
Abstract
Artemis is a single-stranded endonuclease, deficiency of which results in a radiation-sensitive form of severe combined immunodeficiency (SCID-A) most effectively treated by allogeneic hematopoietic stem cell (HSC) transplantation and potentially treatable by administration of genetically corrected autologous HSCs. We previously reported cytotoxicity associated with Artemis overexpression and subsequently characterized the human Artemis promoter with the intention to provide Artemis expression that is nontoxic yet sufficient to support immunodevelopment. Here we compare the human Artemis promoter (APro) with the moderate-strength human phosphoglycerate kinase (PGK) promoter and the strong human elongation factor-1α (EF1α) promoter to regulate expression of Artemis after ex vivo lentiviral transduction of HSCs in a murine model of SCID-A. Recipient animals treated with the PGK-Artemis vector exhibited moderate repopulation of their immune compartment, yet demonstrated a defective proliferative T lymphocyte response to in vitro antigen stimulation. Animals treated with the EF1α-Artemis vector displayed high levels of T lymphocytes but an absence of B lymphocytes and deficient lymphocyte function. In contrast, ex vivo transduction with the APro-Artemis vector supported effective immune reconstitution to wild-type levels, resulting in fully functional T and B lymphocyte responses. These results demonstrate the importance of regulated Artemis expression in immune reconstitution of Artemis-deficient SCID.
Introduction
A
Clinical trials have demonstrated the effectiveness of gene transfer into autologous hematopoietic stem cells (HSCs) for treatment of adenosine deaminase (ADA)-deficient SCID and X-linked SCID. 9 –15 The success of these trials demonstrates that ex vivo gene transfer can be an effective treatment for genetic deficiency, a compelling argument for genetic correction of other forms of SCID, including SCID-A. Two independent groups reported the correction of murine models of SCID-A by transplantation of genetically modified HSCs. 16,17 In both studies, Artemis-deficient animals were transplanted with HSCs that had been transduced with a lentiviral vector encoding human Artemis regulated by the human phosphoglycerate kinase (PGK) promoter, resulting in reconstitution of B and T lymphocyte compartments. 16,17 Surprisingly, Mostoslavsky and colleagues reported lack of lymphoid reconstitution in RAG-1-deficient animals transplanted with SCID-A HSCs that had been transduced using lentiviral vectors encoding human Artemis regulated by the stronger cytomegalovirus (CMV) or elongation factor-1α (EF1α) promoter. 16
We subsequently demonstrated that overexpression of Artemis after lentiviral transduction is associated with cytotoxicity, a halt in cell cycle progression, and fragmentation of genomic DNA ultimately resulting in apoptosis. 18 These results, along with the previous reports demonstrating incomplete immune reconstitution of SCID-A after ex vivo transduction with an exogenous promoter, 16,17 emphasize the importance of providing Artemis expression at a level that is nontoxic and yet sufficient to correct the SCID-A T−B− phenotype. Accordingly, we isolated and characterized the human Artemis promoter (APro) as a sequence extending 1 kilobase upstream from the human Artemis translational start site on human chromosome 10. 19 Ex vivo transduction of murine bone marrow with an APro-regulated green fluorescent protein (GFP) lentiviral vector conferred GFP expression at a significantly reduced level in comparison with control mice transplanted with EF1α-GFP-transduced marrow and supported GFP expression in all hematopoietic lineages that persisted in secondary transplant recipients. 19 These results established the usefulness of this promoter for providing reliable, moderate-level gene expression in hematopoietic cells. 19
In this study, we evaluated the effect of promoter strength on immune reconstitution after ex vivo lentiviral transduction of the Artemis coding sequence in a murine model of SCID-A. Previous studies of ex vivo lentiviral correction of Artemis deficiency 16,17 used a SCID-A mouse model exhibiting leaky T lymphocyte development, evident from low numbers of single- and double-positive thymocytes and CD4+ T cells in peripheral blood. For our study, we used a murine model of SCID-A that is nonleaky and thus more accurately models the human SCID-A clinical presentation and phenotype. 20 We also bred our SCID-A model onto both CD45.1 and CD45.2 congenic backgrounds, allowing us to quantitatively track donor engraftment at the cellular level. Bone marrow from Artemis-deficient mice was transduced with lentiviral vectors regulating the Artemis coding sequence using the moderate-strength human PGK promoter, the strong human EF1α promoter, or the human Artemis promoter (APro) and then transplanted into congenic, irradiated SCID-A recipients. We found that both APro-Artemis and PGK-Artemis transduction supported effective engraftment of T and B lymphocytes to normal levels. Furthermore, animals treated with APro-Artemis demonstrated a restored response to in vivo antigen challenge and in vitro mitogen stimulation. In contrast, EF1α-Artemis-treated animals exhibited reduced engraftment potential, were unable to repopulate the B lymphoid compartment, and lacked the ability to class switch and display antigen-specific IgG. These results suggest the necessity for near endogenous levels of Artemis to achieve functional immune reconstitution on ex vivo complementation of Artemis deficiency, demonstrating the importance of transgene regulation. These studies also validate the effectiveness of the human Artemis promoter in restoring immune function in SCID-A mice and, potentially, in human SCID-A.
Materials and Methods
Plasmids
Lentiviral vector plasmids pCSIIEG, 21 pCSEPuro, 18 pOK/EF1α-Artemis (including the EF1α intron 1 sequence), and pOK/PGK-Artemis 18 have been previously described. For construction of pOK/APro-Artemis, the EF1α promoter was excised from pOK/EF1α-Artemis with AgeI, yielding pOK/Artemis. The APro fragment was excised from pCR2.1/APro 19 with AgeI and ligated into the AgeI site of pOK/Artemis directly upstream of the murine Artemis coding sequence (Fig. 1A). Luciferase expression plasmids pAPro-Luc (pGL3-APro 19 ) and pPGK-Luc (pGL3-based 22 ), and the simian virus 40 (SV40)-regulated plasmid pGL3-C (Clontech, Palo Alto, CA) have been previously described. Plasmid pEf1α-Luc was assembled by ligation of a 1102-bp XhoI fragment (including the EF1α promoter) isolated from pDL2G 23 into pGL3-Basic at the XhoI site upstream of the luciferase coding sequence.
