
Abstract
For long it has been recognized that tumor necrosis factor alpha (TNFa) has anticancer characteristics, and its use as a cancer therapeutic was proposed already in the 1980s. However, its systemic toxicity has limited its usability. Oncolytic viruses, selectively cancer-killing viruses, have shown great potency, and one of their most useful aspects is their ability to produce high amounts of transgene products locally, resulting in high local versus systemic concentrations. Therefore, the overall magnitude of tumor cell killing results from the combination of oncolysis, transgene-mediated direct effect such as TNFa-mediated apoptosis, and, perhaps most significantly, from activation of the host immune system against the tumor. We generated a novel chimeric oncolytic adenovirus expressing human TNFa, Ad5/3-D24-hTNFa, whose efficacy and immunogenicity were tested in vitro and in vivo. The hTNFa-expressing adenovirus showed increased cancer-eradicating potency, which was shown to be because of elevated apoptosis and necrosis rates and induction of various immune responses. Interestingly, we saw increase in immunogenic cell death markers in Ad5/3-d24-hTNFa-treated cells. Moreover, tumors treated with Ad5/3-D24-hTNFa displayed enhanced presence of OVA-specific cytotoxic T cells. We thus can conclude that tumor eradication and antitumor immune responses mediated by Ad5/3-d24-hTNFa offer a new potential drug candidate for cancer therapy.
Introduction
O
Since 1975, it has been known that tumor necrosis factor alpha (TNFa) has anticancer properties, and it is possibly the most tumor cell toxic molecule produced by the human body. 7,8 In fact, it has been proposed to be the “active ingredient” in Coley's toxin, the first tumor immunotherapy used in the late 1880s. 8,9 In initial clinical studies performed with recombinant TNFa in the 1980s, its antitumor properties were confirmed, but severe systemic toxicity was also encountered. 10,11 Thus, systemic use was soon abandoned and its only current clinical use is in isolated limb perfusion. 12,13
TNFa is a multifunctional cytotoxic molecule that has multiple properties of a powerful anticancer agent. It can induce tumor cell death by apoptosis and necrosis. It is also an inflammatory and immunostimulatory cytokine that can stimulate release of other cytokines and activate and recruit immune cells. 14,15 It is also documented that TNFa has synergistic actions with radiation by increasing the apoptotic and necrotic cell death and vascular damage. 16,17 We hypothesized that an oncolytic, replicating virus expressing human TNFa would achieve high expression of the transgene in the tumor, thus providing all the beneficial effects of TNFa in the tumor but avoiding the systemic toxicity.
TNFerade (GenVec Inc.) is a nonreplicating adenovirus vector with radiation-inducible promoter, which has progressed up to phase III clinical trial of pancreatic cancer patients. While the approach was shown to be safe and produced some activity, the phase III clinical trial failed because of too low efficacy. 18,19 The problem may have been too low production of TNFa at the tumor site. Therefore, the oncolytic platform proposed here is appealing, and its self-amplifying nature solves the problem of low TNFa at the tumor. Because TNFa production is linked to virus replication, which occurs only at the tumor, the tumor-to-systemic circulation ratio of TNFa is predicted to be further increased over a nonreplicating platform.
In this study we generated a novel oncolytic adenovirus expressing human TNFa and studied its efficacy and immunogenicity in vitro and in vivo. We show that Ad5/3-D24-hTNFa is a potent antitumor drug that can induce tumor cell killing by apoptosis and oncolysis as well as by inducing antitumoral immune responses in the tumor site.
Materials and Methods
Cell lines
Human transformed embryonic kidney cell line 293, lung cancer cell line A549, embryonic retinoblast cell line 911, a hormone refractory breast cancer cell line MDA-MB-435, and a TNFa-sensitive mouse fibroblast cell line WEHI-13VAR were obtained from the American Type Culture Collection (ATCC). A head-and-neck squamous cell carcinoma (HNSCC) cell line UT-SCC8 was purchased from PromoCell GmbH. A highly metastatic human prostate cancer cell line PC-3 MM2 was obtained from Isaiah J. Fidler, MD Anderson Cancer Center (Houston, TX). The human bladder cancer EJ cells were provided and authenticated by A.G. Eliopoulos, University of Crete Medical School and Laboratory of Cancer Biology (Heraklion, Crete, Greece). B16-OVA, a mouse melanoma cell line expressing chicken ovalbumin, was kindly provided by Professor Richard Vile at Mayo Clinic (Rochester, MN). All cell lines were cultured under recommended conditions.
Adenoviruses
To create the oncolytic Ad5/3-Δ24-hTNFa, we constructed pTHSN-hTNFa, which contains the human TNFa transgene in the E3 region of the adenoviral genome deleted for 6.7K/gp19K. To construct the pTHSN-hTNFa, pORF-hTNFa (InvivoGen) was digested with SgrAI and MheI and then ligated with BsiWI and MfeI-linearized pTHSN. pAdEasy-1.5/3-Δ24-hTNFa was generated by homologous recombination in Escherichia coli BJ5183 cells (Qbiogene Inc.) between FsbI-linearized pTHSN-hTNFa and SfrI-linearized pAdEasy-1.5/3-Δ24, 20 a rescue plasmid containing the serotype 3 knob and a 24 bp deletion in E1A. The genome of Ad5/3-Δ24-hTNFa was released by PacI digestion and subsequent transfection of 911 cells. The virus was propagated on A549 cells and purified on cesium chloride gradients. The viral particle concentration was determined by OD260 reading, and standard TCID50 (tissue culture infectious dose 50) assay on 293 cells was performed to determine infectious particle (pfu) titer. Virus was characterized by PCR and restriction enzyme analysis. The control viruses Ad5/3-Δ24, 21 Ad5/3-Luc1, 22 and the wild-type Ad300Wt (ATCC VR-5) have been described elsewhere.
hTNFa expression analysis
The expression levels of the hTNFa transgene were examined from the supernatants of Ad5/3-D24-hTNFa-infected (10 VP/cell) human cells lines by FACSArray using a CBA Human Soluble Protein Flex Set for human TNFalpha and Human Soluble Protein Master Buffer Kit (BD Biosciences) according to manufacturer's instructions. BD FACSArray Bioanalyzer, BD FACS Array System software, and FCAP Array v1.0.2 software (BD Biosciences) were used for data analysis. From the infected (100 VP/cell) murine cells, hTNFa levels were measured using a human TNFa ELISA kit (KHC3011; Invitrogen).
