotherapy.
Introduction
T
Adenoviruses are the most used vectors in human gene therapy trials. Since they are nonintegrative viruses, ex vivo strategies of re-engineering target cells are excluded, and most of the gene therapy protocols require in situ injection or systemic injection of therapeutic vectors in the bloodstream. This latter way of administration suffers from several drawbacks, in particular a high liver uptake of the vector, and consequently a poor availability for target cells or tissues. Alternative strategies have been proposed to overcome this hurdle, including the design of vector mutants or chimeras, but the results have been somewhat disappointing [reviewed in (Coughlan et al., 2010)].
Recent breakthrough in the understanding of HAdV-C5–host interactions has shown that the vector particle accumulation in the liver is the result of HAdV-C5 binding to human blood coagulation factor X (FX) via the hexon capsomers, followed by the interaction of the HAdV-C5–FX complexes to heparan sulfate proteoglycan (HSPG) molecules that are present in a high concentration at the surface of the Kupffer cells (Vigant et al., 2008; Waddington et al., 2008; Alba et al., 2010). To overcome this limitation, different strategies have been envisaged: (1) the use of HAdV serotypes (or HAdV-C5-based chimeric vectors), which are naturally (or artificially) nonbinders of blood coagulation factors (Waddington et al., 2008); (2) mutations in the wild-type HAdV-C5 hexon to prevent FX binding (Bradshaw et al., 2012; Doronin et al., 2012); and (3) the design of molecules that compete with FX for hexon binding (Khare et al., 2012; Duffy et al., 2013). Although relatively less studied, the latter strategy has recently regained some interest, after the observation that FX can shield HAdV-C5 from the attack by the innate immune system (Xu et al., 2013).
In this study, we investigated the effects of using a synthetic peptide (the GLA domain of human FX consisting of a 40-mer polypeptide biotinylated at its C-terminus [GLAmim]) mimicking the FX-GLA domain as a competitor to FX during HAdV-C5-mediated cell transduction. We found that although GLAmim was capable of binding to the hexon protein with a high affinity (in the nanomolar range), it was not able to compete with FX for hexon binding. Surface plasmon resistance (SPR) and cell transduction experiments suggested that FX and the GLAmim peptide bound to separate sites on the hexon capsomer. Three-dimensional reconstruction of cryoelectron microscopy images of GLAmim–adenovirion complexes taken at high resolution demonstrated that a GLAmim-induced extra density is at the periphery of the hexon molecule, whereas full-length FX has been shown to bind to its central cavity. The extra density location coincided with highly variable loop 1 (HVR-1), a highly accessible and variable loop of the hexon capsomer, which was recently reported to be involved in scavenger receptor recognition (Khare et al., 2012). Using an in vivo mouse tumor model, we found that the systemic administration of HAdV5-Luc in complex with a bifunctional GLAmimRGD peptide resulted in a modest but significant liver detargeting, but efficient vector targeting to αβ-integrin-overexpressing tumor cells. Our results with the FX-derived GLAmim peptide and a bivalent targeting peptide including GLAmim open the path to new perspectives and new strategies in the design of rational adenoviral vectors for biotherapy.
Materials and Methods
Cells and virus
HeLa cells (European Cell Culture Collection) were cultured in Dulbecco's modified essential medium (DMEM) supplemented with 10% fetal calf serum, 2 mM L-glutamine (Glu), and 100 U/ml penicillin and 100 μg/ml streptomycin. HAdV5Luc, a replicative HAdV-C5 vector carrying the luciferase gene driven by CMV promoter inserted in the E3 region (Mittal et al., 1993), was used in all experiments. For luciferase quantification, 48-well plates (Falcon) were seeded with 2×104 cells, 1 day before infection. Wells were infected at 100 vp/cell in 200 μl of medium without serum for 1 hr. Five hours postinfection, cell lysate was recovered in 100 μl cell lysis buffer (Promega) and luciferase activity was quantified using the Luciferase assay system (Promega) on a Victor2 (Wallac) luminometer. Results were given as the mean of triplicates with standard deviation. For experiments with FX, virus samples were preincubated for 30 min at room temperature with either 8 or 0.08 μg/ml of human FX (Cryopep) directly or after a previous preincubation with 10 μg/ml of GLAmim peptide.
GLAmim peptide
The GLAmim was synthesized by GENPEP and purified by high-performance liquid chromatography. GLAmim contained 12 γ-carboxyglutamate residues at their exact positions in human FX. A biotin tag (GLAmim), or the tripeptide sequence RGD (GLAmim/RGD) was added at the C-terminus with a free NH2 group. The GLAmim sequence is depicted in Fig. 1A.
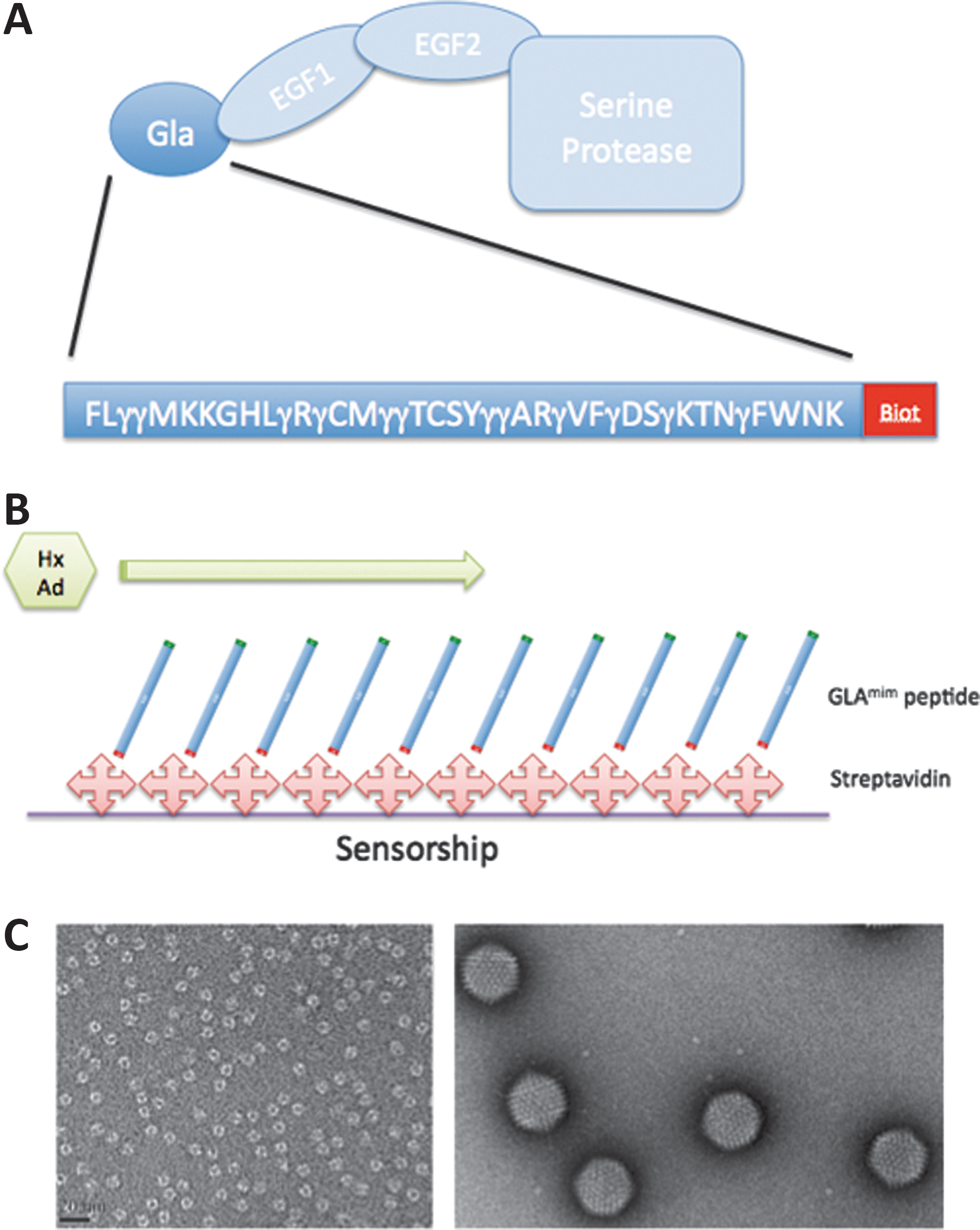
Presentation of the partners.
