
Abstract
Human adenoviruses are the most widely used vectors in gene medicine, with applications ranging from oncolytic therapies to vaccinations, but adenovirus vectors are not without side effects. In addition, natural adenoviruses pose severe risks for immunocompromised people, yet infections are usually mild and self-limiting in immunocompetent individuals. Here we describe how adenoviruses are recognized by the host innate defense system during entry and replication in immune and nonimmune cells. Innate defense protects the host and represents a major barrier to using adenoviruses as therapeutic interventions in humans. Innate response against adenoviruses involves intrinsic factors present at constant levels, and innate factors mounted by the host cell upon viral challenge. These factors exert antiviral effects by directly binding to viruses or viral components, or shield the virus, for example, soluble factors, such as blood clotting components, the complement system, preexisting immunoglobulins, or defensins. In addition, Toll-like receptors and lectins in the plasma membrane and endosomes are intrinsic factors against adenoviruses. Important innate factors restricting adenovirus in the cytosol are tripartite motif-containing proteins, nucleotide-binding oligomerization domain-like inflammatory receptors, and DNA sensors triggering interferon, such as DEAD (Asp-Glu-Ala-Asp) box polypeptide 41 and cyclic guanosine monophosphate–adenosine monophosphate synthase. Adenovirus tunes the function of antiviral autophagy, and counters innate defense by virtue of its early proteins E1A, E1B, E3, and E4 and two virus-associated noncoding RNAs VA-I and VA-II. We conclude by discussing strategies to engineer adenovirus vectors with attenuated innate responses and enhanced delivery features.
Introduction
V
A major bottleneck in molecular therapy is a shortage of efficient and nontoxic delivery agents. Human adenoviruses (HAdVs) are the most widely used agents in gene therapy, largely because of their high efficiency in gene transfer and deep knowledge of their infection biology (
One of the major biological obstacles in gene therapy is that host cells contain intricate viral detection mechanisms that activate inflammatory or cytotoxic responses directed against viruses. This innate immunity is based on a large variety of well-studied inducible factors, such as proteins, lipids, or RNA (for reviews, see Pichlmair and Reis E Sousa, 2007; Schoggins and Randall, 2013). More recently, it was shown that mammalian cells (besides plant and insect cells) have antiviral RNA interference (Maillard et al., 2013). Mammalian cells accumulate small 22-nucleotide RNAs from viral replication intermediates and guide them to the argonaute proteins to eliminate viral RNA. Collectively, innate immunity steers the organism to adaptive immunity, which is pathogen specific, and comprises selective antibodies. Both innate and adaptive immunity generally antagonize viral efficacy in gene therapy (reviewed in Janeway and Medzhitov, 2002; Fejer et al., 2011; Russell et al., 2012), although the treatment of aggressive forms of cancer by therapeutic viruses can be enhanced by the inflammatory host response (for reviews, see Wong et al., 2010; Russell et al., 2012). Here we summarize the current knowledge of the mechanisms that lead to inflammation and innate immunity in cells inoculated with HAdV.
Early Signaling: Mobilizing Cell Defense
The outcome of virus–cell interactions ranges from productive or persistent infection to no infection where the virus is completely rejected. Permissive cells support virus replication and produce progeny viruses, as their defense is outpowered by the virus. In many instances, productive infections are cytolytic, and in the case of cancer cells oncolytic, and the cells die. If cellular defense out-powers the virus, cells are nonpermissive and do not produce infectious progeny virus. Such infections are abortive. If a set of viral genes is incompletely transcribed or translated, the infection is restrictive. This can lead to persistent or in certain cases transforming infections, where viral DNA is maintained but progeny virus usually not produced or if so, at low levels.
Infection can be tuned by signaling during entry and this can impact cell death by apoptosis, necrosis, or pyroptosis, as well as innate signaling with pro- or antiviral effects (Greber, 2002; Faure and Rabourdin-Combe, 2011; Mercer and Greber, 2013). Cell death signals emerge from viral engagement of death receptors, signaling during uncoating, and postentry events (for reviews, see Lamkanfi and Dixit, 2010; Danthi, 2011; Agol, 2012; Kaiser et al., 2013). Innate immune responses comprise intrinsic mechanisms, which directly restrict viral replication and assembly, therefore leading to nonpermissiveness of the cell (Yan and Chen, 2012).
Extrinsic innate immunity impairs infection by indirect mechanisms, which involve signaling to elicit an antiviral state. Extrinsic modulators include Toll-like receptors (TLRs), C-type lectin receptors, RIG-I like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), cytosolic DNA sensors, and effector molecules. Antiviral effects can occur, for example, through engagement of cell surface or endosomal pattern recognition receptors (PRR), such as mannose receptor, dendritic cell-specific ICAM grabbing nonintegrin (DC-SIGN), or defensins disrupting bacterial membranes or binding to viral capsids (Buck, 2008; Sato et al., 2009). PRRs trigger complex intracellular signaling cascades and type 1 interferon (IFN) production, and eventually lead to an antiviral state of the host cell. The following sections discuss how host innate defense senses HAdV, and how this triggers innate immune responses.
HAdV Entry: A Gain-of-Function Process for the Virus
Adenovirus
Adenoviruses are icosahedral, non-enveloped double-stranded (ds) DNA viruses infecting both dividing and quiescent cells in a species-specific manner. HAdVs comprise more than 55 types, and are the most frequently used vector in human gene therapy (Smith et al., 2010b). According to hemagglutination and genome sequences, there are seven species, HAdV-A, B, C, D, E, F, and G (Harrach et al., 2011). They are part of the genus Mastadenovirus. HAdVs are considered to be nononcogenic in humans. They maintain episomal genomes in the nucleus without integration into host DNA (Harui et al., 1999). HAdV infections in immunocompetent individuals typically cause respiratory or gastrointestinal symptoms and are self-limiting. Infections can, however, be fatal in immunodeficient hosts or newborns (Echavarria, 2008). Yet, to date there are no effective drugs for the treatment of HAdV infections. Even the well-established nucleoside inhibitor cidofovir has low clinical efficacy (Lenaerts and Naesens, 2006; Skevaki et al., 2011; Greber et al., 2013).
HAdVs are highly stable outside of cells, which is a great advantage for gene therapy (see Fig. 1A). The crystal structure and high-resolution cryo-EM structures for HAdV-C are available, providing a solid basis for rational engineering (Liu et al., 2010; Reddy et al., 2010). Despite their great thermal and chemical stability (Buckland and Tyrrell, 1963; Tuladhar et al., 2012), HAdV-C2/5 have evolved remarkably high efficacies for cell entry and uncoating of their DNA (for reviews, see Greber et al., 1994; Greber and Way, 2006; Puntener and Greber, 2009; Suomalainen and Greber, 2013; Suomalainen et al., 2013; Wolfrum and Greber, 2013). Notably, HAdVs are well known to activate innate immunity by virtue of their pathogen-associated molecular patterns (PAMPs), such as capsid or DNA, and HAdV infection, and this leads to production of IFNs and inflammatory cytokines (Bruder and Kovesdi, 1997; Suomalainen et al., 2001; Tibbles et al., 2002; Basner-Tschakarjan et al., 2006; Hartman et al., 2007b; Nociari et al., 2007; Lutschg et al., 2011).

Schematic illustration of HAdV and key steps in HAdV entry.
Entry
The entry of HAdV into nonimmune cells is initiated through binding of the fiber knobs to entry receptors and attachment factors (see Fig. 1B) (Arnberg, 2012; Wolfrum and Greber, 2013). Entry pathways into immune cells, such as macrophages and dendritic cells, may be different and could be modulated by cytokines or antibodies, and availability of low-affinity, high-avidity receptors, such as scavenger receptors (Meier et al., 2005; Fejer et al., 2011; Khare et al., 2012; Mercer and Greber, 2013). Entry of HAdV-C2/5 into polarized epithelial cells from the apical side (facing the airways) is enhanced by cytokines and chemokines, including interleukin 8 and tissue necrosis factor alpha (Lutschg et al., 2011). The cytokines increase the availability of CAR and integrin receptors, which allows HAdV-C to enter along the well-described pathways involving clathrin-mediated dynamin-dependent endocytosis (Wang et al., 1998; Meier et al., 2002; Gastaldelli et al., 2008). The species B HAdVs use CD46 or desmoglein-2 as their major receptors, and engage in macropinocytosis for infectious entry (Gaggar et al., 2003; Sirena et al., 2004; Amstutz et al., 2008; Hall et al., 2009; Kalin et al., 2010; Wang et al., 2011; Trinh et al., 2012). How the CD46 pathway relates to the observation that HAdV-B suppresses IFN-γ-triggered production of the proinflammatory cytokine interleukin 12 is unknown (Iacobelli-Martinez et al., 2005).