Biofunctionality assay
hTNFalpha expressed by the Ad5/3-D24-hTNFa virus was tested using WEHI-13VAR mouse fibroblast cells, which are highly sensitive to TNFa when the assay is performed in the presence of 500 ng/ml actinomycin D. Supernatants from Ad5/3-D24-hTNFa and Ad5/3-D24-infected A549 cells 48 hr after infection were added onto WEHI-13VAR cells and cultured in actinomycin D-containing media. After 24 hr, MTS cell viability test was performed.
In vitro tumor cell-killing assay
Tumor cells were seeded at 1.0×104 cells per well on 96-well plates. On the next day, viruses were diluted in growth media with 2% fetal calf serum, and cells were infected for 1 hr at 37°C and then incubated in 5% FCS-containing media at 37°C for 3–5 days. Cell viability was determined by MTS according to the manufacturer's protocol (Cell Titer 96 AQueous One Solution Cell Proliferation Assay; Promega).
Analysis of apoptotic and necrotic cells
In the in vitro experiment, cells were plated onto 6-well plates, 2×105 cells/well. Cells were infected with 10 VP/cell of virus (or PBS for mock). The amount of apoptotic and necrotic cells was measured 48 hr postinfection (p.i.) with a TACS Annexin V-FITC kit (Trevigen Inc.) and BD Accuri C6 flow cytometer (BD Biosciences) according to manufacturer's instructions. In the in vivo experiment, tumors were smashed through a 70 μm cell strainer to obtain a single-cell suspension and the cells were cultured in 10% RPMI-1640 medium overnight. Cells were stained and analyzed with the TACS Annexin V kit as above.
Immunogenicity of cell death
Calreticulin exposure
Cells in triplicates were infected for 2 hr with 100 VP/cell. Twelve hours (human cells) or 48 hr (mouse cells) later, cells were harvested and stained with 1:1000 diluted rabbit polyclonal Anti-Calreticulin antibody (ab2907; Abcam) for 40 min at 4°C and then with 1:100 diluted Alexa Fluor 488 IgG as secondary antibody (A21206; Invitrogen) for flow cytometric analysis.
Adenosine triphosphate release
Cells in triplicates were infected for 2 hr with 100 VP/cell. Supernatants were collected after 12 hr (human cells) or 48 hr (mouse cells) and analyzed with ATP Determination Kit (A22066; Molecular Probes, Invitrogen).
HMGB-1 release
Cells in triplicates were infected with 100 VP/cell. Twenty-four hours later, supernatant was collected and HMGB-1 was measured with an ELISA kit (ST51011; IBL International GmbH).
Animal experiments
All animal experiments were reviewed and approved by the Experimental Animal Committee of the University of Helsinki and the Provincial Government of Southern Finland. Mice were obtained from Scanbur at 3–5 weeks of age and quarantined at least for 2 weeks before the study. Health status of the mice was frequently monitored, and as soon as signs of pain or distress were evident, they were euthanized.
For the efficacy experiment, human xenografts were established by injecting 5×106 PC-3 MM2 cells subcutaneously into the flanks of 6-week-old male NMRI nu/nu mice (N=6 per group). Tumors (2 tumors per mouse, ∼5×5 mm in diameter) were injected intratumorally with a volume of 50 μl for four times on days 0, 1, 4, and 5 with 108 VP/tumor, and control tumors were injected with PBS only. The formula (length×width2×0.5) was used to calculate the tumor volumes. Mice were euthanized when a tumor reached an average diameter of 15 mm.
The in vivo immunogenicity of the virus was tested in immunocompetent, 5-week-old C57BL/6 female mice (N=7 per group). An amount of 1×105 B16-OVA cells were injected subcutaneously on shaved flank of the mice (one tumor per mouse). In the “mild”-regimen experiment, virus was injected intratumorally at 108 VP/tumor on days 0, 1, 4, and 5, and in the “high”-regimen experiment, the same amount of virus was given every other day (in total of 7 times) starting when tumors reached the size of ∼4×4 mm.
Immune cell analysis
Tumors, spleens, and lymph nodes of treated mice were collected, smashed through a 70 μm cell strainer, and cultured overnight in 10% RPMI-1640 medium. Single-cell suspensions were stained with fluorochrome-conjugated monoclonal antibodies and analyzed using a FACSAria flow cytometer (BD Biosciences) and FlowJo software (TreeStar). FITC-conjugated Rat Anti-Mouse CD8 (BD Biosciences), PE-conjugated Rat Anti-Mouse CD19 (BD Biosciences), and APC-conjugated SIINFEKL MHC-I pentamer (Proimmune) were used.