Isolation of HAdV-C5 virions and hexon capsomers
HAdV-C5 virions were recovered from HAdV-C5-infected HeLa cells, and purified by a conventional method including a double cycle of CsCl banding (Franqueville et al., 2008). The native, trimeric hexon protein was recovered from the cell lysate and purified by a three-step procedure, as previously described (Boulanger and Puvion, 1973; Molinier-Frenkel et al., 2000, 2002). Purification was checked by conventional sodium dodecyl sulfate polyacrylamide gel electrophoresis and Coomassie blue staining, and Western blot analysis using rabbit antibodies against antihexon or anti-HAdV-C5 whole virion, as previously described (Franqueville et al., 2008; Corjon et al., 2011).
Negative-staining and transmission electron microscopy
HAdV-C5 virions and hexon capsomers were quality controlled by negative-staining and transmission electron microscopy (TEM). The standard mica-carbon preparation was used with the protein at 0.1 mg/ml or virion diluted 10 times after CsCl purification. Samples were stained by uranyl acetate and observed under a Phillips CM12 electron microscope at 120 kV.
SPR experiments
All SPR experiments were performed in a running buffer composed of HBS buffer (GE Healthcare) supplemented by 2 mM CaCl2 (HBS-Ca) at a flow rate of 5 μl/min on a BIAcore 3000 instrument (GE Healthcare). Surface immobilization was done by the standard EDC-NHS activation (GE Healthcare) for 10 min followed by injection of the ligand either streptavidin or human FX at 1 μg/ml in 10 mM acetate buffer pH 4.5 for 10 min (4,100 and 4,200 RU, respectively). Blocking was done by a 10 min inactivation with 1 M ethanolamine. For GLA experiments, biotinylated GLAmim peptide was injected at 10 μg/ml for 10 min in HBS-Ca (1,080 RU). When FX was used as the ligand (4,200 RU), the negative controls for background subtraction consisted of EDC-NHS inactivated flow cell. When GLAmim peptide was used as the ligand, negative controls used streptavidin-coated flowcell. In all experiments (done in triplicate), surface regeneration was done by a two-time injection of 10 mM HCl for 2 min. Between two injections, there was a stabilization period in HBS-Ca for 15 min.
Cryo-EM
HAdV-C5 virions were incubated in 10 mM Hepes buffer pH 7.4 supplemented with 2 mM CaCl2 and a 10-fold molar excess of GLAmim peptide. After 1 hr incubation, the excess peptide was eliminated by dialysis. Four microliters of the sample (either HAdV-C5 or HAdV-C5–GLAmim) was loaded onto a Quantifoil R2/1 holey grid (Quantifoil Micro Tools GmbH), vitrified using a Mark IV vitrobot (FEI). The frozen grid was transferred into a Polara electron microscope working at 300 kV. The images were taken under low-dose conditions (<20 e−/Å2) and with a nominal magnification of 31,000 on KODAK SO-163 films. The negatives were developed in full-strength D19 developer for 12 min.
Image analysis
Negatives were screened for astigmatism and drift by optical diffraction, and only those (33 negatives in total for HAdV-C5–GLAmim, 30 for HAdV-C5 alone) showing information up to 7 Å were digitized using a Photoscan TD scanner with a pixel size of 7 mm (2.25 Å per pixel at the specimen level). The particles were selected interactively using x3d (Conway and Steven, 1999) boxed into 447×447 pixel squares and corrected for the contrast transfer function effect as previously described (CTFMIX) (Fabry et al., 2005). Particle origin and orientation were determined with the model-based PFT2 programs using a previously determined and scaled to the right size 3D structure of HAdV-C5 as a starting model (Fabry et al., 2005). The final reconstructions were calculated using EM3DR2 (Fuller et al., 1996) with a total of 4,762 particles (about 66% of the total) for HAdV-C5–GLAmim and 3,648 particles (about 66% of the total) for HAdV-C5. The resolution of the final maps was estimated to be about 9 Å by Fourier shell correlation (not shown).
Animal tumor model
All animal experiments were conducted in agreement with the Principles of Laboratory Animal Care (NIH Publication No. 86-23, revised 1985). HEKb3 is a cell line derived from the original HEK-293 cell line (human embryonic kidney cells) stably expressing the β3 integrin subunit. HEKβ3 cells were cultured at 37°C in a humidified 95% air–5% CO2 atmosphere in DMEM supplemented with 10% fetal bovine serum. HEKβ3 were harvested, washed, and resuspended in phosphate buffered saline at 107 cells/200 μl for subcutaneous implantation in the right posterior limb of anesthetized (isoflurane) NMRI nude mice (female, 6 weeks old).