Endosomal escape of HAdV to the cytosol is important in triggering an innate response, although the spectrum of host factors supporting this important step is incompletely known. The endosomal escape process is not spontaneous but requires specific changes in the viral structure. It is linked with the first steps of virus uncoating triggered by receptor motility on the cell surface (Helmuth et al., 2007; Burckhardt and Greber, 2009; Burckhardt et al., 2011). This leads to structural changes at the vertices of the capsid and exposes the internal membrane lytic protein VI, which in turn facilitates the escape of the virus from an early endosome (Wiethoff et al., 2005; Wodrich et al., 2010; Burckhardt et al., 2011). It should be emphasized here that the escape of both HAdV-B and HAdV-C serotypes is independent of endosomal pH, as recently demonstrated by a direct single-cell, single-virus penetration assay (Suomalainen et al., 2013). This study used three different classes of chemical inhibitors to neutralize endosomal pH: the vacuolar proton pump inhibitor bafilomycin A, the protonophore niclosamide, and the lysosomotropic proton buffer ammonium chloride (Matlin et al., 1981; Bowman et al., 1988; Jurgeit et al., 2012). Earlier studies suggested that HAdV uses low pH for penetration and uncoating, for example, based on the observation that incubation of viruses with low salt, EDTA, and low pH for several hours leads to the dissociation of the pentons from the capsid (Laver et al., 1969). The observation that low endosomal pH is not involved in HAdV infection, however, does not exclude that other ions in endosomes are important for the penetration process. This has been suggested by observations that HAdV infection is sensitive to inhibitors of the sodium/potassium ATPase (Seth et al., 1987), the sodium/proton exchanger (Meier et al., 2002; Amstutz et al., 2008; Kalin et al., 2010), and the lysosomotropic agent ammonium chloride (Greber et al., 1993; Suomalainen et al., 2013), but not inhibitors of the vacuolar proton ATPase (Perez and Carrasco, 1994).
The notion that infection is independent of endosomal pH is compatible with earlier results that the initial steps of virus uncoating, the shedding of the fibers, and the exposure of the membrane lytic protein VI as well as protein VI-mediated membrane lysis are independent of low pH (Greber et al., 1993; Wiethoff et al., 2005; Suomalainen et al., 2013). This means that the virus does not need to visit an acidic endosome to be infectious. In fact, residing in a late endosome or lysosome bears the risk of degradation, as shown for the endosome escape-defective HAdV-C2 mutant TS1 (Greber et al., 1996; Imelli et al., 2009).
Infectious virus reaches the cytosol, and uses dynein/dynactin and microtubule-based transport to reach the nuclear membrane (Suomalainen et al., 1999, 2001; Leopold et al., 2000; Mabit et al., 2002; Strunze et al., 2005; Bremner et al., 2009; Gazzola et al., 2009; Wodrich et al., 2010; Engelke et al., 2011). It then docks to the nuclear pore complex and activates a kinesin-mediated capsid disruption program (Wisnivesky et al., 1999; Trotman et al., 2001; Strunze et al., 2011). Although most of the particles are disrupted during this process, only a minor fraction of the viral DNA is imported into the nucleus, and as much as 50–90% stays behind in the cytosol with large cell-to-cell variability (Wang et al., 2013). This suggests that the nuclear pore complex is a bottleneck for viral DNA import into the nucleus.
HAdV vectors: a short glimpse
Therapeutic HAdVs are genetically attenuated, or if wild-type viruses are used, particular conditions preclude unintended virus replication and shedding to the environment (Lichtenstein and Wold, 2004). HAdV can be readily engineered as replicating or nonreplicating particles, and can be produced in high amounts under good manufacturing practice (GMP) (Lusky, 2005), using established cell lines with a wide range of complementing properties (Kovesdi and Hedley, 2010). The first-generation HAdV vectors were derived from early region 1 (E1)-deleted wild-type viruses, mainly HAdV-C2/5. In addition to E1 deletion, second-generation HAdV vectors were constructed with inactivated E2, E3, or E4 regions (Rein et al., 2006). Helper-dependent gutless vectors had the entire viral genome deleted, except for the inverted terminal repeats that are crucial cis-acting elements for DNA packaging and replication (Ostapchuk and Hearing, 2003; Raty et al., 2008). Gutless viruses were designed to minimize the expression of viral genes, and thereby facilitate long-term expression of therapeutic transgenes (Kreppel and Kochanek, 2004). Yet, even the gutless viruses elicit innate and adaptive immune responses that are directed against components of the vector or the therapeutic gene products (Schiedner et al., 2003; Stilwell et al., 2003).
Innate responses elicited by viral DNA invariably shape the adaptive, pathogen-specific immune response. The adaptive immune response comprises virus-specific antibodies, which can neutralize the virus and limit the success of gene therapy. Interestingly, the prevalence of antibodies against HAdVs varies largely depending on the serotype (Aste-Amezaga et al., 2004). For example, the widespread serotypes HAdV-C2/5 have a seroprevalence of 82% and 35%, whereas HAdV-B35 has close to 0%. Hence, different HAdV serotypes may be uniquely suited for gene therapy. Nonetheless, HAdV-C2/5 are more widely used than any other serotype in the clinics, despite their high seroprevalence (Toth et al., 2010; Yamamoto and Curiel, 2010; Greber et al., 2013; Wolfrum and Greber, 2013). The major argument for pushing HAdV-C2/5 into clinical applications has been that their biology is well understood, and their seroprevalence can eventually be overcome by engineering strategies (see below). In the next sections, we highlight factors and mechanisms that control innate immunity against HAdV in the plasma membrane, endosomes, and the cytosol.
Soluble factors: local and systemic defense
A major quest in gene therapy is targeting the cells of interest by systemic applications of the vector. Upon intravascular injection, HAdV is normally filtered out of circulation before reaching its intended targets. Vector sequestration occurs by clotting factors and Kupffer cells, sinusoidal endothelial cells or hepatocytes of the liver, immunoglobulins (Igs) and defensins, or the complement system (for reviews, see Haisma and Bellu, 2011; Khare et al., 2011). The soluble factors implicated in HAdV infection are depicted in Fig. 2.

Adenovirus-induced host innate responses. The most prominent cellular innate signaling pathways elicited during HAdV entry comprise lectin receptors (LRs), toll-like receptors (TLRs), inflammasome signaling comprising AIM2-like receptors (ALRs), NOD-like receptors (NLRs), and RIG-I-like receptors (RLRs), autophagy, and interferon (IFN) signaling. In addition, defensins, intracellular antibodies, and most importantly DNA sensors cGAS and DDX41 together with the adapter STING provide crucial innate defense against HAdV. The virus antagonizes innate defense by early proteins of the E1, E3, and E4 regions, as well as by VA-RNAs.
Clotting factor X and the liver
Many HAdVs bind the blood coagulation factor X (FX), and this is essential for liver transduction in mice (see Fig. 2, upper right) (Kalyuzhniy et al., 2008; Vigant et al., 2008; Waddington et al., 2008). For HAdV-C5, binding of FX is of high affinity and occurs through solvent-exposed hypervariable loops of the viral capsid protein hexon. Recently, FX interaction with the HAdV-C5 hexon was modeled using high-resolution cryoelectron microscopy and led to identification of the T423-E424-T425 amino acid motif in hypervariable region 7 as critical for FX binding to virus. Furthermore, a single amino acid substitution, T425A, completely abrogated FX binding to HAdV-C5 (Doronin et al., 2012). This FX-binding-ablated virus failed to infect hepatocytes when injected in mice. FX acts as a bridge for the virus to bind to particular classes of heparin sulfate proteoglycans on hepatocytes (Bradshaw et al., 2010).