Irradiation experiments
The combination of the viral treatment and radiotherapy was tested in vitro and in vivo. Cells were irradiated 24 hr after virus infection with 4, 8, or 10 Gy. Mice received 2×2 Gy whole-body irradiation. The irradiation was given 24 hr after virus injections. The cells in 96-well plates were placed in a plastic phantom filled with water while the mice were kept awake in standard plastic cages. Irradiation was performed with a medical linear accelerator (Clinac 600 C/D; Varian Medical Systems) in a 6 MV photon beam using a dose rate of about 4 Gy/min (400 MU/min). Cell viability was measured after 6 days (or 15 days for B16-OVA) with an MTS cell viability test. Uniformity of dose distribution within the cells and mice was maximized by performing the irradiation through a water-equivalent dose build-up layer of about 2 cm.
Statistical analysis
Statistical significance was determined by unpaired, two-tailed Student's t-test using GraphPad Prism 6 (GraphPad Software, Inc.). Differences in tumor growth were analyzed using two-tailed Student's t-test (for immunocompetent mouse experiments) or with the nonparametric Mann–Whitney test (for immunocompromised mouse experiments). Kaplan–Meier survival curves and statistical analysis (log rank statistics) were also performed using GraphPad Prism 6. Results are presented as mean±SD or as mean±SEM for tumor growth. All p-values were two-sided and considered statistically significant when ≤0.05. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Results
In vitro bioactivity of human TNFa expressed by an oncolytic virus
We generated a new virus construct Ad5/3-D24-hTNFa. It is a serotype 5 adenovirus with serotype 3 knobs on its fibers, a 24 bp deletion in its E1A region, which makes it selective to retinoblastoma protein defective cells (most tumor cells), and it expresses human TNFa from its E3 gp19K region (Fig. 1a).

In vitro characterization of the Ad5/3-D24-hTNFa virus.
The expression of human TNFa from the virus was measured in two human cell lines (A549 and 293 [featuring transgenic E1A expression]) at several time points after virus infection (Fig. 1b). The amount of hTNFa increased by time up to 659 ng/ml in A549 cells up to 279 ng/ml in 293 cells 72 hr after infection with Ad5/3-D24-hTNFa. In the murine B16-OVA cell line, the expression levels of hTNFa were much lower, rising up to 83 pg/ml 72 hr after infection. The production of hTNFa after Ad5/3-D24-hTNFa infection started around 24–36 hr after the infection in human cells and 48 hr after in mouse cells.
The biological functionality of the hTNFa produced by the virus was tested in a TNFa-sensitive cell line, a mouse fibroblast cell line WEHI-13VAR. These cells die in the presence of TNFa and actinomycin. As shown in Fig. 1c, the cell viability of Ad5/3-D24-hTNFa-infected cells decreased with correlation to virus dose. These results prove that the viral construct is functional and that the hTNFa produced by the virus is also biologically active.
Ad5/3-D24-hTNFa shows increased cell-killing potency and immunogenicity in vitro
The oncolytic potency of the virus was tested in four different human cancer cell lines (PC-3 MM2, A549, UT-SCC8, and MDA-MB-435) (Fig. 2a). The MTS cell viability assays show that Ad5/3-D24-hTNFa kills tumor cells even faster than the control virus Ad5/3-D24 in all cell lines (p<0.01, <0.001, <0.05, and <0.05, respectively).

In vitro efficacy of Ad5/3-D24-hTNFa.
TNFa is known to induce apoptotic and necrotic cell death in high concentrations, and so we studied the ability of the Ad5/3-D24-hTNFa virus to kill tumor cells by these mechanisms. We measured the amount of Annexin-V positive, that is, early apoptotic cells, and propidium iodide (PI), that is, late apoptotic/necrotic cells, 48 hr after virus infection in three different cell lines (A549, PC-3 MM2, and B16-OVA) (Fig. 2b). The levels of both the early and the late apoptotic/necrotic cells were elevated in all tested cell lines.
The increased exposure of calreticulin on cell surface and elevated adenosine triphosphate (ATP) and high-mobility group box 1 (HMGB1) release are indicative of immunogenic cell death that has been shown to be one important mechanism of chemotherapy-related 23 as well as oncolytic virus-induced cell death. 5,24 When these three indicators are elevated, the tumor cell death can be assumed to activate antitumor immune responses (immunogenic cell death). We tested in three human tumor cell lines (PC-3 MM2, A549, and EJ) and in mouse melanoma cell line (B16-OVA) the amount of these indicators after virus infection. ATP release was found to be significantly increased in all human cell lines (p<0.05, <0.01, and <0.001, respectively) when the cells were treated with Ad5/3-D24-hTNFa, compared with Ad5/3-D24 (Fig. 3a). Also, the calreticulin exposure on the cell surface of Ad5/3-D24-hTNFa-treated cells and the HMGB1 release were elevated compared with controls, but not significantly in all cell lines (Fig. 3b and c).