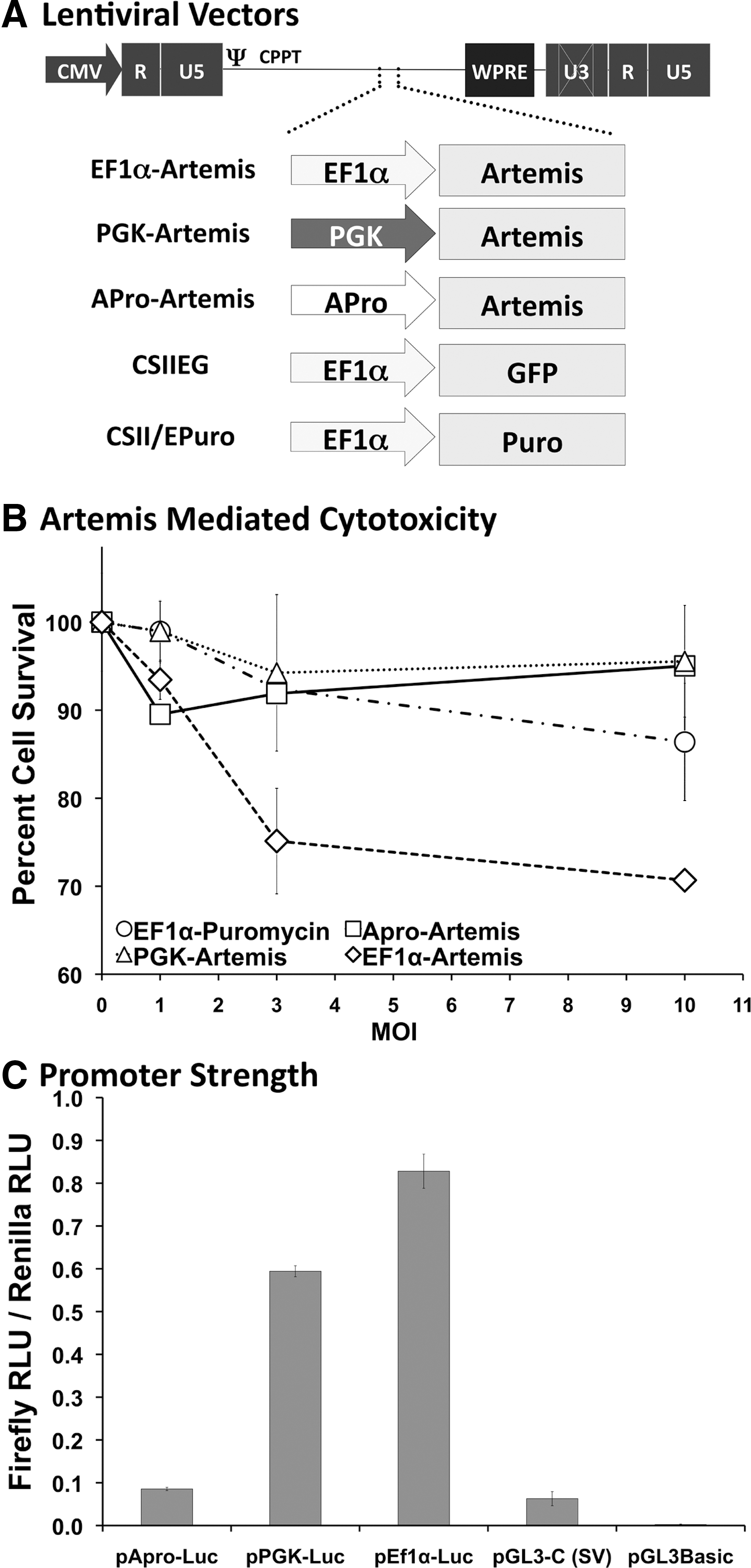
Lentiviral vector constructs and Artemis-associated cytotoxicity.
Mammalian cell culture and transfection
HEK 293T and murine NIH 3T3 thymidine kinase (TK)− cells were routinely cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic at 37°C and 5% CO2. HEK 293T cells (3×106/6-cm plate) were transfected using Lipofectamine 3000 (Life Technologies/Thermo Fisher Scientific, Carlsbad, CA) with 2 μg of test firefly luciferase-encoding plasmid along with 25 ng of phRL-CMV standardizing plasmid encoding Renilla luciferase. Two days later cells were harvested and lysates assayed for both firefly and Renilla luciferases, using a Berthold Lumat luminometer (Berthold Detection Systems, Bad Wildbad, Germany) and the Dual-Luciferase reporter assay system (Promega, Madison, WI).
Preparation and titering of lentiviral vectors
Vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped lentiviral vectors were generated as described.
23,24
Briefly, 1.4×107 HEK 293T cells were seeded into poly-
Vector titers were determined by transduction of NIH 3T3 TK− cells in the presence of Polybrene (8 μg/ml). Forty-eight hours later, DNA was extracted from the exposed cells and vector copy number was determined by quantitative PCR for the integrated lentiviral strong stop sequence or for the GFP sequence as previously described. 23 Concentrated vector titers ranged from 108/ml to 109/ml. CSIIEG was also titered for GFP expression by flow cytometry as previously described. 23
Ex vivo lentiviral transduction and bone marrow transplantation
All procedures were reviewed and approved by the University of Minnesota Institutional Animal Care and Use Committee. A murine model of Artemis deficiency backcrossed onto C57BL/6 background and exhibiting no leakiness 20 was maintained under specific pathogen-free conditions at the University of Minnesota and further bred onto both CD45.1 and CD45.2 congenic backgrounds. CD45.2 and CD45.1 C57BL/6 mice were obtained from the National Cancer Institute (Frederick, MD). Animals were provided food and water ad libitum.
Bone marrow was flushed from the hind limbs of donor mice into DMEM supplemented with heparin (10 U/ml), 10% FBS, and 1% Pen Strep (Invitrogen, Carlsbad, CA). Red blood cells were lysed with ammonium chloride hemolysis buffer (0.8% NH4Cl with 0.1 mM EDTA) (StemCell Technologies, Vancouver, BC, Canada), and then the nucleated cells were washed with 1×phosphate-buffered saline (PBS) and rendered into a single-cell suspension in transduction medium (complete StemPro-34 SFM medium with supplements [Invitrogen]) supplemented with 2 mM
For secondary transplantation, marrow samples were collected from individual primary recipients as described previously and the whole marrow sample was divided equally and infused into three secondary irradiated C57BL/6 (800 rads, X-irradiation source) recipients.
Flow cytometry
Whole blood was collected from the submandibular vein, treated with ammonium chloride hemolysis buffer (0.8% NH4Cl with 0.1 mM EDTA) (StemCell Technologies), washed, and then the leukocytes were pelleted and resuspended in staining buffer (1×PBS, 1% FBS, and 0.002% sodium azide) plus fluorochrome-conjugated monoclonal antibodies recognizing CD45.1, CD45.2, B220 (B lymphocytes), CD3e (T lymphocytes), CD4 (helper T lymphocytes), CD8α (cytotoxic T lymphocytes), NK1.1 (natural killer cells), Gr-1 and CD11b (myeloid lineages). Fluorochrome staining and GFP expression (for CSIIEG-transduced cells) were assayed on an LSRII instrument. Isotype staining was done to provide an internal control and to determine appropriate gating. Data were collected using CellQuest Pro (BD Biosciences, San Jose, CA) and analyzed using FlowJo (Tree Star, Ashland, OR) software. Total lymphocyte number was determined by Hemavet (Drew Scientific, Dallas, TX) analysis of whole blood. Total B cells and T cells were calculated as the product of the percentage of B cells, CD4+ T cells, and CD8+ T cells times the total number of donor lymphocytes.