Another possibility is that FX shields the virus from attack by the complement system (Xu et al., 2013). IgM antibodies and the complement system are well known to interact with HAdV-C5 and trigger inflammatory cytokine-mediated reactions (Cichon et al., 2001; Shayakhmetov et al., 2005; Carlisle et al., 2009a). However, complement-mediated HAdV elimination is most likely more complex in vivo, and may involve particular cell types, besides modification of the virus with complement factors, such as complement factor C3 (Tian et al., 2009). For example, the temperature-sensitive HAdV-C2 mutant TS1, which fails to uncoat and enter the cytosol, did not elicit the complement cascade upon intravenous injections in mice unlike wild-type HAdV, although antibodies were binding to the TS1 capsid presumably similarly as to the wild-type HAdV-C2 (Tian et al., 2009). Likewise, evidence indicates that canine adenovirus did not activate the human complement system in vitro, although it was recognized by cross-reacting antibodies in human sera (Perreau et al., 2007). This suggested that virus interactions with the cells are critical for triggering complement in vivo. Intriguingly, canine adenovirus and TS1 both visited late endosomes in their entry pathways unlike HAdV-C2/5 (Greber et al., 1996; Salinas et al., 2009; Suomalainen et al., 2013). Recently, it was shown that high levels of Igs, including IgM, negatively correlated with HAdV-C5 transduction of hepatic cells in different mouse strains (Khare et al., 2013). In animals lacking Kupffer cells, HAdV-C5 transduction was high, even in the presence of Ig, and partial reconstitution of IgM into Rag knockout animals reduced HAdV transduction of hepatic cells. These data suggested a model where IgM mediates the clearance of HAdV-C5 by Kupffer cells.
Igs and tripartite motif-containing protein 21: extracellular and intracellular defense
Antibodies, in particular Igs, protect against lethal infections by viruses, including HAdVs (Moore et al., 2004). They emerge mainly from plasma cells, marginal zone B cells, and other innate B cells, and are directed against specific epitopes of viral proteins or other biologicals. Igs normally recognize their targets in extracellular space, block their biological functions, and direct them to degradation in immune cells for antigen presentation. However, in some instances, antibody inhibition against viruses is mediated by just a single antibody per virion. The inhibition occurs postadsorption to cells, or depends on IFN (see Fig. 2, lower left) (Wohlfart, 1988; Vrijsen et al., 1993; Burdeinick-Kerr et al., 2007). It was later shown that a nonreplicating HAdV-C5_dE1 loaded with antibodies can access the cytosol of nonimmune cells, and there the virus–antibody complex recruited tripartite motif-containing protein 21 (TRIM21) to the Fc portion of an IgG or IgM (Mallery et al., 2010). Similar results were recently reported for a replicating mouse adenovirus (Watkinson et al., 2013). The cytosolic antibody receptor TRIM21 is a RING finger E3-ubiquitin ligase of a family of nearly 100 tripartite motif genes in the mammalian genome. It acted together with the host AAA ATPase valosin-containing protein and dismantled the viral capsid, thereby enabling virus presentation to the proteasome, and blocking infection (Hauler et al., 2012).
Importantly, TRIM21 has been shown to protect wild-type mice from lethal challenge with mouse adenovirus (Vaysburd et al., 2013). Protection involved upregulation of TRIM21, and TRIM21 stimulated IFN response and proinflammatory cytokines through nuclear factor-kappa B (NF-κB), activator protein 1 (AP1), and IFN regulatory factor (IRF) 3, IRF5, or 7 (Mcewan et al., 2013). Interestingly, TRIM21-mediated innate immunity was triggered by both DNA and RNA viruses, as well as bacteria. This suggests that the TRIM21–antibody machinery is unusually broad in detecting danger signals. It may act independently of other PAMP receptors, or at least upstream of them. Regardless, the machinery for intracellular antibody-mediated degradation of PAMPs is present in most human tissues, and represents an example of encapsulated immunity as opposed to systemic immune surveillance.
Defensins: for local defense
Another line of defense that acts locally rather than systemically are defensins, which are abundant antimicrobial peptides, that occur in high concentrations at micromolar to millimolar ranges in extracellular fluids of nasal, lung, or vaginal epithelia (reviewed in Lehrer and Lu, 2012). Defensins are effective against viruses, as originally shown for herpes viruses, vesicular stomatitis virus, and influenza virus with cell supernatants from human neutrophils (Ganz et al., 1985; Daher et al., 1986; Wilson et al., 2013). Later it was shown that defensins also protect against nonenveloped viruses by directly binding to HAdV or human papilloma virus and blocking viral uncoating or signaling (Buck et al., 2006; Smith and Nemerow, 2008). Defensins are small cationic peptides of 30–40 amino acids. Humans express a broad range of α- and β-defensins. α-defensins are mostly expressed from human neutrophils but also monocytes/macrophages, B and T cells, and immature dendritic cells (Selsted and Ouellette, 2005), whereas β-defensins are released from epithelial cells in skin and mucosal tissue (Pazgier et al., 2006).
The α-defensin human defensin 5 inactivates HAdV-C by binding to intrinsically disordered regions of the viral capsid involving the RGD loops of penton base at the fivefold icosahedral axis (see Fig. 2, lower left) (Flatt et al., 2013). This interferes with the dynamics of the capsid and blocks the release of the membrane lytic protein VI from the capsid (Smith and Nemerow, 2008; Smith et al., 2010a; Snijder et al., 2013). At present, it is not known if HAdV infections induce the expression of defensins, as has been reported for RNA viruses or cells transfected with poly (I:C), implicating cytosolic detection of double-stranded RNA as a trigger for defensin induction (reviewed in Wilson et al., 2013). Future research is needed to reveal more of the intricate mechanisms by which enteric and neutrophil defensins modulate HAdV infections.
Toll-like receptors
HAdVs are also controlled by membrane-bound proteins. The mammalian homologs of the Drosophila TLRs are a class of PRRs detecting and responding to PAMPs and triggering innate immune reactions (Beutler et al., 2006; Kawai and Akira, 2011; Thompson et al., 2011). There are 10 human TLRs and 12 murine TLRs. Some TLRs are predominantly on the plasma membrane, such as TLR1, 2, 4, 5, and 6, and others in endosomal compartments, for example, TLR3, 7, 8, 9, and 10. All human TLRs require the adaptor myeloid differentiation primary response gene 88 (MyD88) for innate signaling, although at different extent (Takeda and Akira, 2004). Transcription profiling of plasma cells and liver from mice inoculated intravenously with HAdV-C2 showed that a large fraction of the genes that were transcriptionally upregulated depended on MyD88, suggesting that at least one TLR senses HAdV-C2 and signals through MyD88 in a mouse model (Hartman et al., 2007b). This was confirmed in cell cultures (Hartman et al., 2007a). The TLR response also activates NF-κB, MAP kinases, and IRFs.
Specifically, TLR9 was found to sense HAdV-B in peripheral blood mononuclear cells and plasmacytoid dendritic cells (pDCs) (see Fig. 2, top right) (Sirena et al., 2004; Iacobelli-Martinez and Nemerow, 2007). TLR9 detects nonmethylated CpG-rich DNA. Since CAR tropic HAdVs were not sensed by TLR9 in these experiments, it is possible that CAR plays no role in pDCs and that other uptake and signaling pathways specific for HAdV types are used in pDCs.
The production of proinflammatory cytokines was also determined in primary macrophages inoculated with helper-dependent (gutless) HAdV-C5 (Cerullo et al., 2007). TLR9 knockout mice had a reduced innate response to helper-dependent HAdV-C5 upon intravenous injection of the vector. In addition to TLR9, TLR2 also contributes to innate responses against HAdV. TLR2 detects triacylated lipoproteins from bacteria. TLR2 knockout mice showed reduced NF-κB activation and humoral responses to HAdV vectors (Appledorn et al., 2008). Notably, MyD88 knockout was, however, not sufficient to silence acute and adaptive responses to HAdV, indicating that other mechanisms than TLR signaling are important in innate and adaptive responses to HAdV (Fejer et al., 2008).
In addition, there is evidence that HAdV-C complexed with FX activates innate immunity through TLR4, and mounts an IL1β inflammatory response (Doronin et al., 2012). Interestingly, a HAdV-C variant ablated in FX binding failed to trigger the inflammasome response, but triggered other innate responses. This suggests that innate immune reactions depend on both the nature of the vector and soluble factors attached to the vector. It remains to be determined if differential responses are connected to trafficking pathways, such as endocytic uptake or subcellular location of subviral structures in immune or nonimmune cells (Mercer and Greber, 2013; Wang et al., 2013), or if blood factors bound to pathogens have direct immune signaling potential.