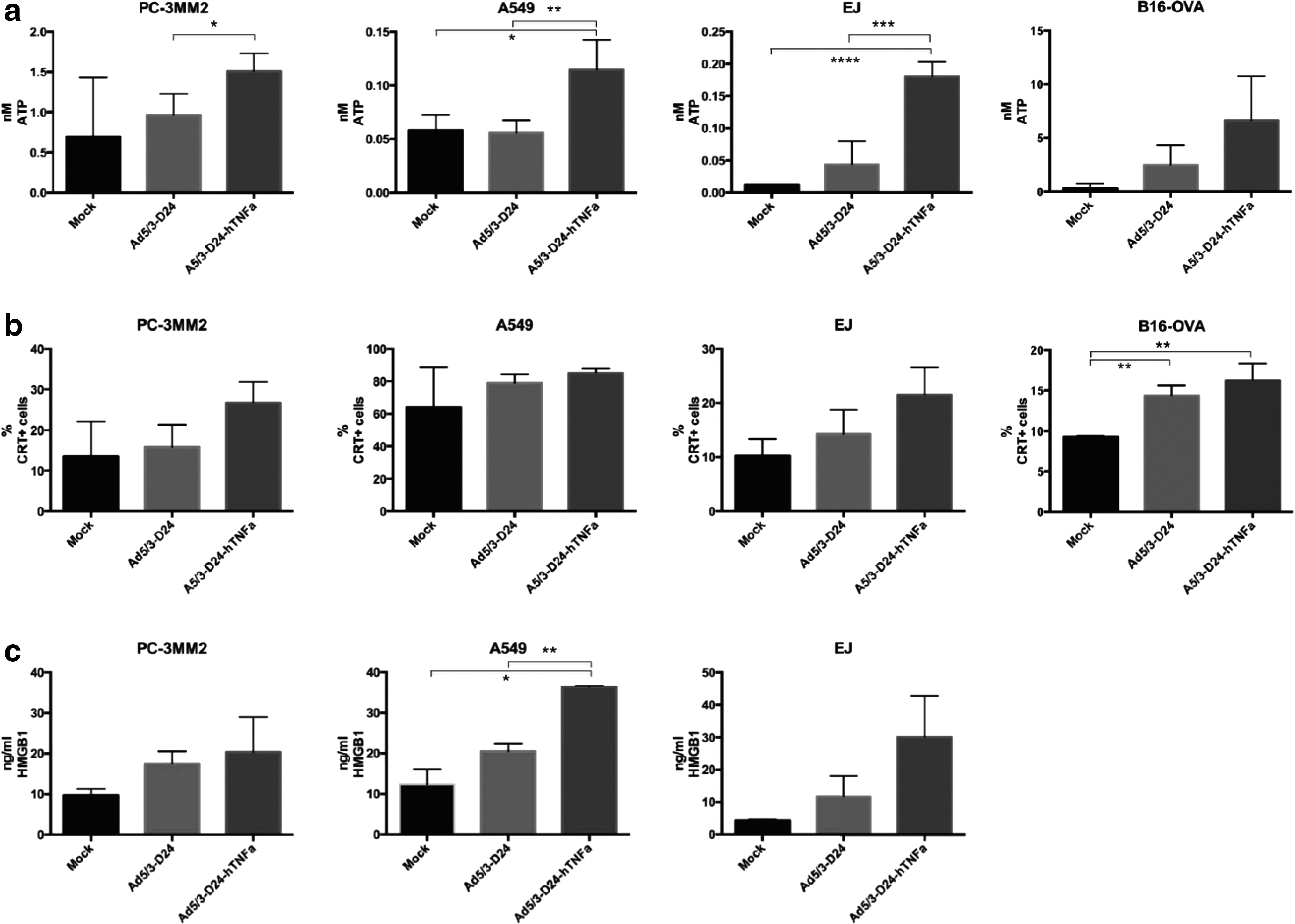
Immunogenic cell death induced by Ad5/3-D24-hTNFa.
Arming the adenovirus with hTNFa results in an enhanced antitumor efficacy in a xenograft model of prostate cancer
The oncolytic activity of Ad5/3-D24-hTNFa was tested in a prostate cancer xenograft model. Nude mice bearing PC-3 MM2 tumors in the flanks were treated intratumorally with 1×108 VP/tumor of viruses (or PBS) on days 0, 1, 4, and 5 and the tumor growth was followed over time. We observed that, even with this mild regimen of virus, the tumor growth was significantly (p<0.001 at day 40) reduced in the Ad5/3-D24-hTNFa-treated mice compared with the control virus (Fig. 4a). The survival of the mice treated with the same regimen was also followed. The group of mice treated with Ad5/3-D24-hTNFa had a significantly longer survival compared with others (p=0.0002, log rank statistics) (Fig. 4b). The local production of hTNFa from the virus in mice treated in a same way was measured from the tumors, which were collected 10 days after the first virus treatment. The mean amount of human TNFa in the Ad5/3-D24-hTNFa tumors was 878 pg/ml (SD±665) (Fig. 4c). Also, the amount of apoptotic and necrotic cells was determined from the tumors (Fig. 4d).