In vivo bioluminescence imaging
Subcutaneous HEKβ3 tumor growth was followed by caliper measurements once a week. When tumor size reached about 500 mm3 (about 8 weeks), the mice were randomized and divided into three groups. Aliquots of HAd5-Luc vector (4×1010 vp/mouse) alone or in combination with GLAmim or GLAmimRGD peptide (10 μg/ml in 10 mM Hepes buffer pH 7.4–2 mM CaCl2) were injected (100 μl) in the tail vein after a 30 min incubation at room temperature. The theoretical number of peptide binding sites is about 3×1013 for 4×1010 vp. Considering that the GLAmim or GLAmimRGD peptides have a molecular mass around 6 kDa, the dose of 1 μg per injection corresponded to 1×1014 molecules, equivalent to a threefold excess over the theoretical binding sites on the vector.
Luciferase expression was monitored by in vivo bioluminescence 24, 48, and 72 hr after the intravenous injection. Vigil mice were injected intraperitoneally with 150 mg/kg of D-Luciferin (Promega) just before anesthesia (isoflurane 4% for induction and 1.5% thereafter). Five minutes after luciferin administration, bioluminescence images were acquired with an IVIS KINETIC (Perkin Elmer). Quantitative analysis was carried out using Living image software (Perkin Elmer). The results were expressed as the number of photons per second (ph/sec).
Results
GLAmim peptides, HAdV-C5 virions, and hexon capsomers
The sequence of the polypeptide selected to mimic GLAmim corresponding to amino acid residues 4–43 of the human FX amino acid sequence (Fig. 1A). The rationale for the design of a GLAmim polypeptide lacking the N-terminal tripeptide A-N-S was to use a minimal GLA domain including the 12 conserved γ-carboxyglutamate residues common to 2 hexon binders, the human FIX and FX. The N-terminal A-N-S tripeptide is not conserved in FIX and FX, and is replaced by Y-N-S-G in FIX. By contrast, the 12 γ-carboxyglutamate residues are strictly conserved in human FX and FIX, and conserved to more than 75% in other human GLA domain-containing proteins, such as FVII, prothrombin, protein C, and protein S (Sunnerhagen et al., 1995).
Two different polypeptides were synthesized: (1) The princeps molecule described above (biotinylated GLAmim) used in SPR studies, so that the GLAmim molecules will react with surface-coated streptavidin with the desired orientation for maximum accessibility (Fig. 1B). (2) For cell targeting assays with a dual-specificity peptide (viral and cellular), the tripeptide RGD was added to the C-terminus in lieu of the biotin radical, as the ligand of cellular integrins (GLAmimRGD). HAdV-C5 virions and hexon capsomers used for the SPR analysis with the GLAmim peptide were both tested for their integrity and purity by negative staining and electron microscopy (Fig. 1C).
SPR analysis of GLAmim–hexon interaction
The interaction of the GLAmim peptide to hexon proteins was first validated by SPR analysis. The biotin-tagged GLAmim peptide of 6 kDa was immobilized on a streptavidin-coated CM4 sensor chip, with its N-terminus oriented upward, as depicted in Fig. 1B. The control flowcells used for background subtraction were coated with streptavidin alone. The hexon protein was injected at different concentrations ranging from 30 to 120 nM in the presence of CaCl2 (Fig. 2A). The dose–response sensorgrams obtained showed a rapid association rate (k
a=1.9×105 M/sec) and a slow dissociation rate (k
d=3.3×10−3/sec). Fitting of these sensorgrams using BIAeval software gave an equilibrium constant of K
D=17 nM for the binding reaction of hexon capsomer to the GLAmim peptide (Fig. 2A). A highly efficient regeneration of the sensor chip, with complete release of hexon capsomers from the flowcell, was achieved using 10 mM EDTA for 2 min (Supplementary Fig. S1; Supplementary Data are available online at
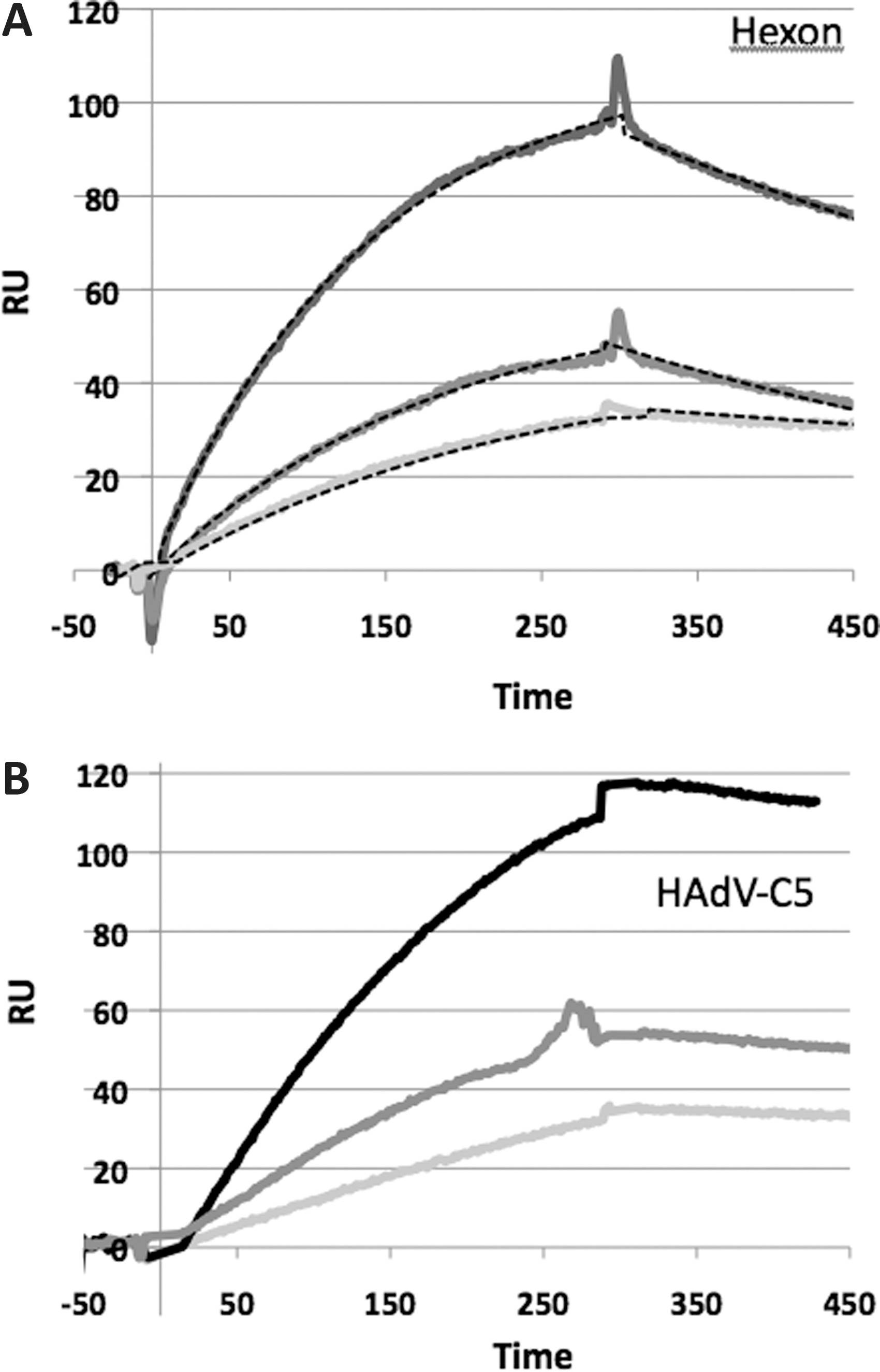
SPR analysis of the binding of hexon capsomers and HAdV-C5 virions to surface-immobilized GLAmim.