Lectin receptors
Lectin receptors (LRs) are a heterogeneous family of PRRs responding to DAMPs typically through direct binding to sugars of the pathogen. LRs are soluble proteins that can be released to the extracellular space, such as galectins (Gals) that bind to mannan sugars, or they are anchored in the plasma membrane, for example, the mannose-receptor dectin-1 (Geijtenbeek et al., 2004; Cerliani et al., 2011). LRs are frequently found on immune cells, such as conventional and pDCs, and are implicated in signaling crosstalk with TLRs, which is thought to enhance immunity (reviewed in Kawai and Akira, 2011; Osorio and Reis E Sousa, 2011).
Two LRs have been implicated in HAdV infection, sialic-acid-binding Ig-like lectins (Siglecs) and Gals (Fig. 2, top left). Siglecs are trans-membrane proteins involved in innate and adaptive immune responses. Similar but not identical to the human Siglec-8, the fiber knob of canine adenovirus was found to bind to terminal sialic acid on complex sugars containing galactose and N-acetyl glucosamine, although Siglec-8 and canine adenovirus fiber knob do not share sequence similarity (Rademacher et al., 2012). It can be speculated that sialic acid is an attachment site for canine adenovirus, similar to earlier reports that HAdV-C2/5 binds to sialic acid residues of heparin sulfate proteoglycans, although the functional implication remains unknown (Dechecchi et al., 2000, 2001). Possibly, the sialic acid residues on the cell surface exert an inhibitory effect on HAdV infection. For example, it was reported that expression of Muc1, which is an O-glycosylated membrane protein and part of the protective mucous barrier on the epithelial surface, reduced the infection of Madin Darby Canine Kidney cells with HAdV-C (Arcasoy et al., 1997). This inhibition was abrogated by treatment of cells with sialidase, thus suggesting that extracellular sialic acid residues inhibit HAdV infection.
The most prominent members of endosomal LRs implicated in HAdV infection are Gals. Gals are a family of β-galactoside-binding proteins with domains for carbohydrate recognition. Gals function in innate immunity and surveillance of innate immune processes (Rabinovich and Toscano, 2009). They are normally localized in intracellular compartments or the cytosol, and can be secreted by a nonconventional mechanism independent of a leader peptide (Seelenmeyer et al., 2005; Schneider et al., 2010). Interestingly, Gal3 puncta have been shown to colocalize with incoming HAdV-C5, and in some cases these colocalization events were also positive for exposed protein VI (Maier et al., 2012). This, together with experiments where mCherry-tagged Gal3 was ectopically expressed, was interpreted to suggest that Gal3 detected galactose sugar residues on ruptured endosomal membranes during HAdV-C5 entry. Whether these membranes were broken or not has remained unknown, however. Nevertheless, it is possible that the colocalization of Gal3 with HAdV-5 involved vesicular transport, for example, endosomal or plasma membrane-localized Gal3. Regardless of how Gal3 colocalized with HAdV-C5, proteomics analyses showed that both Gal1 and Gal3 were strongly downregulated in human lung epithelial cells upon infection with HAdV-C5 or B3 (Trinh et al., 2013). This reinforces the notion that HAdV drastically alters the function of Gals. It remains to be seen if Gals are degraded, or released from infected cells by nonconventional secretion. It is noteworthy that also newly synthesized penton base and fiber proteins in HAdV-C2-infected A549 cells are secreted by a nonconventional mechanism, and this secretion has been suggested to aid virus shedding from polarized epithelial cells (Walters et al., 2002; Trotman et al., 2003).
Cytosolic DNA: triggering inflammasomes
Besides TLRs and LRs, most mammalian cells have TLR-independent mechanisms to detect cytosolic viral DNA. These pathways can be proinflammatory and independent of IRFs. They enhance antiviral defense, and involve NLRs, the core of the inflammasomes.
Myeloid cells derived from granulocyte precursors in the bone marrow or the spinal cord contain a multiprotein complex of NLR family proteins, the so-called NLR-pyrin domain (PYD)-containing protein (NLRP) inflammasomes (reviewed in Tschopp et al., 2003; Bauernfeind and Hornung, 2013). NLRP inflammasomes further consist of NACHT, leucine-rich repeat (LRR), and pyrin domains-containing protein 1 (NALP1); apoptosis-associated speck-like protein (ASC); caspase 1; and yet other proteins. They comprise the classical NLRP1 and absent in melanoma 2 (AIM2), a PYHIN protein that leads to caspase 1 activation and maturation of IL1β. The AIM2-like receptors (ALR) sense double-stranded DNA via the HIN200 domain of AIM2, and interact with the Caspase-1 adaptor protein via a PYD domain (Hornung et al., 2009). Inflammasome activation triggers inflammatory responses via NF-κB signaling; converts pro-IL1β and pro-IL18 into IL1β and IL18, respectively; and can lead to DNA fragmentation, membrane pore formation, and eventually cell death by pyroptosis (reviewed in Fink and Cookson, 2005).
NLRs are cytosolic DNA-sensing proteins. They are composed of a central nucleotide-binding (NOD or NACHT) domain responsible for ATP-dependent self-oligomerization, a C-terminal LRR domain that senses the presence of a ligand and a variable N-terminal interaction domain that mediates protein–protein interactions, mainly via a CARD or PYD. NOD1–4 contain a CARD domain, NOD5 lacks an N-terminal domain, and the NALPs have a PYD domain. Signaling downstream of NLRs leads to the formation of inflammasomes, and this may involve microtubules as suggested for NLRP3 (Misawa et al., 2013). In addition, NLRs cooperate with TLRs to regulate inflammatory and apoptotic responses.
In HAdV-infected myeloid cells, two types of inflammasomes are activated, AIM2 and NALP3 (Fig. 2, lower right). HAdV DNA is sensed through AIM2 (Stein and Falck-Pedersen, 2012; Stein et al., 2012), the TRAF family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK1) (Nociari et al., 2007), the NALP3-ASC-caspase-1 complex (Muruve et al., 2008), or a yet unknown cytosolic DNA sensor in conventional DCs (Zhu et al., 2007). HAdV-C5 activation of the NLRP3 inflammasome was shown in THP1 cells conditioned with phorbol esters, and this led to the production of IL1β and the release of lysosomal cathepsin B to the cytosol (Barlan et al., 2011a,b). Interestingly, cathepsin B release did not correlate with lysosomal localization of the virus, suggesting that cathepsin relocalization occurs by an indirect mechanism.
The inflammasome was triggered not only in myeloid cells but also in the skin. HAdV-C5_dE1 vector application or liposome-mediated transfection of purified HAdV-C5_dE1 DNA to full skin ex vivo or in vivo, or into HaCaT or HKT cells led to the expression of inflammatory cytokines and type 1 IFN-β (Steinstraesser et al., 2011; Schulte et al., 2013). This was dependent on the DNA sensors AIM2, NALP3, and the RNA sensor MDA5. The transient knockdown of AIM2, NALP3, MDA5, and, to a small extent, also the DNA-dependent activator of IRFs (DAI) increased viral expression of GFP from the major cytomegalovirus promoter. This suggests the feasibility of HAdV gene transfer into immunosuppressed skin.
Cytosolic DNA: Pathway to Interferon
DNA sensors: definition and function
Foreign nucleic acids can be a major insult to the integrity and hereditary programs of cells. A major question is how cells distinguish between self and nonself nucleic acids. One point of distinction is recognizing structural features; for example, cytosolic double-stranded RNA with 5′-triphosphate groups is sensed by RLRs, such as RIG-I (Weber et al., 2013). Another point of distinction is the localization of the nucleic acid. Extracellular nucleic acids are detected by membrane-associated TLRs, such as TLR3, 7, and 8 binding to RNA, and TLR9 to double-stranded DNA. But for cytosolic DNA, the distinction by localization is not generally true, since DNA sensing occurs in both the nuclear and the cytosolic compartment, as shown for herpes virus (Li et al., 2012; Orzalli et al., 2012). It remains possible that innate signaling from cytosolic DNA occurs on specialized cytoplasmic structures or organelles.
The cell requires sensors for self versus nonself distinction of DNA. Sensors can be proteins with receptor function binding to DNA. These sensors induce an IFN or inflammatory cytokine responses upon DNA exposure.