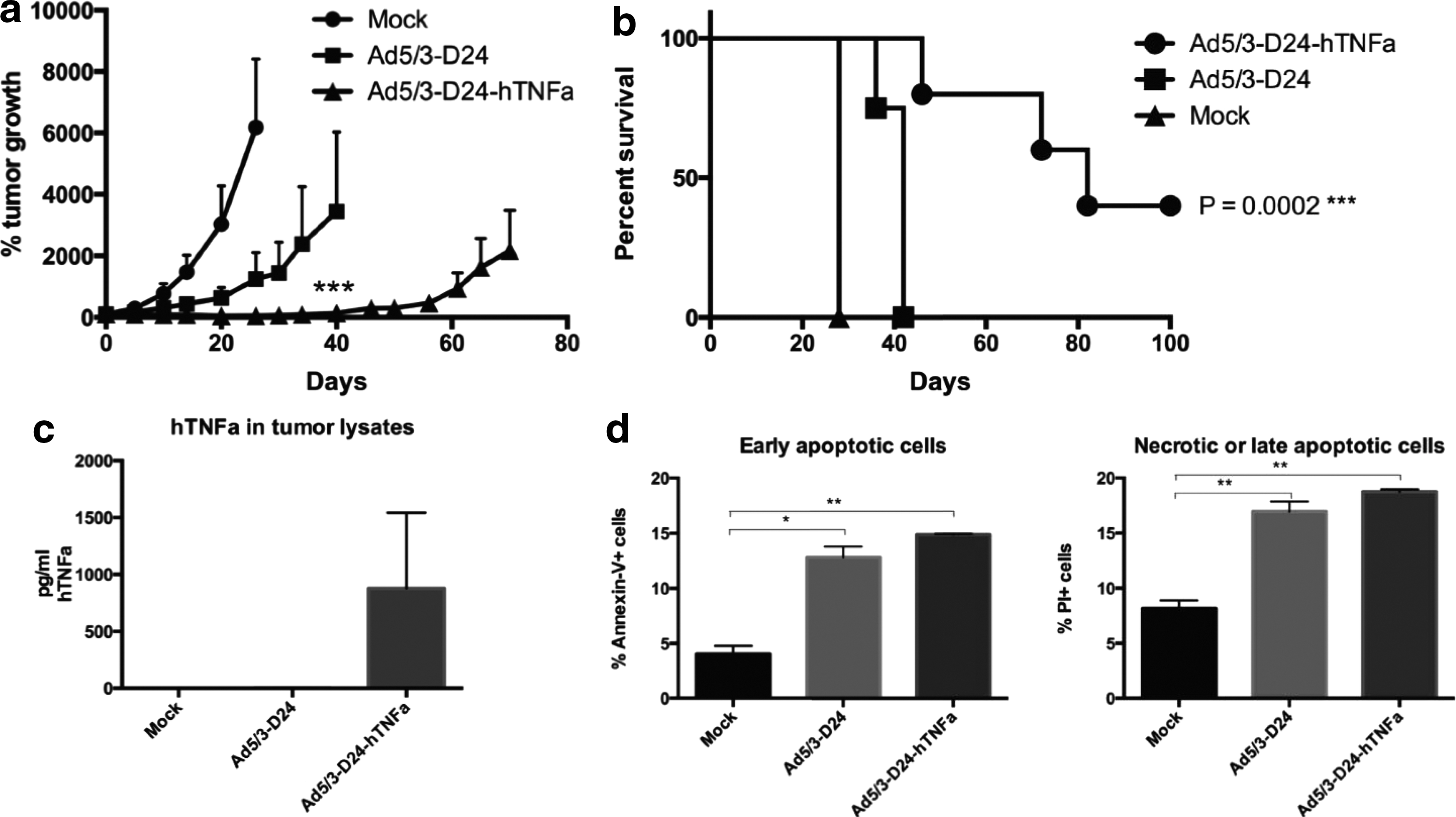
In vivo efficacy of Ad5/3-D24-hTNFa.
We also collected serum from the mice on days 4, 8, and 10 after the first virus injection and checked the levels of human TNFa in blood circulation and also in tumor at day 10. We observed no significant differences in the blood levels compared with control mice (Supplementary Fig. S1; Supplementary Data are available online at
Ad5/3-D24-hTNFa displays increased efficacy and enhanced antitumor immune responses in vivo in a syngeneic melanoma model
To show the immunological effects of Ad5/3-D24-hTNFa, we performed in vivo experiments with immunocompetent C57BL/6 mice bearing syngeneic B16-OVA melanoma tumors. We tested two different regimens of virus, mild and high. In the mild regimen the mice were treated intratumorally with 1×108 VP/tumor on days 0, 1, 4, and 5, whereas in the high regimen the mice received virus every other day. The tumor growth was shown to be reduced with both regimens in the Ad5/3-D24-hTNFa-treated group compared with Ad5/3-D24-treated group (p<0.0001 and <0.01, respectively) (Fig. 5a and b). However, there was no difference between tumor growth of mice receiving mild or high regimen of virus.
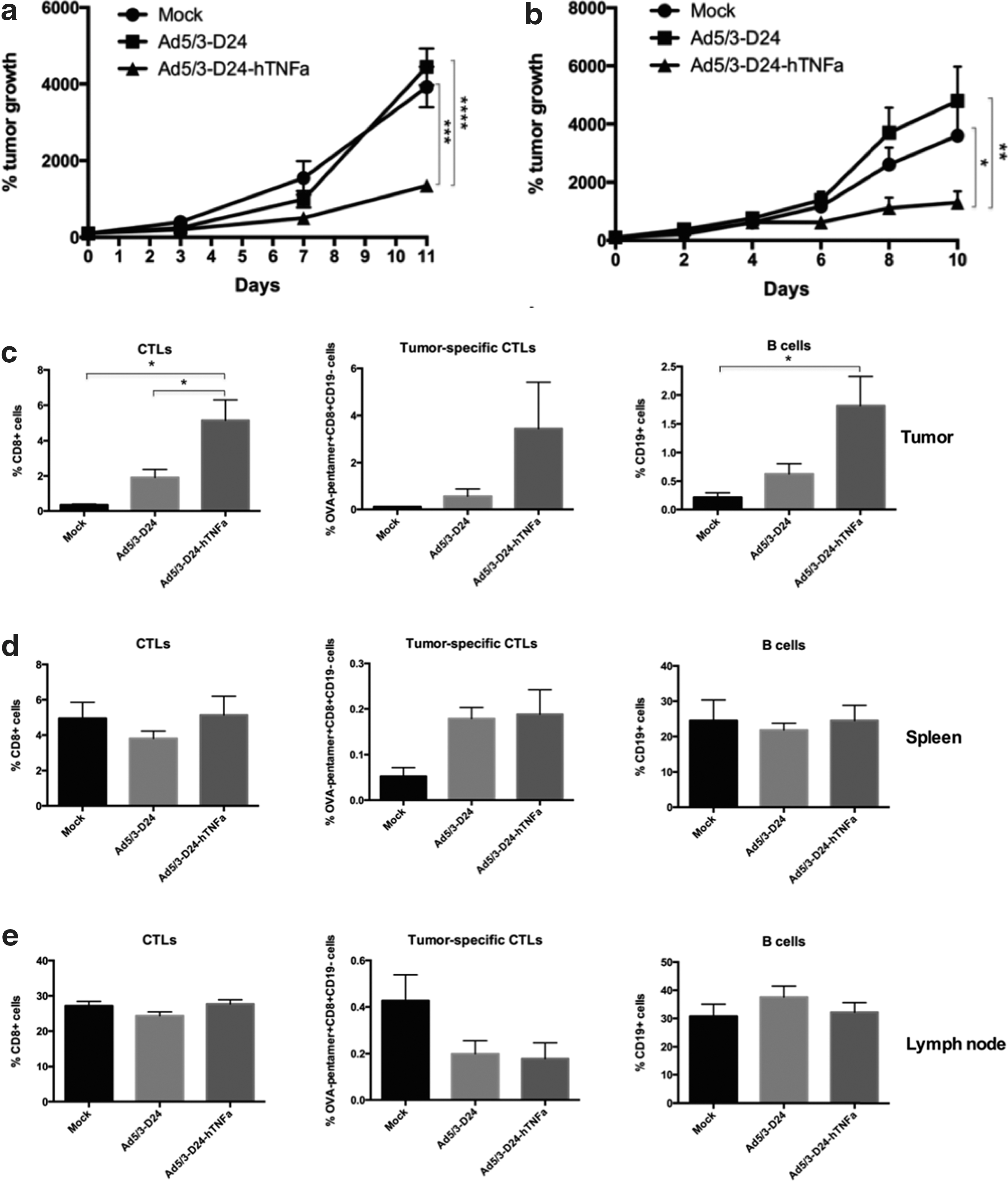
Effects of the expressed hTNFa in immunocompetent mice. C57BL/6 mice bearing syngeneic B16-OVA tumors were treated intratumorally with PBS (mock) or with 1×108 VP/tumor of Ad5/3-D24 or Ad5/3-D24-hTNFa virus
We then characterized the immune response in tumors, spleens, and lymph nodes of these mice (Fig. 5c, d, and e, respectively). The organs were collected 9 days after the first treatment. The amount of CD8+ T cells (effector T cells) was increased in the TNFa-virus-treated tumors compared with mock- and Ad5/3-D24-treated tumors (p<0.05). Also, the amount of CD19+ cells (B cells) was significantly elevated in the Ad5/3-D24-hTNFa-treated tumors compared with mock (p<0.05). Because we used the B16-OVA tumor model that expresses chicken ovalbumin (OVA), a foreign and thus easily recognizable protein in mice, we were also able to assess the amount of OVA-specific CD8+ T cells (tumor-specific CTLs) by using an OVA-peptide-specific pentamer. We observed an increase in the amount of these OVA-specific T cells in the Ad5/3-D24-hTNFa-treated tumors, but the differences were not significant.