The interaction of HAdV-C5 virions with immobilized GLAmim was also investigated by injection of different concentrations of HAdV-C5 virions, ranging from 1 to 4×1010 vp/ml. HAdV-C5 binding was detected for all the concentrations tested, and the intensities of the signals correlated with the different virus concentrations (Fig. 2B). Again, the return to the baseline occurred upon EDTA regeneration, but required a longer injection time (5 min) compared with free hexon capsomers. This was consistent with an avidity effect that could take place between the 12 hexon trimers forming the adenoviral facets and the GLAmim peptide-coated surface of the sensor chip. This avidity effect was evident on sensorgrams recorded at the end of injections: the absence of dissociation observed for the whole virus suggested a very strong interaction between the virion and the immobilized GLAmim, likely because of multivalent interactions between the hexon facets and the sensor chip surface. The SPR analysis therefore validated the functionality of the GLAmim peptide in terms of specific interaction with the HAdV-C5 capsid, via a high-affinity binding to the hexon capsomers.
Competition between GLAmim and FX for hexon binding: SPR analysis
Hexon protein
We next explored the capacity of the GLAmim peptide to compete with FX for binding to the isolated or capsid-incorporated hexon protein, using an SPR setup in which human FX was immobilized onto the sensor chip. As control, a solution of the hexon protein at different concentrations (ranging from 0.8 to 66 nM) was injected. As expected from our previous study (Corjon et al., 2011), the sensorgram showed an efficient binding of hexon to FX (Fig. 3A). Surprisingly, when the hexon solution at the same concentrations was preincubated with GLAmim at 10 μg/ml (1.6 μM) before injection, a significant increase in the intensity of the SPR signal was observed (Fig. 3A). The higher signal observed in the presence of GLAmim was not an effect caused by the peptide mass. Whatever the stoichiometry of hexon-bound GLAmim to hexon, that is, 1 GLAmim molecule (6 kDa) to 1 hexon trimer (330 kDa) or 1 GLAmim to 1 hexon monomer (110 kDa), the ratio of peptide-to-hexon protein would only represent 5.5–1.8% of the total mass. Moreover, GLAmim was not the limiting factor in this experiment, and when lower concentrations of GLAmim (down to 16 nM) were used, the enhancing effect on the binding of hexon to FX was still observed (Fig. 3B).

SPR analysis of the binding of hexon capsomers to immobilized FX, in the presence or absence of GLAmim.
Interestingly, the enhancing effect of GLAmim on hexon binding was higher at lower hexon concentrations (≥10-fold), and progressively decreased in a hexon dose-dependent manner (Fig. 3B and C). In the sensorgrams, an SPR signal of hexon binding to FX was detectable for hexon concentrations below 2.4 nM in the presence of GLAmim, whereas no signal was observed for the same hexon concentrations in the absence of GLAmim (Fig. 3A). Collectively, our data suggested that the affinity of hexon toward FX was higher in the presence of GLAmim compared with its binding affinity in the absence of GLAmim: a value of about 2 nM has been reported in previous studies (Vigant et al., 2008; Waddington et al., 2008).
HAdV-C5 virions