Besides TLRs and inflammasome-associated DNA sensing, a range of cytosolic proteins have been implicated in protecting cells against double-stranded DNA, as shown, for example, in early studies with HAdV and myeloid cells (see Fig. 2, lower left) (Nociari et al., 2007, 2009; Zhu et al., 2007; Fejer et al., 2008). Cytosolic proteins implicated in IFN signaling upon DNA challenge include DAI, DNA-dependent protein kinase (DNA-PK) sensing linear double-stranded DNA and repairing DNA double-stranded breaks, RNA polymerase III converting cytosolic DNA into double-stranded RNA for RLR signaling, IFN-γ-inducible protein 16 (IFI16, also known as p204), a member of the Pyrin family, as well as DEAD (Asp-Glu-Ala-Asp) box polypeptide 41 (DDX41), and cyclic guanosine monophosphate–adenosine monophosphate synthase (cGMP-AMP synthase, short cGAS) (for reviews, see Weitzman et al., 2010; Rathinam and Fitzgerald, 2011; Ferguson et al., 2012; Xiao and Fitzgerald, 2013).
So far, there are only three proteins that appear to fulfill the strict definition of a DNA sensor: IFI16, DDX41, and cGAS. For HAdV, DDX41 and cGAS have been implicated. It is possible that viral interference blocks particular DNA-sensing pathways, or that features of HAdV-DNA camouflage recognition. For example, the covalent attachment of the terminal protein to the 5′ ends could prevent DNA-PK activation, or core protein VII could block IFI16 (Zhao et al., 2009; Karen and Hearing, 2011).
RNA polymerase III
The adenoviral genome encodes two RNA molecules, virus associated RNA I (VA-I) and VA-II. VA RNAs block the IFN-induced protein kinase R (PKR), which relieves protein synthesis inhibition in the antiviral state (Ma and Mathews, 1996). The VA-I and VA-II genes are transcribed by host RNA polymerase III (Pol III) into short noncoding RNAs of 157 and 158 nucleotides, respectively, and both have extensive secondary structures (Akusjarvi et al., 1980). Upon transfection, VA-I and VA-II were found to bind to RIG-I, a DEAD box helicase that binds to 5′-triphosphorylated double-stranded RNA (see Fig. 2, lower right) (Minamitani et al., 2011; Weber et al., 2013). HAdV-C5_dE1 induced a biphasic production of type 1 IFN (IFN-β) in human gastric cancer NU-GC-3 cells (Minamitani et al., 2011). Silencing of RIG-I or IRF3, or UV-inactivation of the virus reduced the late response at 48–60 hr postinfection, whereas the early response at 12–24 hr postinfection was not affected. This suggests that RIG-I and IRF3 are not required for a type 1 IFN response in early infection, but triggered by late events, which coincide with the expression of VA-I and VA-II. This makes it unlikely that HAdV DNA is subject to transcription by Pol III. This notion is supported by the observation that the immediate early HAdV transactivator E1A, which is present throughout the early phase of infection, blocks Pol III transcription (Sollerbrant et al., 1993).
For vaccinia virus and herpes virus infections, cytoplasmic Pol III was reported to transcribe double-stranded viral DNA to 5′-triphosphorylated RNA, which was sensed by RIG-I and turned into a type 1 IFN response through mitochondrial antiviral signaling protein (MAVS), also called IFN-β promoter stimulator (IPS1)/virus-induced signaling adapter (VISA)/Cardif (Chiu et al., 2009). RIG-I (or MDA5) interaction with MAVS occurs via a caspase recruitment domain (CARD), and activates the protein kinases IKKA and IKKB, and NF-κB translocation to the nucleus. In cell types defective of other DNA-sensing pathways (presumably cGAS/stimulator of IFN genes [STING]), the Pol III inhibitor ML-60218 blocked the production of type 1 IFN upon exposure to herpes viruses or Legionella bacteria, suggesting that Pol III can be part of an innate mechanism in the cytosol.
DDX41
DDX41 is a member of the DExD box family of ATP-dependent helicases, and a cytosolic DNA sensor in myeloid dendritic cells that works together with STING (Zhang et al., 2011). The sensor function depends on the Walker A and B motifs and directly binds to STING, and this is required for signaling together with DDX41 binding to DNA. Upon stimulation of cells with poly (dA:dT), STING relocates from the endoplasmatic reticulum (ER) to a vesicular compartment where it colocalizes with DDX41. Together, STING and DDX41 signal through TBK1, mitogen-activated protein kinases, and NF-κB, and trigger an IFN response. Knockdown of DDX41 in mouse DCs by RNA interference strongly reduced the type 1 IFN production upon challenge of cells with HAdV, similar to the knockdown of STING (see Fig. 2, lower left) (Zhang et al., 2011). Accordingly, the knockdown of DDX41 in RAW 264.7 cells reduced the levels of phosphorylated IRF3 after inoculation with replication-defective HAdV-C5 (Stein and Falck-Pedersen, 2012). The role of IRF7 was not addressed in this study, although IRF7 was critical for type 1 IFN induction by HAdV in mice (Fejer et al., 2008). Collectively, the data suggest that DDX41 is a cytoplasmic sensor that detects HAdV DNA, and is involved in triggering an antiviral DNA response in certain cells.
cGAS
cGAS is a nucleotidyl-transferase involved in sensing cytosolic DNA. Similar to IFI16, cGAS recognizes the DNA via the sugar backbone. This leaves open the possibility that cGAS also detects cellular DNA in the cytosol (Jin et al., 2012; Gao et al., 2013). cGAS catalyzes the formation of cyclic guanosine monophosphate–adenosine monophosphate (cGMP-AMP, short cGAMP) from ATP and GTP in a DNA-dependent manner (Sun et al., 2013). cGAMP was identified in Vibrio cholerae bacteria, where it functions in chemotaxis and colonization (Davies et al., 2012). In mammalian cells, cGAMP binds the adaptor protein STING with high affinity, and leads to the activation of TBK1 and IRF3 and the production of IFN-β (Ablasser et al., 2013a; Zhang et al., 2013). The cGAS/STING pathway is active in epithelial, endothelial, and myeloid cells, and is probably the most prominent pathway for protecting cells from untypical DNA. Interestingly, the transfer of the cGAMP second messenger between cells via gap junctions confers bystander effects from infected to uninfected neighboring cells (Ablasser et al., 2013b).
Recently, it was shown that HAdV DNA is sensed by cGAS that triggers a major IFN response in murine RAW 264.7 macrophage-like cells (see Fig. 2, lower left) (Lam et al., 2014). This response is likely related to the observation that incoming HAdV-DNA is delivered not only to the nucleus, but also to the cytosol (Wang et al., 2013).
STING: downstream effector and signaling hub
In order to establish an innate immune response, upstream sensors amplify signals by engaging downstream cascades. These cascades involve adaptor molecules, MAVS, MyD88, TIR-domain-containing adapter-inducing IFN-β, or STING (also known as trans-membrane protein 173). This triggers activation of transcription factors, for example, NF-κB, AP1, IRF3/7, or signal transducer and activator of transcription 1/2 (STAT1/2), and eventually leads to a type 1 IFN response. However, this response is not guaranteed, since the sensors and downstream effector proteins are often expressed in a cell type-specific manner, and viruses actively interfere with the signaling cascade. For example, it was shown that replication-defective HAdV-C5_dE1 is efficiently sensed in RAW264.7 macrophages, as indicated by phosphorylation of IRF3 and STAT1/2, but did not elicit a response in FL83B hepatocytes (Stein et al., 2012). The reason for lack of DNA signaling in the hepatocytes was apparently the lack of STING, as shown by ectopic expression of STING and RNA interference. STING localizes to the ER in close association with mitochondria (Ishikawa and Barber, 2008). STING senses cGAMP, dimerizes, and thereby activates type 1 IFN through TBK1-mediated phosphorylation of IRF3 and STAT6. STING can be activated by binding to DDX41, DAI, or IFI16. IFI16 was shown to directly bind viral DNA, and STING was recruited to IFI16 after DNA stimulation (Unterholzner et al., 2010). Phosphorylated IRF3 and STAT6 dimerize and enter the nucleus for transcriptional gene activation (Tanaka and Chen, 2012). Importantly, STING can be downregulated by the E3-ubiquitin ligase ring finger protein 5 (Zhong et al., 2009).
It is interesting to note that HAdV-C5 was reported to induce necrosis of liver CD68-positive macrophages, independent of STING, through a mechanism involving IRF3 upstream of transcription (Di Paolo et al., 2013). This necrosis pathway involved the permeabilzation of endosomes or the plasma membrane, as suggested by the observation that the endosome-escape defective HAdV-C2_TS1 did not induce necrosis (Imelli et al., 2009; Di Paolo et al., 2013). In this scenario, IRF3 is not acting on STING, or triggering apoptosis or IFN signaling, but rather involved in necrotic cell death, which may be a pathway not involving STING.