Combining Ad5/3-D24-hTNFa with radiotherapy results in enhanced tumor growth control and increases apoptotic and necrotic cell death of tumor cells
It is known that TNFa can sensitize cells to radiation. 9 Therefore, we wanted to test whether the combination treatment with a TNFa-expressing virus and radiation would result in enhanced tumor cell killing. First, we tested the effect of combination treatment of virus and radiation in vitro with three cell lines (A549, PC-3 MM2, and B16-OVA) (Supplementary Fig. S2). The irradiation seemed to have no effect on the cell viability regardless of the amount of irradiation given, and there were no significant difference between TNFa-expressing virus and the control virus in any of the cell lines.
In vivo, we tested the sensitizing effect of hTNFa produced by the virus on irradiated tumors. First, we performed an experiment with a prostate cancer xenograft model (nude mice bearing PC-3 MM2 tumors) (Fig. 6a) and then with a syngeneic melanoma model in immunocompetent mice (C57BL/6 mice bearing B16-OVA tumors) (Supplementary Fig. S3). The mice received 1×108 VP/tumor of virus (or PBS) on days 0, 1, 4, and 5, and on days 2 and 6 they received 2 Gy whole-body irradiation. In the xenograft model, we saw clear reduction in tumor growth in the Ad5/3-D24-hTNFa-treated group compared with the Ad5/3-D24 group (p<0.01 at day 34), which indicates that TNFa produced by the virus has some sensitizing effect for irradiation. In the immunocompetent model, the tumor growth was significantly reduced (p<0.05) in the Ad5/3-D24-hTNFa-treated group compared with the mock-treated group, but there was no significant difference between Ad5/3-D24-hTNFa-treated and Ad5/3-D24-treated groups at day 11.