Similar SPR analysis was then performed using HAdV-C5 virions and immobilized FX. Preincubation of GLAmim with virions significantly enhanced their binding to FX, ∼3-fold for the low virus input (Fig. 4A, left panel), compared with 1.2-fold for the higher virus input (Fig. 4A, right panel). Again, this effect cannot be explained by an effect of the peptide mass, which was negligible compared with the whole virus particle. These results confirmed those obtained with isolated hexon capsomers.
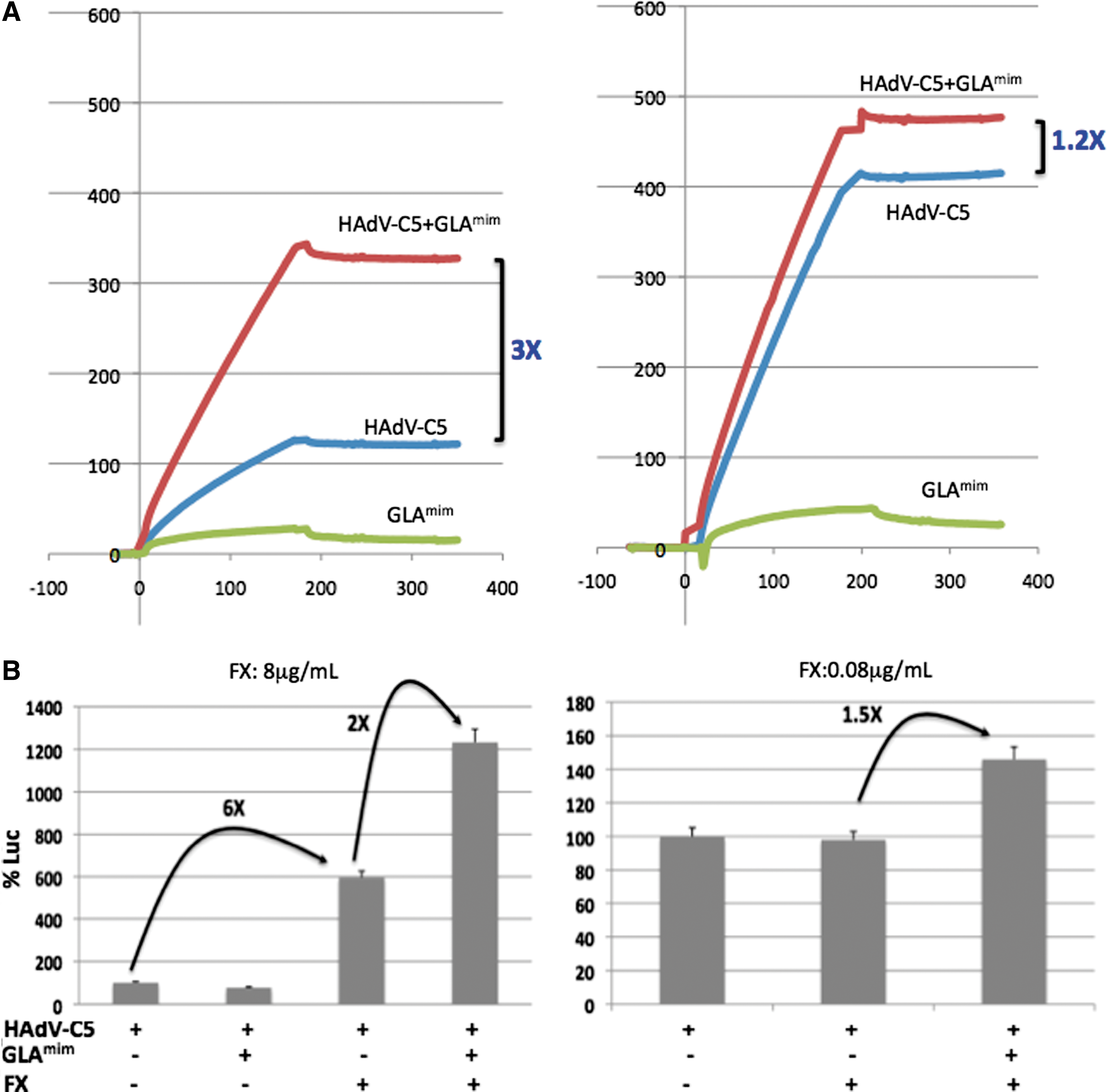
Influence of GLAmim on the binding of HAdV-C5 virions to immobilized FX and influence on the HAdV5-Luc vector-mediated transduction.
Competition between GLAmim and FX for hexon binding: cellular assays
Our next experiment was designed to determine whether GLAmim could have an effect, positively or negatively, on HAdV-C5-mediated cell transduction in the presence of FX at physiological concentrations. HeLa cells were incubated at 37°C for 1 hr with constant inputs of HAdV5-Luc vector, complexed or not with their ligands. HAdV5-Luc was preincubated with FX at physiological (8 μg/ml) or subphysiological (0.08 μg/ml) concentrations, alone or with variable GLAmim concentrations. Cell transduction efficiency was then evaluated from the level of luciferase activity at 5 hr postinfection. At this early time, the genome replication had not started, and only the efficacy of the cellular uptake of the vector was measured.
As expected, incubation of HAdV5-Luc with FX at the physiological concentration resulted in a sixfold increase in the luciferase expression (Fig. 4B, right panel). However, preincubation of HAdV5-Luc with GLAmim before FX at the physiological concentration resulted in an extra twofold increase in the luciferase signal (Fig. 4B, right panel). This was consistent with the SPR data showing an enhancing effect of GLAmim on the FX–hexon interaction. FX used at the subphysiological concentration showed no detectable effect on HAdV5-Luc-mediated cell transduction (Fig. 4B, left panel). Importantly, a significant enhancing effect on luciferase expression was detected upon GLAmim addition (1.5-fold; Fig. 4B, left panel). This result confirmed the SPR data (refer to Figs. 3C and 4B), and indicated that the presence of GLAmim peptide made detectable the hexon–FX interaction, which would otherwise remain undetectable with FX alone.
Cryo-EM analysis
We next examined HAdV-C5-GLAmim complexes by cryo-EM, in comparison with HAdV-C5 alone (Fig. 5). The rationale for this analysis was that the identification of the binding sites of GLAmim on the viral capsid might help to elucidate the molecular mechanism of the effect of this peptide on hexon– and adenovirion–FX interaction. Strong extra densities, absent from control HAdV-C5, were clearly visible on the hexon capsomers of the HAdV-C5-GLAmim complexes (Fig. 5B and C). Interestingly, three extra densities were found per hexon capsomer (Fig. 5D). More importantly, these extra densities were located at the periphery of the hexon capsomer and not in the cavity of hexon, where FX had been previously localized (Waddington et al., 2008; Irons et al., 2013) (Fig. 5B and D). The HAdV-C5 capsid 3D map has been deposited in the Macromolecular Structure Database (Accession No. EMD-2568).
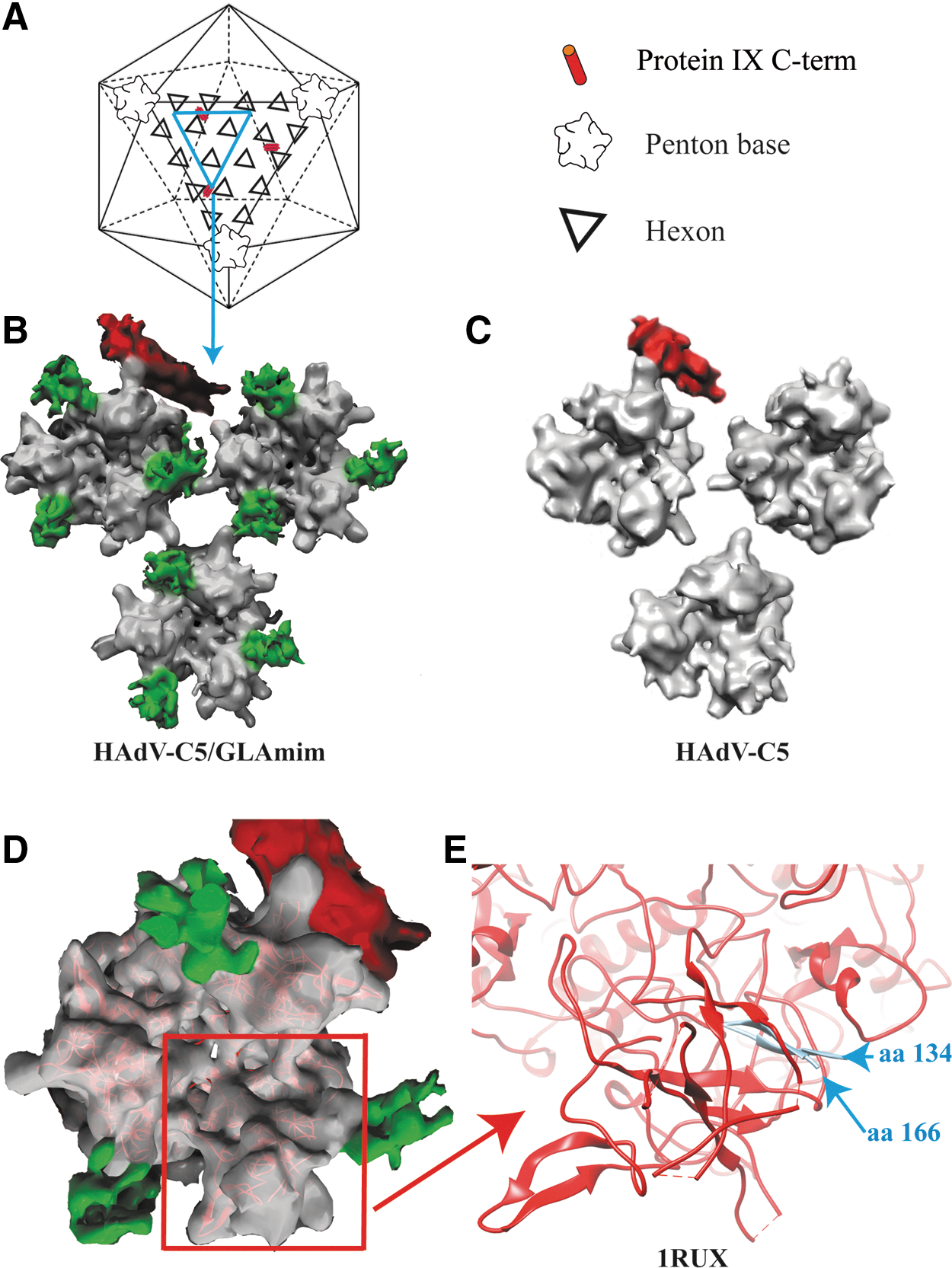
Three-dimensional (3D) reconstruction of HAdV-C5–GLAmim complexes from Cryo-EM images.