Autophagy: proviral or antiviral?
Beyond controlling inflammasomes, TBK1 plays important roles in triggering innate immune responses against double-stranded DNA, which critically depends on STING (Saitoh et al., 2009). STING is an ER-associated membrane protein. Upon sensing cytosolic DNA, STING moves from the ER to the Golgi, and then associates with TBK1 on punctate structures in the cytoplasm, which contain the autophagy-related gene 9a (ATG9a). The structures lack ATG5 and ATG7, suggesting that they are not double-membrane autophagosomes. Cells with depleted ATG9a have enhanced STING-TBK1 complexes, and aberrantly high activation of innate immunity upon sensing cytosolic DNA, and TBK1 association in this compartment is key for an IFN response. Hence, nonconventional autophagy membrane trafficking downtunes innate immunity upon DNA sensing.
Autophagy also negatively regulates the secretion of IL1β downstream of inflammasome activation, downtunes inflammatory responses, and potentially enhances infection (Deretic et al., 2012). Autophagy augments unconventional secretion of signal-peptide-lacking proteins (Nickel and Rabouille, 2009; Dupont et al., 2011), but it is unknown if it accounts for the loss of Gal1 and Gal3 from HAdV-infected cells (see Fig. 2, lower left) (Trinh et al., 2013).
Classical autophagy eliminates long-lived organelles and other cytosolic substrates by isolating and delivering them to lysosomes (for reviews, see Munz, 2011; Deretic et al., 2012; Randow and Munz, 2012). Autophagy was originally found to be upregulated under nutrient starvation in order to recycle cellular constituents and maintain homeostasis (for a historical review, see Yang and Klionsky, 2010). There are three major forms of autophagy: chaperone-mediated autophagy, micro-autophagy, and macro-autophagy. Chaperone and micro-autophagy directly deliver substrates into lysosomes, whereas macro-autophagy engulfs cytosolic substrates with a double-lipid membrane, and these structures then fuse with lysosomes. Engulfing bacteria or viruses by macro-autophagy is termed xenophagy (from Greek “strange-eating”), and limits infection (reviewed in Levine et al., 2011).
The autophagosomal isolation membrane around cytoplasmic contents is formed by recruitment of the class III phosphatitylinositol-3-OH kinase complex at ER–mitochondria contact sites (Hamasaki et al., 2013). This consists of Vps34, Vps15, beclin-1 (ATG6), and ATG14, which recruits further effector proteins for the generation of the isolation membrane. Elongation of this structure is mediated by two ubiquitin-like conjugation systems. First, ATG12-ATG5 is produced by the E1-like activity of ATG7 and E2-like activity of ATG10 together with ATG16L1. Second, E1- and E2-like activities of ATG7 and ATG3, respectively, lead to the conjugation of ATG8 homologs, for example, microtubule-associated protein 1A/1B-light chain 3 (LC3), with phosphatidyl-ethanolamine (Levine et al., 2011). These serve for the elongation of the structure and loading of cargo that is bound to LC3-interacting proteins, such as p62 or Alfy (Bjørkøy et al., 2005). Subsequently, the ATG5-ATG12-ATG16L1 complex dissociates from the outer phagosomal membrane upon completion of the compartment. Eventually, the autophagosome matures by fusion with late endosomes and lysosomes in a Rab7-GTPase-dependent manner, and lysosomal hydrolases degrade luminal contents as well as the inner autophagosomal membrane (Mizushima et al., 1998; Jager et al., 2004).
Normally, airway cells are under high oxygen pressure, which induces adaptive autophagy (Ryter and Choi, 2010). Respiratory pathogens, such as HAdV, have likely adapted to take advantage of such conditions. Indeed, it has been shown that adaptive autophagy, for example, induced by starvation in airway cell cultures, enhanced the expression of early HAdV-C2 genes and virus production (see Fig. 2, lower left) (Zeng and Carlin, 2013). Conversely, inhibition of autophagy decreased viral yields, possibly by lowering the recycling of nutrients (Rodriguez-Rocha et al., 2011). It is possible that autophagy-mediated infection enhancement is associated with fusion of early endosomes with autophagosomes. Some of the fused compartments, the so-called amphisomes, were positive for HAdV-C2, suggesting that HAdV-C2 may use amphisomes to break free into the cytosol (Zeng and Carlin, 2013). This aspect of infection could be enhanced by autophagy, and deserves further attention.
In other instances, autophagy was found to be induced, for example, in human glioma cells inoculated with a second-generation HAdV-C5 vector dl922–947 (Mcneish et al., 2005). dl922–947 has a small E1A deletion in the conserved region 2, and a deletion in the E3B locus (Heise et al., 2000). When autophagy was reduced with broad-range inhibitors, such as chloroquine or 3-methyladenine, the cytotoxic effects of dl922–947 were enhanced in cell cultures and mouse xenograft models (Mcneish et al., 2005). Cancer cells may use autophagy to enhance their survival, and defend against HAdV vectors. It is possible that E3B tunes autophagy, and E3B is missing in dl922–947. E3B is also known to contain RIDα (receptor internalization and downregulation α), an integral membrane protein of early and late endosomes (Crooks et al., 2000). It acts as a GTP-Rab7 mimic interacting with Rab7 effectors, such as Rab7-interacting lysosomal protein or oxysterol-binding protein-related protein 1 (Shah et al., 2007). Interestingly, RIDα was shown to rescue the cholesterol storage phenotype of Niemann–Pick disease type C mutant fibroblasts, and is involved in lipid droplet formation (Cianciola and Carlin, 2009; Cianciola et al., 2013). One can envisage that a combination of autophagy-inducing compounds together with HAdV may enhance oncolytic efficacy of viral therapies (Rodriguez-Rocha et al., 2011; Cheng et al., 2013).
Countering the IFN response: HAdV early proteins and noncoding RNAs
The early proteins of HAdV, including proteins from the E1, E3, and E4, are the best studied host innate response antagonists (reviewed in Weitzman and Ornelles, 2005). In addition, the VA-RNAs antagonizing PKR and the structural protein VI have been described in attenuating innate antiviral response (Burgert et al., 2002; Schreiner et al., 2012). For a schematic representation, see Fig. 2.
E1A proteins
Early analyses have indicated that multiple HAdV genes interfere with host immunity. Of particular note is the E1A protein, the immediate early viral transactivator. E1A is transcribed and alternatively spliced soon after arrival of the viral DNA genome in the nucleus. E1A proteins encoded by 9S, 12S, and 13S mRNAs exert a large array of effects, including control of the cell cycle, apoptosis, immune evasion, tumorigenesis, and viral gene expression (for reviews, see Berk, 1986; White, 1993; Burgert et al., 2002; Frisch and Mymryk, 2002). E1A changes the epigenetic program of the cell within just a few hours of infection (Ferrari et al., 2008; Horwitz et al., 2008).
E1A potently blocks type 1 IFN-inducible gene expression (see Fig. 2, upper left) (Ackrill et al., 1991; Gutch and Reich, 1991; Kalvakolanu et al., 1991). The inhibitory activity of E1A depended on the conserved region 1 (CR1) domain. In addition, E1A blocks the induction of HLA class II genes by type 2 IFN-γ, and IFN-β mRNA in response to double-stranded RNA, and this involves a block in transcription complex formation (Kalvakolanu et al., 1991). Specifically, E1A targeted the IFN-alpha-stimulated transcription factor 3 (ISGF3) consisting of Stat1, Stat2, and p48 by inhibiting p300 and/or cAMP response element-binding protein (CREB)-binding protein (CBP) (Bhattacharya et al., 1996). P300/CBP is targeted by E1A, and this leads to repressed Stat2 transactivation. Another mechanism by which E1A blocks IFN-stimulated gene (ISG) transcription is by interference with histone 2B mono-ubiquitination through the ubiquitin ligase RNF20/hBRE1, which is necessary for ISG transcription (Fonseca et al., 2012). E1A also activates viral transcription by recruiting the scaffold protein hPAF1 to RNF20/hBRE1, and this boosts viral infection (Fonseca et al., 2013).
Furthermore, E1A interacts with the 20S and 26S proteasome, in particular the immunoproteasome, which emerges from regular proteasomes upon IFN-γ treatment (Berhane et al., 2011). E1A also interferes with the presentation of peptides to the immunoproteasome by interacting with the MECL1 of the immunoproteasome, and downregulating MECL1 expression. This interception of innate immunity reduces antigen presentation on infected cells and enhances the survival. Collectively, all this illustrates the great versatility of E1A, which is an intrinsically disordered protein that works as a major functional hub in a context-dependent manner (Ferreon et al., 2013).