Determination of lentiviral integrant copy number in peripheral blood
At various time points, DNA was extracted from whole blood, using Whatman FTA Elute Micro Cards (Fisher Scientific, Pittsburg, PA) according to the manufacturer's instructions. Cells stained with anti-IgM (B lymphocytes) and anti-CD3e (T lymphocytes)–fluorochrome were sorted on a BD FACSDiva instrument into IMDMEM plus 10% FBS. Isotype staining was used to determine appropriate gating. DNA was extracted from the sorted cells or from total peripheral blood mononucleocytes upon sacrifice, using a ZymoBead genomic DNA kit (Zymo Research, Cambridge Bioscience, Cambridge, UK). Whole genome amplification was performed on DNA extracts from the sorted cells, using an illustra GenomiPhi V2 DNA amplification kit (GE Healthcare, Piscataway, NJ). Vector copy number was determined by quantitative PCR for the lentiviral strong stop sequence as described. 23
In vivo KLH challenge
Test animals were immunized by intraperitoneal injection of 100 μg of NP-KLH (4-hydroxy-3-nitrophenylacetyl hapten-conjugated keyhole limpet hemocyanin) (Biosearch Technologies, Novato, CA) and boosted 5 weeks later. Before immunization and 1 week after the boost injection, sera were collected, aliquoted, and stored at −20°C for analysis by enzyme-linked immunosorbent assay (ELISA). Microtiter plates (Nunc, Rochester, NY) were coated with 0.5 μg of NP-BSA (4-hydroxy-3-nitrophenylacetyl hapten-conjugated bovine serum albumin) (Biosearch Technologies) in coating buffer (0.05 M carbonate–bicarbonate, pH 9.6) and incubated overnight at 4°C. Coating solution was aspirated from the plate and each well was washed three times in Tris–saline (50 mM Tris-buffered saline [TBS], pH 8.0; 0.05% Tween 20), incubated with blocking solution (50 mM TBS, pH 8.0; 1% BSA) for 30 min, washed three times, and then incubated for 1 hr with test serum samples serially diluted in sample buffer (50 mM TBS, pH 8.0; 1% BSA, 0.05% Tween 20). The wells were washed five times and then supplemented with horseradish peroxidase (HRP)-conjugated secondary antibodies diluted in sample buffer (1:100,000 dilution of anti-IgM–HRP and 1:150,000 dilution anti-IgG–HRP; both antibodies from Bethyl Laboratories, Montgomery, TX). After 1 hr, the plates were washed five times and then supplemented with HRP enzymatic reaction components, using a TMB 20-component microwell peroxidase substrate kit (Kirkegaard & Perry, Gaithersburg, MD). Reaction products were quantified with a Biotech SpectraMax plate reader at 450 nm.
Splenocyte stimulation
Spleens were harvested from mice on sacrifice and brought to a single-cell suspension in RPMI plus 10% FBS. The spleen cell suspension was treated with ammonium chloride hemolysis buffer, washed twice in RPMI plus 10% FBS, and resuspended in complete RPMI medium (supplemented with 1% Pen Strep and 10% FBS). Microtiter plates were treated with anti-CD3e antibody diluted in 1×PBS to 0, 2, 5, and 10 μg/ml, added to wells in triplicate, and incubated at 37°C for 4 hr. The wells were washed twice with 1×PBS and then 4×105 splenocytes were added to each well and incubated at 37°C and 5% CO2 for 48 hr. For concanavalin A (Con-A) stimulation, splenocytes were plated at 5×105 cells per well. The plate was supplemented with Con-A (Amersham Biosciences, Piscataway, NJ) diluted in complete RPMI medium to yield final concentrations of 0, 2.5, 5, and 10 μg/ml, and then incubated at 37°C and 5% CO2 for 48 hr. After incubation, proliferation was assayed with a CellTiter 96 nonradioactive cell proliferation assay (MTT) kit (Promega) and quantified with a Biotech SpectraMax plate reader at 570 nm. Results are reported as proliferation index, calculated as the absorbance observed in the presence of mitogen divided by absorbance observed for samples incubated without mitogen.
Statistical analysis
Data were statistically evaluated either by unpaired Student t test when variance was compared between two groups or by analysis of variance (ANOVA) when measuring three or more levels for variability, using Prism 4 software (GraphPad Software, San Diego, CA), with p<0.05 considered significant.
Results
Innate regulation of Artemis prevents Artemis-mediated cytotoxicity
Previously, we reported that overexpression of Artemis results in apoptosis due to DNA fragmentation and a halt at G1 in the cell cycle. 18 We subsequently characterized the 1-kb APro segment 19 as a weak promoter both in vitro and in vivo with the intention of employing this endogenous element to regulate Artemis expression in lentiviral vector-transduced cells. To determine the effect of promoter strength on Artemis-mediated cytotoxicity and the potential for correction of SCID-A by lentiviral transduction, an APro-regulated Artemis vector was compared with other vectors previously generated and characterized that contain the Artemis coding sequence regulated by the strong EF1α promoter and the more moderate-strength PGK promoter (Fig. 1A). Dose-dependent cell survival was assessed in 3T3 cells 5 days posttransduction with increasing amounts of APro-, PGK-, and EF1α-regulated Artemis lentiviral vectors, with an EF1α-regulated puromycin resistance vector serving as a transduction control (Fig. 1B). Similar to our previous report, 18 in a 5-day survival study mouse 3T3 cultures transduced with both EF1α-Puro and PGK-Artemis remained viable at all vector doses (p>0.05 at an MOI of 10); however, a dose-dependent decrease in cell survival was observed for cultures transduced with EF1α-Artemis at increasing multiplicities of infection (p<0.05 vs. all other groups; Fig. 1B). In contrast, we observed that cultures transduced with the APro-Artemis vector remained viable even at increasing multiplicities of infection, demonstrating that innate regulation of Artemis obviates a cytotoxic response (p<0.05 vs. EF1α-Artemis; p>0.05 vs. all other groups; Fig. 1B). In a side-by-side comparison with the EF1α and PGK promoters, we found that the level of expression conferred by the Artemis promoter was reduced by 6- to 8-fold after transfection of luciferase-encoding constructs into human 293T cells (Fig. 1C).