When compared with the X-ray structure of hexon crystals, the extra densities were found to be located in the invisible regions assigned to the HVR1 loops (Fig. 5E). Although the HVR1 loop structure has been solved neither by X-ray crystallography nor by cryoEM image analysis, our three-dimensional reconstruction strongly suggested that the extra densities visible on the hexon capsomers were because of GLAmim peptides, either directly or indirectly. In the latter case, the GLAmim peptide would induce some rigidity and/or reorganization of the secondary structure of the intrinsically disordered HVR1 loop.
Hepatotropism and tumor targeting in vivo in a mouse model
As the GLAmim peptide interacted with HAdV-C5 at 720 copies/virion, we next investigated if a bivalent peptide comprising of two entities, the viral ligand GLAmim on the N-terminal side, coupled to a cell surface ligand on the C-terminal side, could be used to redirect the HAdV5-Luc vector to a specific cell target. To this aim, a tripeptide RGD was added to the exposed C-terminus of the GLAmim peptide (GLAmimRGD), and the HAdV5-Luc–GLAmimRGD complex was tested in vivo in a mouse tumor model, in which mice were engrafted with HEKβ3 cells overexpressing human αβ-integrins. When the tumor size reached about 500 mm3, the mice were intravenously injected with HAdV5-Luc alone or with HAdV5-Luc preincubated with GLAmim or GLAmimRGD. The level of luciferase expression was monitored by noninvasive whole-body imaging over 3 days, and quantitatively determined in the liver and grafted tumor (Supplementary Fig. S3 and Fig. 6A).
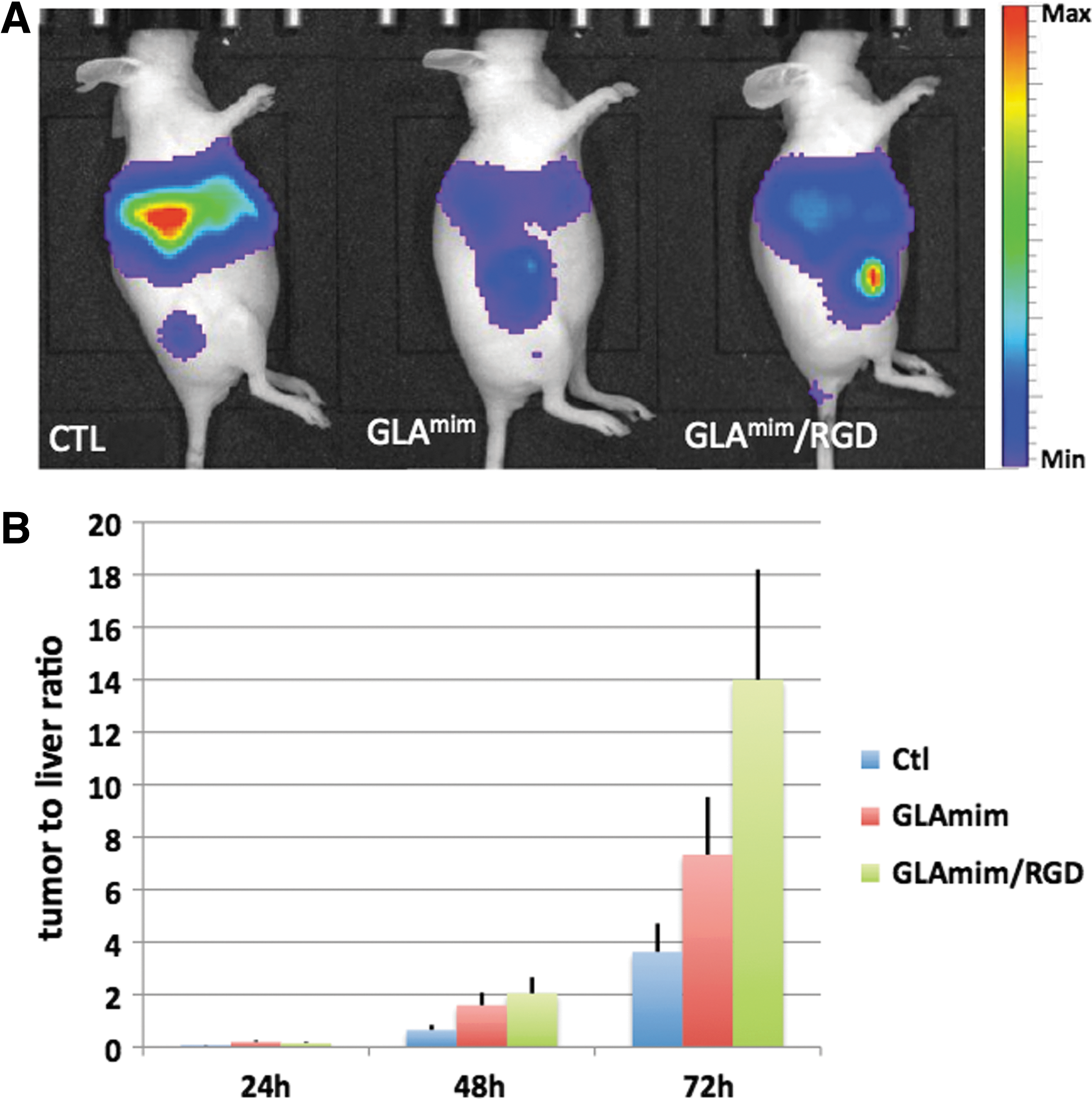
Influence of GLAmim peptides on the tissue tropism of a HAdV-C5-based vector in a mouse tumor model. Three groups of mice were engrafted in the right posterior limb with HEK-β cells. Each mouse received an aliquot (4×1010 vp) of HAdV5Luc vector (alone or complexed with GLAmim or GLAmimRGD peptides) by injection in the tail vein.