E1B proteins
In addition to E1A, a significant number of other HAdV gene products modulate host immune responses, and thereby help the virus to persist in an infected host (Mahr and Gooding, 1999; Wold et al., 1999). Most prominently, the E1B-19K and E1B-55K proteins expressed early in infection antagonize E1A-induced p53-mediated apoptosis (Sabbatini et al., 1995; Teodoro and Branton, 1997). In addition, E1B-55K interferes with the induction of IFN-inducible genes, as E1B-55K null viruses are exquisitely sensitive to type 1 IFN (Chahal and Flint, 2012; Chahal et al., 2012). The transcriptional repression mechanism by E1B-55K occurs through the tumor suppressor protein p53, which interacts with E1B-55K (Chahal et al., 2013).
In addition, E1B-55K together with the E4 open reading frame 6 (E4orf6) protein, which is an E3-ubiquitin ligase, triggers proteasome-mediated degradation of defense factor death-domain-associated (Daxx) (Schreiner et al., 2010, 2013a). Daxx restricts viral gene expression by forming a complex with the ATP-dependent helicase (ATRX). The degradation of Daxx thereby relieves a viral transcription block, and allows viral gene expression. It was also reported that the virion protein VI inhibited Daxx (Schreiner et al., 2012). Since protein VI is rapidly degraded during virus entry and the mode of Daxx inhibition by protein VI does not seem to involve Daxx degradation, the stoichiometry of incoming protein VI does not match that of Daxx, particularly since Daxx is induced by IFN (Greber et al., 1993; Gongora et al., 2001; Burckhardt et al., 2011; Schreiner et al., 2012).
E1B-55K also works in complexes with E4 proteins to block antiviral innate reactions. Together with E4orf3, E1B-55K relocates the Mre11-Rad50-Nbs1 (MRN) complex and thereby precludes the formation of concatamers and DNA damage signaling during viral replication, thus increasing virus yield from infected cells (Stracker et al., 2002, 2005; Evans and Hearing, 2003, 2005; Carson et al., 2009). Yet another complex of E1B-55K functions in enhancing viral gene expression. E1B-55K-E4orf6 and a cullin-based E3 ubiquitin ligase targets the antiviral factor SPOC1 for degradation by the proteasome (Schreiner et al., 2013b). SPOC1 normally works in DNA damage response.
E3 proteins
E3 proteins are best known for their immunomodulatory functions (reviewed in Burgert et al., 2002; Horwitz, 2004; Lichtenstein et al., 2004). The E3-glycoprotein 19K (E3-gp19K) blocks MHC I transport to the plasma membrane, and thereby reduces the attack of infected cells by leukocytes. E3-gp19K also lowers the cell surface levels of receptors for natural killer (NK) cells, which furthers the survival of infected cells (Mcsharry et al., 2008). E3-14.7K (interference with apoptosis), E3-10.4K (named also receptor internalization and degradation RID-α), E3-14.5K (RID-β), and E3-6.7K block extrinsic apoptosis by downregulation of death receptors, and inhibition of cellular mediators that block the inflammatory and cell survival factor NF-κB. In addition to apoptosis control, RID-α induces a class III PI3-kinase-dependent cholesterol trafficking pathway that leads to the formation of autophagy-like vesicles distinct from late endosomal/lysosomal cholesterol storage compartments (Cianciola and Carlin, 2009). The observation that RID-α controls transport of low-density lipoprotein-cholesterol complexes from endosomes to the ER for cholesterol esterification suggests that RID-α controls aspects of lipid droplet formation (Cianciola et al., 2013). How these unexpected lipid trafficking phenotypes relate to cell death or innate immunity needs to be explored further.
Recently, distinct features of an unusual E3 protein from HAdV-D19, E3-49K, were reported (Windheim et al., 2013). E3-49K is targeted to the secretory pathway and proteolytically cleaved to the soluble fragment E3-sec49K and released from infected cells. E3-sec49K bound to leukocyte CD45, a receptor protein tyrosine phosphatase. It reduced expression of activation markers on NK cells, and inhibited phosphorylation of T cell receptor, suggesting that it has immunomodulating functions on NK cells and T cells, but the exact role of E3-sec49K in natural adenovirus infection is still unknown.
E4 proteins
The E4 region of HAdV-C encodes at least 7 distinct proteins involved in viral late gene expression, nonhomologous end joining, DNA damage response, and apoptosis (reviewed in Weitzman, 2005). For example, E4orf6 together with E1B-55K from HAdV-C induces the selective export of viral late mRNAs from the nucleus to the cytoplasm, and inhibits export of cellular mRNAs (Flint and Gonzalez, 2003). Another E4 protein also cooperates with E1B-55K. E4orf3 together with E1B-55K inhibits the MRN complex, which would otherwise block viral replication (see section E1B).
Independent of this function, E4orf3 inhibits IFN production and disturbs the organization of promyelocytic leukemia protein (PML) bodies (also called PML oncogenic domain, or nuclear domain 10) (Carvalho et al., 1995; Ullman et al., 2007; Ullman and Hearing, 2008; Leppard et al., 2009). PML bodies are mounted by the IFN-induced proteins PML and Daxx, and they bind the HAdV E1A proteins depending on the CR2 region of E1A (Carvalho et al., 1995; Gongora et al., 2001). Since the E4 locus is conserved across many mastadenoviruses, including the human types, it is possible that E4orf3 acts to overcome species specific innate virus restriction.
Domesticating HAdV: An Outlook
Coating the virus
HAdV is the vector of choice for systemic gene delivery because of high stability and efficacy. Nonetheless, HAdVs bind to components of the blood, including erythrocytes, platelets, complement, and coagulation factors; the viruses are sequestered to the liver, taken up into immune cells, destroyed by complement, or trapped by nontarget cells (Jiang et al., 2004; Lyons et al., 2006; Othman et al., 2007; Stone et al., 2007; Carlisle et al., 2009b). For a schematic representation, see Fig. 3.
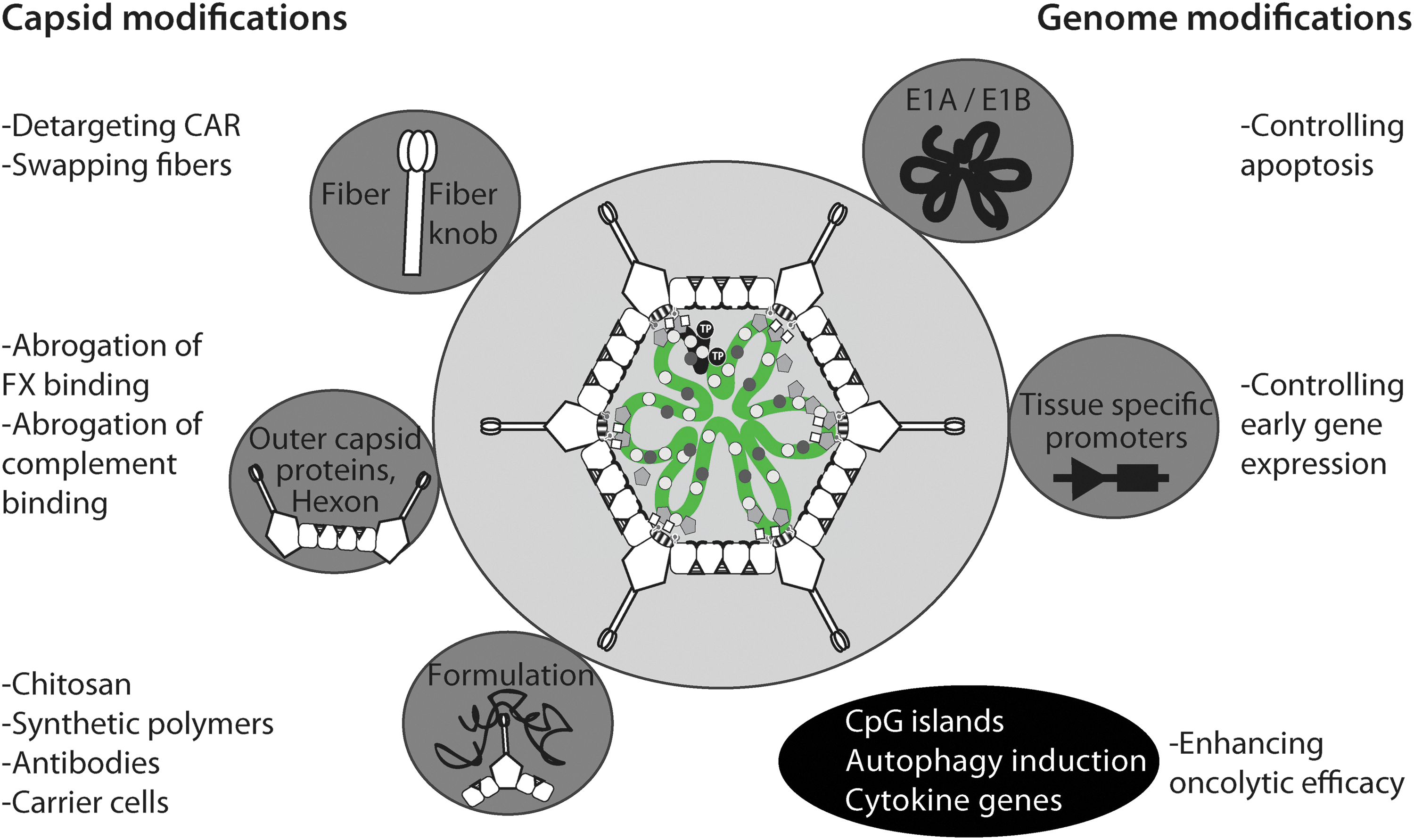
Domesticating adenovirus for gene therapy. To tailor HAdV for clinical purposes, the capsid (left section, gray circles) or genome (right section, gray circles) can be modified. Capsid modifications include swaps of fiber or fiber knob between different HAdV types, hexon modifications, or coating the virus with chemicals, such as synthetic polymers. Genome modifications involve deletion or replacement of viral genes or promoters to enhance or attenuate viral replication or toxicity. The latter is prominently used in oncolytic approaches (lower section, black ellipse) aiming to eliminate diseased tissue. For details, see main text.