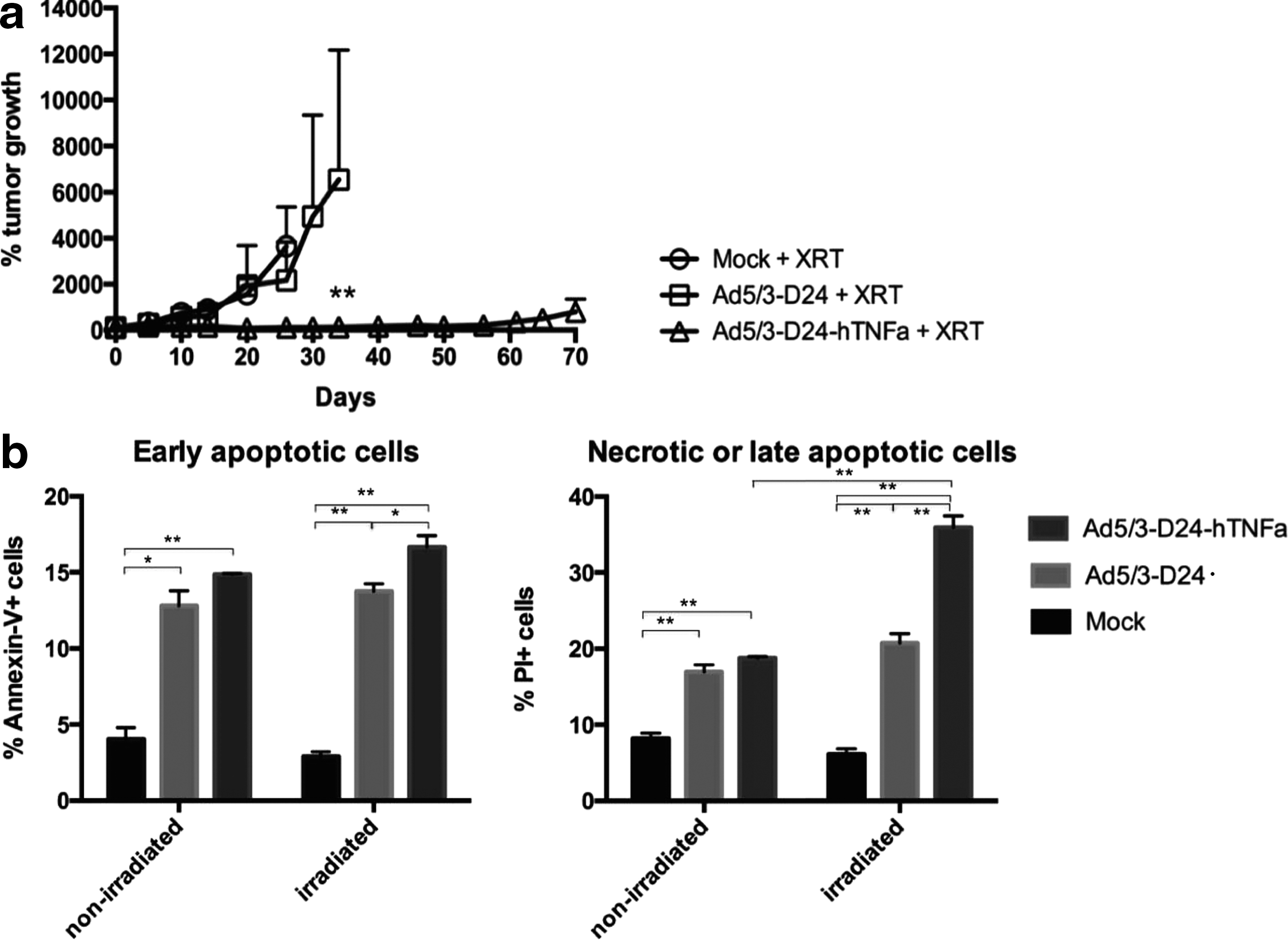
Combination treatment with radiation in prostate cancer xenograft model.
The ability of the Ad5/3-D24-hTNFa+radiation combination to increase apoptotic and necrotic tumor cell death was measured from the virus-treated and irradiated PC-3 MM2 tumors of the xenograft mice at day 8 after the first virus injection. The amounts of early apoptotic (Annexin V+) and necrotic/late apoptotic (PI+) cells were shown to be increased in Ad5/3-D24-hTNFa+irradiation-treated tumors, compared with Ad5/3-D24+irradiation-treated tumors (p<0.05 and <0.01, respectively) (Fig. 6b). We also observed an increase in the amount of late apoptotic/necrotic cells in the irradiated, Ad5/3-D24-hTNFa-treated tumors compared with nonirradiated tumors (p<0.01).
Discussion
We have successfully generated a new chimeric oncolytic adenovirus that expresses human TNFa (Ad5/3-D24-hTNFa). In this report, we demonstrate that this virus is potent in tumor cell killing both in vitro and in vivo because of its oncolytic activity and its ability to induce both apoptotic and necrotic tumor cell death and antitumor immune responses.
TNFa is a multifunctional, powerful cytokine that has critical roles in innate and adaptive immunity. TNFa is known to have antitumor properties in high concentrations. 15,25 It has the ability to induce tumor cell death directly by inducing apoptosis via TNF receptor 1 (TNF-RI) 26,27 and it can induce secretion of blood clotting and adhesion proteins in vascular endothelium, which then can result in vascular thrombosis and necrosis of the tumor tissue. 14 TNFa also induces secretion of other cytokines and chemokines (GM-CSF, IFN-gamma, IL-6, IL-1, and IL-8), which together with the production of endothelial adhesion factors can activate and recruit various immune cells (neutrophils, macrophages, eosinophils, NK-cells, and lymphocytes) to attack the TNF production site (i.e., tumor in this case). 9,15 TNFa was also shown to increase viral entry into tumors by extravasation through a Rho A/Rho kinase pathway, 28 and many cell types are known to become highly susceptible to the cytotoxic effects of TNFa after viral infection. 15
There have been several attempts to treat cancer using TNFa, but most of them have failed because of systemic toxicity of TNFa. 25,29,30 Recent studies have therefore focused on local delivery of TNFa. For example, TNFerade (GenVec, Inc.), a replication-deficient adenovirus with hTNFa and a promoter that can be activated by ionizing radiation, has gone through phase I, II, and III trials. This virus has shown safety and efficacy in preclinical studies and in phase I and II trials, but failed to show reasonably good efficiency in the phase III trial. 17,19
We wanted to utilize the multifold antitumor properties of this cytokine and use it to further improve the tumor-eradicating efficacy of the oncolytic adenovirus. To this end, we developed an oncolytic adenovirus expressing TNFa that would enable introduction of high local concentrations of TNFa in tumor site without the need of systemic dosing of TNFa, which can cause severe toxicity. 11 Because replication entails multiplication of transgene expression, it should therefore result in better utility of antitumor properties of TNFa.