With control HAdV5-Luc alone, the bioluminescent signal was localized massively within the liver (Supplementary Fig. S3A). With the HAdV5-Luc–GLAmim complex, we observed a 2–3-fold lower luminescent signal in the liver, compared with the HAdV5-Luc vector alone. This effect was detected at 24 and 48 hr postinjection, and persisted until day 3. A reduced hepatotropism, compared with the control, was also observed with the HAdV5-Luc–GLAmimRGD complex, but mainly at late times after injection (Supplementary Fig. S3A).
In parallel to its decrease in the liver, the luciferase activity increased significantly over time in the HEKβ3 tumors of mice injected with HAdV5-Luc–GLAmimRGD (∼2-fold), compared with control groups injected with HAdV5-Luc alone or HAdV5-Luc–GLAmim complex (Supplementary Fig. S3B). At each time point, the tumor-to-liver signal ratio was significantly higher in mice administered with HAdV5-Luc complexed with GLAmimRGD, compared with HAdV5-Luc alone or HAdV5-Luc–GLAmim complex (2–4-fold; Fig. 6B). This implied an increased retargeting of HAdV5-Luc to HEKβ3 cells, and a higher susceptibility of αβ-expressing tumor cells to HAdV5-Luc in the presence of GLAmimRGD.
The results of our in vivo experiments were in apparent contradiction with our biochemical and in vitro cellular studies, showing an enhancing effect of GLAmim on the FX–hexon binding and on HAdV5-Luc-mediated cell transduction in the presence of FX. However, recent data suggested the implication of HVR1 in the recognition of scavenger receptors by HAdV-C5 (Khare et al., 2012), suggesting that GLAmim could interfere with scavenger receptors for binding to hexon HVR1. However, to get a full view on the mechanism taking place after HAdV-C5–peptide injections, an immunocompetent model of mice would be required to investigate whether the GLAmim peptide can shield the HAdV-C5 vector and thus prevent the sequential events of (1) nonneutralizing antibody binding, (2) complement activation, and (3) triggering of the innate response, as previously reported (Baker et al., 2013; Xu et al., 2013).
Discussion
After the discovery of the binding of blood coagulation factors to adenoviral vectors during systemic administration, and the resulting sequestration of the vectors within the liver, it appeared necessary to reconsider the conclusions of past clinical trials, and to develop new strategies to overcome this limitation for future biotherapy protocols. Blood coagulation factors bridge the hexon capsomers to ubiquitous cellular receptors, such as HSPGs present in high concentration at the surface of Kuppfer cells. Among the diverse strategies proposed to palliate this inconvenience, the development of molecules blocking this interaction has been less investigated (Chen et al., 2010; Duffy et al., 2013), compared with direct modifications of the coagulation factor binding site on the hexon capsomers (Vigant et al., 2008; Waddington et al., 2008). The present study is in line with the first type of experimental approach.
Intuitively, a peptide mimicking the region of interaction of FX with hexon, that is, the GLA domain, would be the ideal antagonist molecule of FX, as it would compete for the binding site on the hexon capsomer. To this aim, we designed a synthetic 40-mer polypeptide, GLAmim, which mimicked the FX GLA domain including the carboxyglutamate residues at highly conserved positions in all GLA-domain-containing proteins, but lacking the first A, N, and S residues. The GLAmim was tagged with a biotinyl group at its C-terminus, to promote its orientation on BIAcore sensor chips in an adenovirus accessible manner, that is, with its N-terminus exposed (refer to Fig. 1C). Chemical synthesis of GLAmim, a 40-mer peptide carrying 12 carboxyglutamate residues, was a challenge, but the result obtained was a soluble, homogenous peptide that was found to be fully functional in terms of interaction with soluble hexons, or HAdV-C5 virions, based on conventional criteria and assays, such as SPR.
We found that hexon capsomers interacted with immobilized GLAmim in a dose-dependent manner, with a dissociation constant in the nanomolar range (K D≈17 nM). Of note, in the original report (Waddington et al., 2008), FX was found to bind to hexon with a K D of ∼2 nM. The eightfold lower affinity of GLAmim for hexon, compared with that of full-length FX, was explained by the higher association rate of FX (2.3×106 M/sec vs. 1.9×105 M/sec for GLAmim), while the dissociation rates were comparable for FX and GLAmim (4.5×10−3/sec and 3.3×10−3/sec, respectively). HAdV-C5 virions also showed a strong interaction with the GLAmim-coated surface, and the SPR plateau of persistent binding at the end of the HAdV-C5 injection suggested an avidity effect. This avidity effect was not surprising, considering the complexity and the multivalent nature of the viral ligand, which is made of 240 copies of trimeric hexons distributed between 20 capsid facets.
However, no blockage of FX–hexon interaction was observed with a large excess of GLAmim in competition assays in vitro and in cellulo. Instead, GLAmim increased the affinity of hexon for FX, as shown by SPR, and augmented the transduction efficiency of HeLa cells with HAdV5-Luc vector in the presence or absence of FX. These paradoxical results incited us to seek for structural information on the binding of GLAmim to the adenoviral capsid, using cryo-EM. Density map comparison between control HAdV-C5 virions and HAdV-C5–GLAmim complexes showed the presence of three extra densities per trimeric hexon capsomer. Our 3D reconstruction and the stoichiometry of 3:1 differed from the original model of HAdV-C5 virions in complex with full-length FX, which assigned only one FX molecule per trimeric hexon capsomer with a stoichiometry of 1:1 (Waddington et al., 2008).