Engraftment after lentiviral transduction of the Artemis coding sequence
To evaluate the comparative effectiveness of lentiviral transduction in the correction of murine SCID-A, whole bone marrow was harvested from donor CD45.1 SCID-A animals, exposed twice (once overnight and once 20 hr postharvest) to either APro-Artemis, PGK-Artemis, or EF1α-Artemis at an MOI of 30, and then transplanted into CD45.2 recipient SCID-A animals preconditioned with 500 cGy of X-irradiation. This was the lowest effective sublethal dose resulting in donor cell engraftment that did not give rise to clinical signs of irradiation toxicity in control animals not receiving a transplant. In animals preconditioned at lower doses of irradiation (100, 200, or 300 cGy), donor mononuclear cells were observed by 8 weeks posttransplantation in all groups, but lymphocyte repopulation was not observed at 16 weeks posttransplantation (data not shown). Donor-derived lymphocytes emerged in recipient animals preconditioned with 500 cGy by 4 weeks posttransplantation, excluding the control group receiving CSIIEG-transduced SCID-A marrow (Fig. 2). Subsequently, donor cell populations increased over time so that by 16 weeks posttransplantation 100% of animals receiving APro-Artemis-transduced marrow demonstrated donor-derived lymphocytes, whereas 83% of PGK-Artemis-treated animals and 80% of EF1α-Artemis-treated animals demonstrated donor lymphocyte engraftment (Fig. 2). Of the total lymphocyte compartment, PGK-Artemis- and EF1α-Artemis-treated animals contained a lower percentage of donor-derived lymphocytes (77.7 and 87.4% donor lymphocytes, respectively) than animals treated with APro-Artemis (95.1%) (p<0.01; Table 1). In addition, by 16 weeks posttransplantation 80% of animals receiving APro-Artemis-transduced marrow demonstrated a repopulated lymphocyte compartment equivalent to wild-type animals (>2×103/μl), whereas only 66% of PGK-Artemis-treated animals and 40% of EF1α-Artemis-treated animals reached wild-type cellular levels (Fig. 2). Animals transplanted with CSIIEG-transduced SCID-A marrow demonstrated low levels of donor mononuclear cell engraftment, whereas animals transplanted with CSIIEG-transduced wild-type C57BL/6 marrow engrafted to wild-type levels by 8 weeks posttransplantation (Fig. 2).
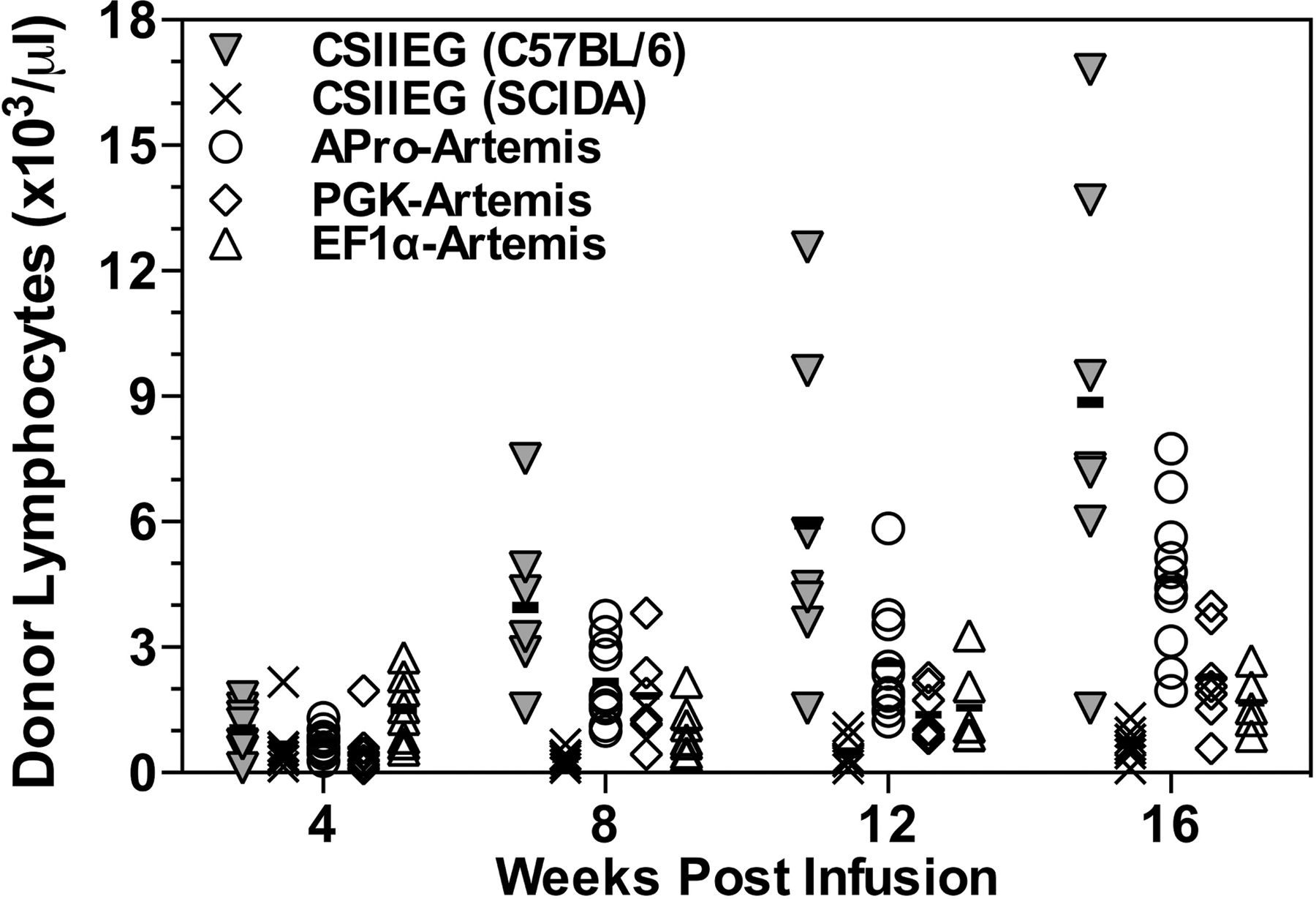
Donor engraftment in SCID-A mice: Time course. After infusion of transduced donor marrow, peripheral blood was collected over a period of 16 weeks to monitor donor lymphoid engraftment and repopulation, and plotted as the number of donor lymphocytes (×103) per microliter. Each symbol represents the results from a single animal for the groups indicated in the key. Bars represent mean values.
GE, genome equivalent; PBMCs, peripheral blood mononuclear cells; VC, vector copies.
Total lymphocyte number exhibited by animals 16 weeks posttransplantation.
Sixteen weeks posttransplantation, donor engraftment was determined.
Quantitative PCR for the lentiviral strong stop sequence was used to determine lentiviral integration frequency in peripheral blood collected 14 weeks posttransplantation and is presented as percent strong stop copies per genome equivalent in peripheral blood mononucleocytes.
Quantitative PCR for the lentiviral strong stop sequence was used to determine vector copy number in peripheral blood cells sorted for CD3+ or IgM+ cells and is presented as the number of viral copies per genome equivalent.
All groups were found to have significant levels of gene marking by qPCR of DNA extracted from peripheral blood 16 weeks posttransplantation (Table 1). APro-Artemis-treated animals exhibited marking at 6.8%, whereas EF1α-Artemis- and PGK-Artemis-treated animals demonstrated 2.7 and 2.0% gene marking, respectively (Table 1). Control SCID-A animals transplanted with CSIIEG-transduced wild-type marrow displayed only 0.6% gene marking, demonstrating a selective advantage for engraftment and repopulation of Artemis-transduced cells (Table 1).