To improve the pharmacokinetics of the virus, different strategies for virus surface modifications have been tested. For example, HAdV-C2 has been coated with soluble fusion protein comprising the extracellular domain of CAR and the constant region of human Ig to target immune cells (Meier et al., 2005). Alternatively, polymers are used to shield immunogenic proteins, such as hexon. For example, coating the surface of HAdV with polyethylene glycol (PEG) is a well-studied modification (e.g., Hofherr et al., 2008; Green et al., 2012). PEG-coated HAdVs elicit less intense immune responses compared with uncoated virus (O'riordan et al., 1999; Croyle et al., 2001). Circulating proinflammatory cytokines, such as IL6, IL12, and TNFα, and liver transduction were reduced in primates receiving PEG-ylated compared with non-PEG-ylated HAdV (Wonganan et al., 2011). Additionally, PEG-ylated HAdV can be functionalized by conjugation of antibodies, and this may increase the targeting of the vector to particular cell types (Kim et al., 2011). Interestingly, PEG-ylation of HAdV-C_dE1/E3/E4 in combination with the anti-inflammatory glucocorticoid methylprednisolone reduced vector uptake into the spleen and nonparenchymal liver cells, and inhibited thrombocytopenia (De Geest et al., 2005). This suggests that vector shielding and downtuning of innate immunity is beneficial for vector applications in murine models.
Besides PEG, other polymers such as N-(2-hydroxypropyl)methacrylamide (HPMA) and chitosan have been successfully tested in preclinical studies (Carlisle et al., 2013; Kwon et al., 2013). The combination of HPMA-coated HAdV with ultrasound gave increased vector delivery to tumors, while reducing liver toxicity compared with naked HAdV in immune deficient mice.
Genetic alterations of the viral capsid
Any surface modification of the vector exclusively affects the first round of infection, but not subsequent rounds of replication. To shield progeny viruses, genetically modified vectors are used. A common strategy has been to swap fibers between immunogenic and nonimmunogenic HAdV types, or exchange immunodominant epitopes on the hexon protein. For example, the fiber of the CAR-tropic HAdV-C5 has been transferred to the capsid core of HAdV-B35, which has one of the lowest levels of seroprevalence (Vogels et al., 2003). Nonetheless, the fiber knob-swapped HAdV-B35_FK5 was more immunogenic than HAdV-B35 in both nonhuman primates and mice (Nanda et al., 2005), suggesting that either the knob of HAdV-C5 or the entry pathway of the CAR-tropic HAdV-B35_FK5 was more immunogenic than the knob or the CD46 pathway of HAdV-B35 (Fleischli et al., 2005). Clearly, further studies are needed to sort out the mechanisms of immune activation by HAdV in animals and humans.
Conclusions
In this review, we have laid out how HAdV is detected by the host innate immune system, and highlighted some of the mechanisms by which the virus antagonizes innate responses. It is clear that HAdV infection affects cell physiology in many ways, including transcriptional profiles and proteomes, and likely also metabolomes and lipidomes. HAdV also breaks the rules of membrane traffic by disrupting organelles, such as endosomes or the nuclear pore complex. It is not unreasonable to expect that further danger signals from HAdVs will be discovered in studies with isolated cells or model animals, such as mice. It should be noted, however, that mouse models have limitations for HAdV infection biology, as mice do not allow HAdV replication, unlike pigs, for example (Jogler et al., 2006). Alternative systems may overcome some of these limitations. For example, human tissue explants with cell type complexities akin to human organs may allow to probe the impact of innate immune factors on HAdV replication and progeny production at single cell resolution in a complex cellular environment.
Future studies will also deal with the contested issue of multiplicity of infection (MOI). Researchers frequently use different MOIs for cell and animal studies or between different cell types. It is important to note that both high and low MOIs occur in lytic HAdV infections, and that viremia indicating high viral load is found in patients (Heemskerk et al., 2005; Yakimovich et al., 2012). It is fundamentally important to define not only the number of viruses added to cells or animals, but also how many viruses actually bind and internalize to cells of interest and hence trigger infection or innate immune reactions. For instance, high MOI may exacerbate particular innate immune responses by saturating limiting host functions that support infection, such as the nuclear pore complex for import of HAdV genomes into the nucleus (Wang et al., 2013). This may enhance the innate response to danger signals, for example, cytosolic viral DNA.
Finally, to address organismic mechanisms of adenoviruses innate immunity, we believe that it is worth considering mouse adenoviruses (MAdVs). For example, MAdVs elicit proinflammatory responses in murine airways similar to HAdV, despite considerable genetic differences between MAdV and HAdV (Meissner et al., 1997; Weinberg et al., 2005; Hemmi et al., 2011).
Outlook
The recent development of methods to study the trafficking of viruses and subviral structures in both immune and nonimmune cells now enables the field to further probe the mechanisms underlying the cell and immune biology of innate responses against HAdV. From such experiments, new procedures using immunostimulatory or immunoreducing treatments for vector applications may emerge. Particular attention will be paid on careful dosing of the virus in order to control innate immune reactions from the host, and to minimize unwanted inflammatory responses to the vector. We also expect that major efforts will be spent on pushing the best understood HAdV-C vectors into clinical trials before other HAdV types with unknown features will be used in humans, although vaccinations with nonhuman adenoviruses are considered to be promising (Ewer et al., 2013). In summary, a balanced mix of in vitro and in vivo studies complemented with clinical data will be essential to tackle the fundamental questions in innate immunity to HAdV. Such approaches will also address other outstanding questions related to innate immunity, for example, how genetically identical cells and organisms can be variably susceptible to virus infections.
Footnotes
Acknowledgments
We thank Dr. Maarit Suomalainen (University of Zurich), Dr. Gyuri Fejer (University of Plymouth, United Kingdom), and Dr. Justin Flatt (Case Western Reserve University, Cleveland, OH) for comments on the article.
The work was supported by a grant from the Swiss National Science Foundation (SNSF 31003A_141222/1 to U.F.G.), and an Initial Training Network grant “AdVance” from the European Union supporting R.H., N.S., J.K., L.K., and A.L. (to U.F.G. and other principal investigators of AdVance, coordinated by Dr. A. Baker, University of Glasgow, United Kingdom).
Author Contributions
R.H. and N.S. wrote the first draft of the article and drew figures; J.K., L.K., and A.L. drafted part of the article; and U.F.G. conceived, coordinated, and wrote the final article.
Author Disclosure Statement
The authors declare that no competing financial interests exist.