The greatest discrepancy between our reconstructed HAdV-C5–GLAmim complex and the original FX-HAdV-C5 model resided in the topography of hexon-bound GLAmim molecules, compared with hexon-bound FX. The FX molecule has been shown to occupy the central cavity of the hexon capsomer (Waddington et al., 2008), and the FX binding involved side chains of amino acids located in the HVR7 loop, including T423, E424, and T425 (Doronin et al., 2012; Irons et al., 2013). In our 3D reconstruction of the HAdV-C5–GLAmim complex, however, the three additional densities surrounded the central cavity and were localized to the undetermined regions in the hexon crystals that were assigned to HVR1 loops (residues 135–165). The extra densities could result from a direct or indirect effect of the GLAmim peptide on HVR1. In the first case, the extra densities would correspond to the GLAmim peptides themselves; in the other case, GLAmim would induce some rigidity and/or secondary structure reorganization of the intrinsically disordered HVR1 loops. Interestingly, the HVR1 loop is the most negatively charged in serotype 5 hexon, with a net charge of −13 amino acids. Upon GLAmim binding, a rigidification/folding of HVR1 loop might offer a better access to the FX protein. This interaction likely required calcium chelation by both GLAmim and HVR1 residues as suggested by the high efficiency of EDTA regeneration in the hexon–GLAmim interaction studied by SPR experiments (Supplementary Fig. S1).
The A-N-S sequence, missing from GLAmim, is highly conserved in FX throughout the vertebrate evolution, and play an important role in coordinating Ca2+ ions, and in stabilizing the N-terminal fold. However, it is unlikely that the lack of these three amino acids is responsible for the differential hexon binding activity between FX and GLAmim for the following reasons: (1) human FIX also binds to hexon, but lacks the N-terminal peptide A-N-S (replaced by Y-N-S-G); (2) likewise, human FVII, another hexon binder (Irons et al., 2013), starts with the A-N-A tripeptide sequence; (3) it has been recently shown that the critical residue for hexon binding is Lys10 (Doronin et al., 2012), whereas the N-terminal hydrophobic cluster is involved in membrane binding (Sunnerhagen et al., 1995).
Our data implied that two types of GLA-binding sites coexist on the same hexon capsomer, displayed by HVR7 and HVR1, respectively. The differential binding of full-length FX molecule versus GLAmim to hexon capsomer remained to be elucidated. One clue might be given by the recent observation that the GLA domains of FX and FVII bind to different sites in the hexon cavity, enabling or not the dimerization of coagulation factors bound to adjacent hexons of the facet edges (Irons et al., 2013). In the same line of hypotheses, the accessibility and/or reactivity of certain amino acid side chains might differ between full-length FX and its isolated GLA domain. Differences in the molecular mass of hexon ligands might also be evoked. Full-length FX molecule with its bulky serine-protease domain of 58 kDa would impair, by steric hindrance, the binding of more than one single FX molecule per hexon, resulting in only 240 copies of FX per virion. In contrast, GLAmim, a small peptide of 6 kDa, could occupy all the available binding sites at the virion surface. In this case, however, the determinants of choice of the HVR1 sites by GLAmim, instead of HVR7 or both HVR1 and HVR7, are unknown and require further studies.
Although our data invalidated our working hypothesis of using GLAmim as an FX antagonist, the development of new therapeutic strategies might be envisaged, based on the specific properties of GLAmim. One major feature and advantage of GLAmim resided in its capacity to bind to the hexon capsomers of HAdV-C5 with high affinity and in high numbers (720 copies of GLAmim per virion), to constitute a mantle of retargeting peptides around the viral capsid. When complexed with HAdV5-Luc, GLAmim appeared to reduce its hepatotropism after systemic administration to mice, suggesting an interfering effect of GLAmim with scavenger receptors for hexon binding via HVR1 (Khare et al., 2012). This decrease implied that FX, HSPGs, and hexon HVR7 were not the only factors controlling the HAdV-C5 hepatotropism, and confirmed the results of a recent study (Khare et al., 2012).
The results of our in vivo experiments were in apparent contradiction with our biochemical and in vitro cellular studies, showing an enhancing effect of GLAmim on the FX-hexon binding and on HAdV5-Luc-mediated cell transduction in the presence of FX. However, recent data suggested the implication of HVR1 in the recognition of scavenger receptors by HAdV-C5 (Khare et al., 2012), suggesting that GLAmim could interfere with scavenger receptors for binding to hexon HVR1.
A similar level of liver detargeting was observed with GLAmimRGD, a bivalent GLAmim-containing peptide terminated by an RGD sequence. More interestingly, GLAmimRGD retargeted the HAdV5-Luc vector to αvβ3-integrin-overexpressing cells in a mouse tumor model in vivo. Thus, in combination with HAdV-C5 vectors mutated in the FX-binding site of HVR7, GLAmim-containing bifunctional peptides might confer two advantages to the mutant vectors, the decrease of adverse effects because of the liver sequestration, and the gain of a new function, that is, a specific cell retargeting. In addition, as ligands of HVR1, GLAmim-derived peptides would also represent valuable biological tools for investigating scavenger receptors exposed at the cell surface.
Our animal experiments were performed using tumor-bearing nude mice. A full view on the mechanism taking place after HAdV-C5–GLA-peptide injection would require an immunocompetent animal model, allowing to investigate whether the GLAmim peptide can shield the HAdV-C5 vector, and thus prevent the sequential events of (1) of nonneutralizing antibody binding, (2) complement activation, and (3) triggering of the innate response, as observed with FX (Baker et al., 2013; Xu et al., 2013).
Footnotes
Acknowledgments
S.S. was supported by the “Bio-Health Computing” European training program. This work used the platforms of the Grenoble Instruct center (ISBG; UMS 3518 Centre National Recherche Scientifique (CNRS)-Commissariat Energie Atomique (CEA)-Université Joseph Fourier (UJF)-European Molecular Biology Laboratory (EMBL)) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology. The electron microscope facility (Polara electron microscope) was supported by the Rhône-Alpes Region, the FRM, the CNRS, the University of Grenoble, and the GIS-IBISA.
The work at the UCBL-Institut National Recherche Agronomique Unité Mixte Internationale-754 was funded in part by the French Foundation for Cystic Fibrosis (Vaincre la Mucoviscidose; Contract VLM-RF2013-0500796). S.S.H. is a scientist of the French Institute of Health and Medical Research (Institut National de la Santé et de la Recherche Médicale, INSERM), and the recipient of a Contrat d'Interface INSERM-Hospices Civils de Lyon (CIF-2008–2013). We are most grateful to Jullien Vollaire and Mélanie Guidetti for their help with animal studies.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.