Immune reconstitution after ex vivo Artemis transduction
After transplantation with transduced SCID-A marrow cells, the B lymphocyte compartment (B220+NK1.1− cells) achieved wild-type levels in groups receiving both PGK- and APro-regulated Artemis (Fig. 3A). On the other hand, there was an absence of B lymphocytes in animals transplanted with EF1α-Art-transduced marrow throughout the 16-week time course of the experiment (Fig. 3A). The majority of treated animals in all three groups had CD3+CD4+ helper and CD3+CD8+ cytotoxic T lymphocyte levels in the normal range, with APro-Artemis-treated animals exhibiting levels close to those achieved by transplantation with wild-type marrow (p>0.05) (Fig. 3B and C).
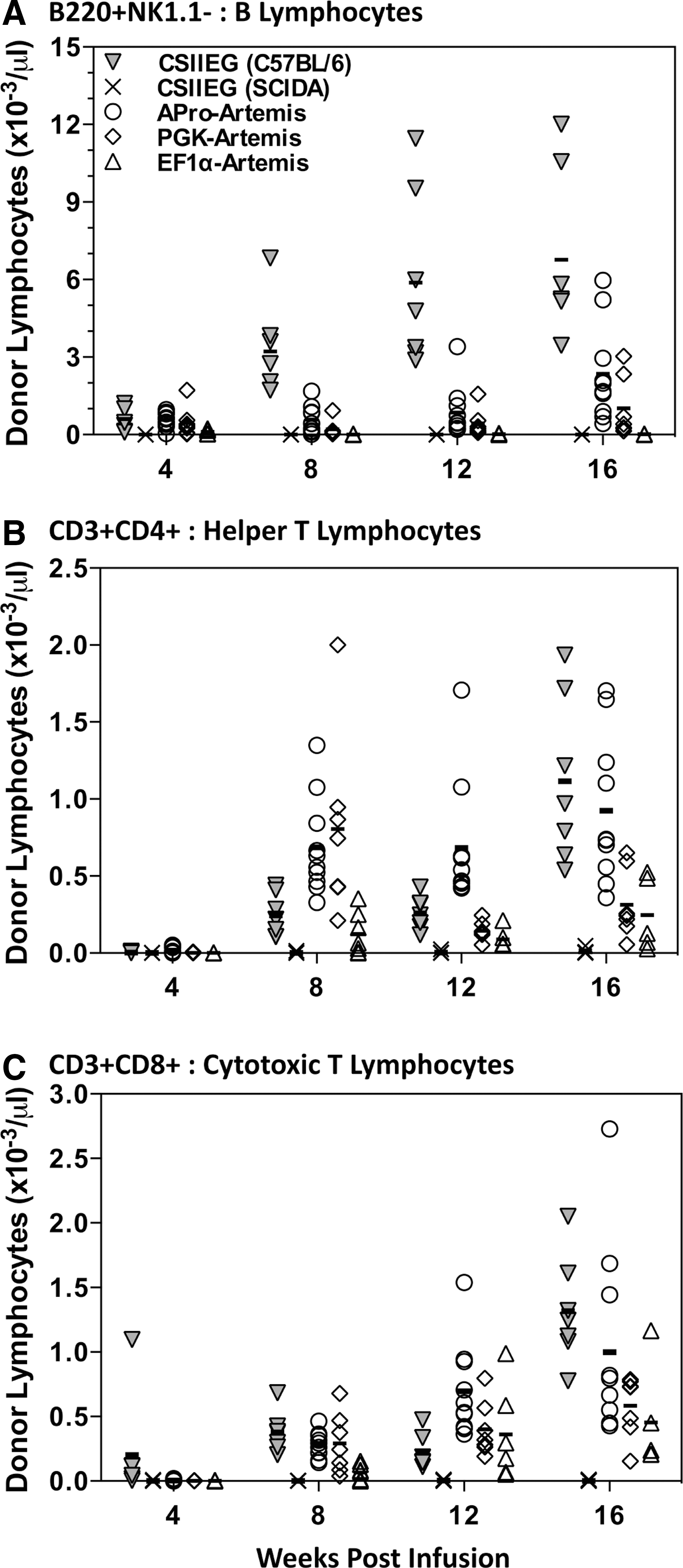
Repopulation of circulating lymphocyte subsets in transplanted SCID-A mice. Peripheral blood was collected over a period of 16 weeks posttransplantation for analysis of lymphocyte populations.
Upon sacrifice, whole blood was drawn from vector-treated animals and from C57BL/6 control animals, sorted for CD3+ and IgM+ populations, and assayed for vector integrants by qPCR analysis of the strong stop sequence. APro-Artemis-treated animals contained an average of 2.1 vector copies per genome equivalent in both CD3+ and IgM+ populations, and PGK-Artemis-treated animals contained an average of 1.7 and 2.8 vector copies per genome equivalent in the CD3+ and IgM+ populations, respectively, demonstrating that all resulting B and T lymphocytes were generated from transduced prelymphoid targets containing at least one lentiviral integrant. EF1α-Artemis-treated animals contained 0.42 vector copies per genome equivalent in the CD3-sorted population, and essentially undetectable vector copies in the IgM-sorted population.
Restored immune function in APro-Artemis-treated animals but aberrant function in EF1α-Artemis-treated animals
At 18 weeks posttransplantation, all animals were challenged in vivo with two injections of 4-hydroxy-3-nitrophenylacetyl hapten-conjugated keyhole limpet hemocyanin (NP-KLH) to evaluate humoral immune response function. One week after the boost injection, all treated groups of animals mounted a significant IgM response against NP-KLH, comparable to wild-type animals (p>0.05) (Fig. 4A). APro-Artemis- and PGK-Artemis-treated animals also exhibited effective class switch and generated an IgG response against the antigen, with only the APro-Artemis-treated group exhibiting a significant response comparable to wild-type animals (p>0.1) (Fig. 4B). Interestingly, EF1α-Artemis-treated animals were unable to generate anti-NP-KLH IgG, demonstrating a failure to class switch and indicating an aberrant immune response against antigen challenge (Fig. 4B).
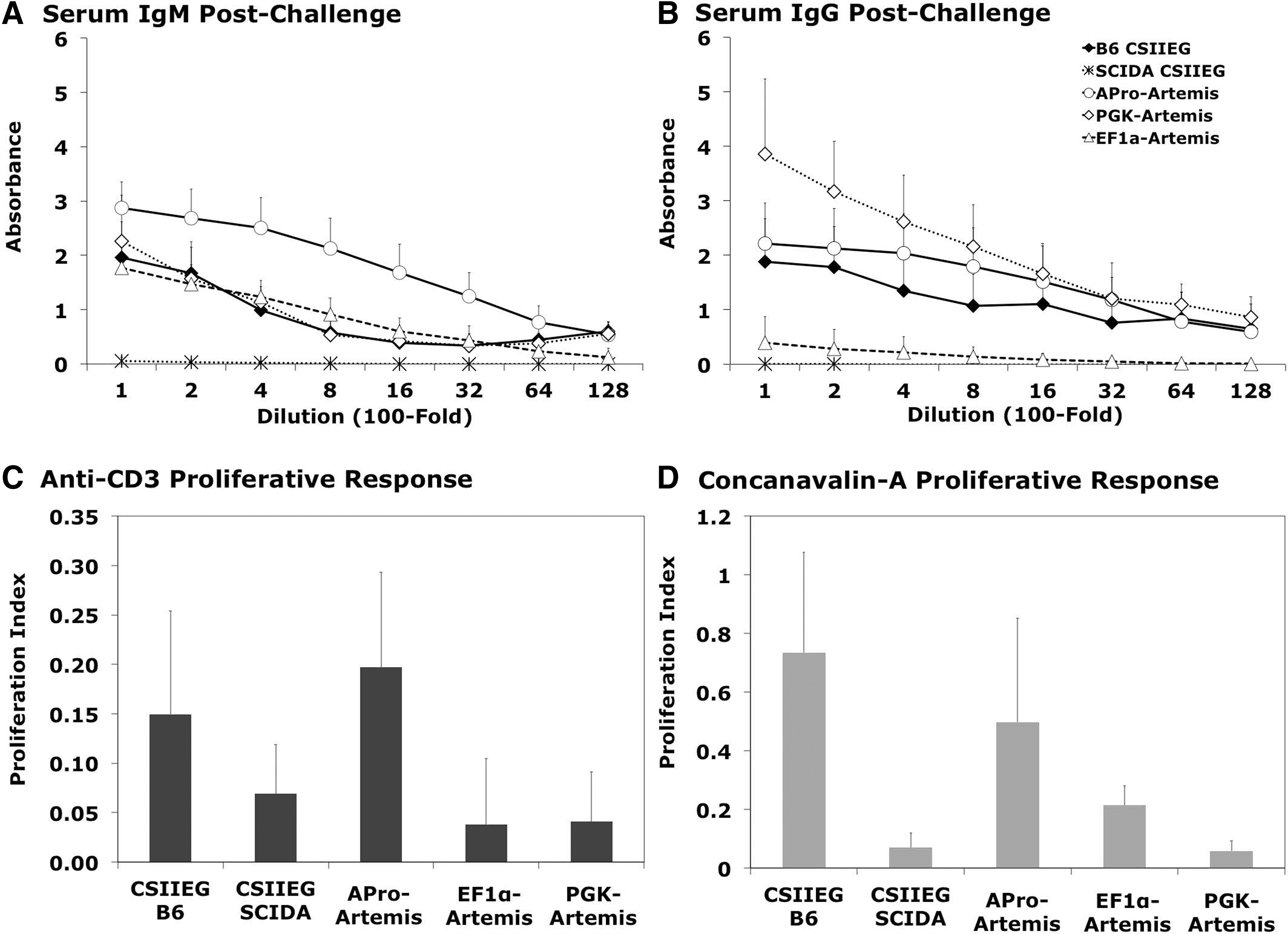
Functional in vivo immune response.
After the in vivo NP-KLH challenge, all animals were killed for further immunophenotyping of cell populations within lymphoid organs. Both APro-Artemis- and PGK-Artemis-treated animals demonstrated wild-type levels of B220+IgM+ B lymphocytes and CD3+CD4+, CD3+CD8+ T lymphocytes in both primary and secondary lymphoid organs: bone marrow (Fig. 5A), thymus (Fig. 5B), spleen (Fig. 5C), and lymph nodes (Fig. 5D). Representative flow plots are shown in Supplementary Fig. S1 (supplementary data are available online at
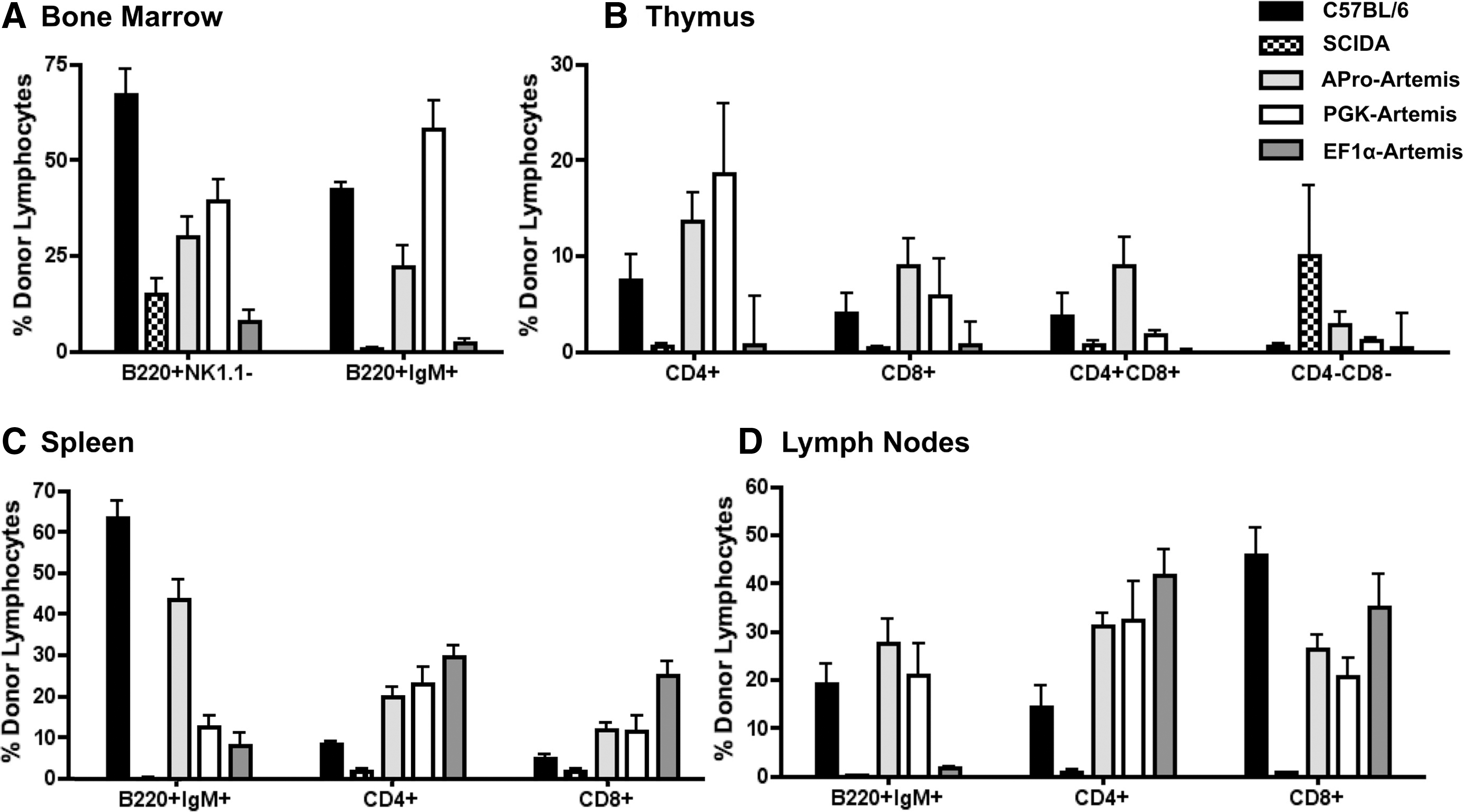
Repopulation within lymphoid organs. Upon sacrifice, primary lymphoid organs were harvested and single-cell suspensions were analyzed by flow cytometry for the presence of B, helper T, and cytotoxic T donor lymphocyte populations of Artemis-treated animals, control C57BL/6 animals, and control SCID-A animals.
To evaluate proliferative responses, splenocytes were prepared from animals on sacrifice and cultured in the presence of either anti-CD3 or Con-A. Splenocytes from APro-Artemis-treated animals exhibited a proliferative response to both stimuli that was similar to that of wild-type C57BL/6 animals (p>0.1), whereas there was essentially no proliferative response of splenocytes from Artemis-deficient control animals (Fig. 4C and D). Animals treated with either PGK-Artemis or EF1α-Artemis demonstrated a markedly reduced proliferative response to either anti-CD3 or Con-A as compared with APro-Artemis-treated animals (p<0.0028 and p<0.028, respectively) (Fig. 4C and D).
Normal immunodevelopment of APro-Artemis- and PGK-Artemis-transduced cells after secondary transplantation
Secondary transplantation was carried out for all treatment groups (APro-Artemis, PGK-Artemis, and EF1α-Artemis) 6 months after primary transplantation of transduced SCID-A marrow. Sixteen weeks later, the majority of animals transplanted with marrow from animals treated with either APro-regulated (Fig. 6A) or PGK-regulated (Fig. 6B) Artemis demonstrated repopulation of both B (B220+NK1.1−) and T (CD3+CD4+ and CD3+CD8+) donor lymphocyte compartments (100% of animals treated with APro-Artemis and 83% of animals treated with PGK-Artemis vectors). Reduced levels of donor CD3+CD8+ cells in secondary recipients of PGK-Artemis-treated animals could be the result of Artemis toxicity. The sustainability of these lymphoid compartments after secondary engraftment demonstrates the capability to maintain Artemis gene expression after transduction into primitive hematopoietic stem cells. In contrast, only 20% of secondary animals receiving EF1α-Artemis-transduced marrow demonstrated repopulation of B lymphocytes and CD3+CD8+ T lymphocytes (Fig. 6C). The failure to repopulate lymphocyte populations in EF1α-Artemis secondary-transplanted animals may be due to Artemis-associated toxicity, either in developing lymphoid cells or in more primitive lymphohematopoietic progenitors.
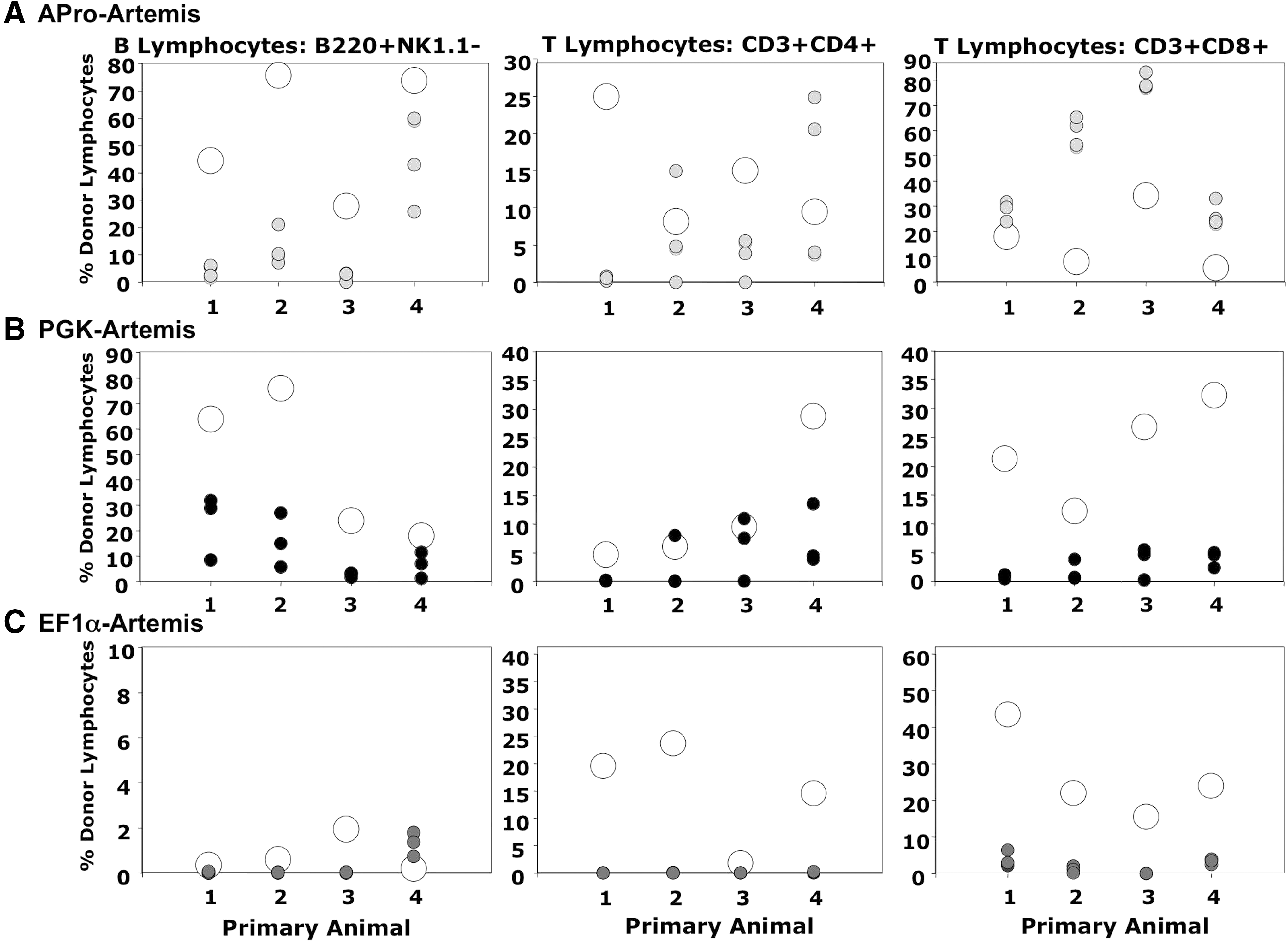
Lymphoid reconstitution after secondary transplantation of transduced marrow. Bone marrow was collected individually from primary recipients treated with
One set of secondary transplant recipients (established from a single EF1α-Artemis-treated primary recipient) exhibited massively enlarged spleens, a condition subsequently characterized histologically as lymphoma with infiltration of several organs. DNA extracted from the marrow and spleen of these animals exhibited only 0.003 to 0.04 lentiviral vector copies per genome equivalent, however, so this was most likely a naturally occurring tumor that arose in the primary Artemis–/– recipient animal that was not associated with lentiviral integration.
Discussion
Regulation of human Artemis expression by its endogenous promoter effectively complemented the Artemis-deficient phenotype, solving the problem of cytotoxicity associated with Artemis overexpression. In contrast, animals treated with an EF1α-Artemis construct exhibited repopulation of the T lymphocyte compartment but an absence of circulating B lymphocytes. In addition, these animals demonstrated an aberrant response to NP-KLH antigen, representative of a dysfunctional immune system. Treatment of SCID-A animals with either PGK-Artemis or APro-Artemis provided complete reconstitution of both B and T lymphocyte compartments. Most importantly, APro-Artemis-treated animals mounted in vivo immune responses against NP-KLH antigen and displayed in vitro mitogenic responses of splenocytes more effectively than PGK-Artemis-treated animals and similar to wild-type animals, thus demonstrating the effectiveness of lentiviral transduction using the natural human Artemis promoter for correction of SCID-A.
Gene transfer is emerging as a promising approach for the treatment of genetic disorders, exemplified by results from clinical trials demonstrating the effectiveness of transplantation using autologous HSCs after ex vivo genetic correction by retroviral transduction for two severe combined immunodeficiencies caused by genetic deficiency of adenosine deaminase and the common γ chain receptor. 10 –14 These studies report long-term engraftment of corrected stem cells in a majority of patients, ultimately resulting in reconstitution of cellular and humoral immunity. However, two independent studies reported adverse events after ex vivo genetic correction of X-linked SCID, 10,12,15 in which 5 of 20 patients developed clonal T cell outgrowth resulting in a leukemia-like syndrome. 26,27 Although insertional oncogene activation was reported in three of the leukemic cases it has also been demonstrated that overexpression of the common γ chain induces cellular proliferation and may have contributed to the T lymphocyte clonal outgrowth. 26 –28 Tighter regulation of the common γ chain gene may thus reduce the risk of oncogenesis resulting from aberrant overexpression.
Achieving expression of Artemis for the correction of SCID-A may present a challenge in transgene regulation similar to what was encountered during the X-linked SCID trial, considering the cytotoxicity associated with Artemis overexpression. We found that overexpression of Artemis upon lentiviral transduction results in genomic shearing, cell cycle arrest, and apoptosis. 18 Considering the endonucleolytic activity of Artemis 29 –32 these results are not surprising, yet they emphasize the importance of providing Artemis expression at a level that is nontoxic and yet sufficient to correct the T−B− phenotype in preclinical studies and in clinical application to human SCID-A. We subsequently reported the isolation and characterization of the endogenous human Artemis promoter as a sequence that effectively mediates gene expression at levels substantially lower than that mediated by the strong EF1α promoter both in vitro and in vivo. 19 Here we extend these results to show lack of cytotoxicity after transduction using a lentiviral vector in which Artemis is naturally regulated. These data establish the effectiveness of APro as a proficient promoter for gene expression in the hematopoietic system, specifically for the regulation of Artemis expression but potentially applicable to other gene products as well.
Consistent with previous reports, we demonstrate that Artemis expression regulated by the moderate-strength PGK promoter resulted in a functional lymphoid compartment; however, it was not reconstituted to wild-type cellular levels and was unresponsive to splenocyte proliferation stimulus. Treatment of SCID-A animals with EF1α-Artemis led to aberrant immune reconstitution with an absence of B lymphocytes detected in the circulation, failure to generate an effective IgG response to NP-KLH, and unresponsiveness of splenocytes to proliferative stimulus. These data suggest that Artemis dysregulation or overexpression may be effecting improper lymphocyte development and repopulation, especially within the B lymphocyte lineage. The presence of B lymphocytes in primary lymphoid organs coupled with the absence of these cells in the peripheral organs and blood of EF1α-Artemis-treated animals suggests an arrest in B cell maturation. One possible mechanism to explain such an arrest would be Artemis-mediated apoptosis during B cell development and maturation. During immunodevelopment, B lymphocytes undergo genomic alteration in the context of V(D)J recombination, activation-induced cytidine deaminase-mediated somatic hypermutation, and class switch recombination, 33,34 all of which require highly active and effective DNA repair machinery. 35 –38 Immature B lymphocytes acquiring an excess number of nonproductive mutations undergo Fas-mediated apoptosis. 39,40 It is tempting to speculate that the coupling of these natural DNA DSB repair-inducing events with the global DNA damage accrued upon Artemis overexpression further sensitizes the cell to apoptotic stimuli. Consistent with this argument, the T lymphocyte population, which does not undergo somatic hypermutation or class switch recombination, was repopulated in EF1α-Artemis-treated mice. In addition, the observed abundance of T cells and absence of B cells may be expected considering that T cells possess a greater propensity to fill a void lymphocyte compartment via homeostatic cytokine signaling than do B cells. What's more, upon sacrifice of the EF1α-Artemis-treated mice, characterization of the splenic B lymphocytes revealed a predominant IgMhigh/IgDlow population (Supplementary Fig. S2), which may suggest an early transitional or marginal B lymphocyte population. 41 –43 Lack of class switch recombination along with the observed B lymphocyte surface marker repertoire indicates incomplete repopulation within the EF1α-Artemis-treated animals by an early transitional thymic-independent B lymphocyte as opposed to a mature thymic-dependent B lymphocyte population. 43,44
Overall, these results underscore the requisite of providing levels of Artemis that avoid cytotoxicity yet allow for successful reconstitution of the lymphoid lineages. We provide an example of how Artemis overexpression results in aberrant lymphoid reconstitution and dysfunctional immune response, and corroborate previously reported results demonstrating complete correction by using a moderately regulated Artemis vector. 16,17 Moreover, we report the complete reconstitution of a functional lymphoid population in SCID-A animals by ex vivo lentiviral gene transfer using a vector in which Artemis expression is regulated by the natural human promoter. These results suggest that the natural levels of Artemis expression achieved via ex vivo lentiviral transduction into hematopoietic stem cells will serve as a clinically relevant and feasible treatment of human SCID-A.
Footnotes
Acknowledgments
The authors thank Dr. Tucker LeBien for helpful discussion. The authors acknowledge the assistance of the Flow Cytometry Core Facility of the Masonic Cancer Center, a comprehensive cancer center designated by the National Cancer Institute, supported in part by P30 CA77598. This work was supported by NIH grant R01 AI063340 to R.S.M., by March of Dimes grant #6-FY-5-84 to M.J.C., and by NIH grant U54 A1082973 to M.J.C.
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.