In this report we demonstrated the functionality and oncolytic potency of our new virus construct, Ad5/3-D24-hTNFa. Arming with TNFa was shown to even increase the tumor cell-killing potency of the virus. This was at least partly because of TNFa-mediated induction of apoptotic and necrotic cell death, which are well-known mechanisms of action of TNFa. 31,32
Recently, we and others have shown immunogenic cell death to be one important mechanism of tumor eradication by oncolytic adenoviruses. 5,24,33,34 Indeed, we observed increase in immunogenic cell death markers (exposure of calreticulin on the cell membrane and release of ATP and nuclear protein HMGB1 in Ad5/3-D24-hTNFa-treated cells). The in vivo efficacy of the virus was demonstrated in a human prostate cancer (PC-3 MM2) xenograft model. Intratumoral administration of Ad5/3-D24-hTNFa resulted in clear tumor reduction, and most of the tumors were fully cured within 40 days after the first virus injection. After that some tumors started growing again, most probably because the treatments were only given during the first 5 days of tumor growth follow-up period and only a mild regimen of virus was administered (4×108 VP/tumor in total). As nude mice lack the T cells, they do not develop memory cells against the tumor, and some tumor cells that are left can start growing again after the effect of the virus disappears. To maintain the tumor-eradicating potency of the virus in immunocompromised animals, repeated administration of the virus would be needed. The efficacy of the hTNFa-producing virus may be because of the general inflammation reaction induced by the TNFa in the Ad5/3-D24-hTNFa-treated mice and activation and recruitment of NK-cells into the treated tumors. 15 NK-cells are shown to have a relevant role in tumor suppression after oncolytic adenovirus treatments. 35 Also we showed some indication that the apoptotic and/or necrotic cell death induced by TNFa could be one of the reasons why Ad5/3-D24-hTNFa is so effective in controlling the tumor growth.
Leakage of high concentrations of TNFa into blood circulation has been shown to lead to severe toxic consequences, like septic shock and even death. 10,11,15 We therefore measured the amount of hTNFa in the serum of the treated mice at several time points. No significant amount of hTNFa in blood circulation was observed after the Ad5/3-D24-hTNFa treatments.
The importance of activation of adaptive immunity in the tumor eradication process is nowadays generally registered, 36 and it is also probably the most important means by which oncolytic virus treatments achieve their long-term tumor suppression. 37 Oncolysis caused by viral replication releases the contents of tumor cells, including tumor antigens, into the surroundings. 38,39 These antigens can be picked up by dendritic cells, which can present tumor-associated peptides to T cells. These activated T cells can then travel to the tumor site and induce tumor cell death. These tumor-specific T cells can subsequently turn into memory cells, which results in long-term immunity against the tumor. Because nude mice lack T cells, we performed studies with immunocompetent C57BL/6 mice with syngeneic melanoma tumors to study the immunological responses induced by the Ad5/3-D24-hTNFa virus in vivo.
Previous studies have shown that mouse TNFa receptor TNF-RI binds both human and mouse TNFa, 15,40 and so human TNFa is active in mice, but with less activity than in humans. 41 –43 In tumor growth follow-up studies, we observed that tumor growth of Ad5/3-D24-hTNFa-treated mice was significantly suppressed compared with the non-TNFa-expressing virus. Because human adenovirus does not replicate in murine cells, tumor suppression is likely to be because of the transgene. Because the B16-OVA cell line is not sensitive to TNFa 44 and it is very aggressive and fast-growing, it is possible that using another murine cell line—more sensitive to TNFa and less aggressive—would result in even more prominent tumor growth eradication. To be able to measure the amount of tumor-specific immunity, we used B16-OVA mouse melanoma cell line in our studies. This cell line expresses chicken ovalbumin, which can be detected by an OVA-peptide-specific MHC-I pentamer. In this way we were able to detect the OVA-specific (antitumor) cytotoxic T cells. TNFa is reported to have the ability to activate and recruit lymphocytes. 15,45 We did indeed show increase in the amounts of immune cells (T and B cells) in the Ad5/3-D24-hTNFa-treated tumors, which is an important factor considering the long-term antitumor suppression.
TNFa has been reported to sensitize cells to radiation. 46,47 Therefore, we conducted studies to test the sensitizing capability of our TNFa-expressing virus together with radiation. We observed some sensitizing effect in a human prostate cancer xenograft model receiving irradiation. We saw clear tumor growth control with the Ad5/3-D24-hTNFa virus compared with the control virus and mock groups. This indicates that, although the oncolytic virus alone is a potent sensitizer for radiation, 48 TNFa adds to the effect. However, in the mouse melanoma model the combination therapy with radiation did not clearly increase efficacy of the virus treatments, probably because of the generally noticed radioresistant nature of melanomas. 49
In summary, we show effective tumor cell killing both in vitro and in vivo, associated with immunogenic, apoptotic, and necrotic cell death and the recruitment of immune cells to the infection site. We also saw potential for combination therapy with radiation. Because TNFa is known to cause selective permeabilization effect on tumor vasculature and to thereby increase the uptake of therapeutic agents into tissues, 9,28 combination therapy with chemotherapeutics would also be worth considering.
Footnotes
Acknowledgments
We thank Sirkka-Liisa Holm, Saila Pesonen, Aila Karioja-Kallio, and Eerika Karli for technical support. This work was supported by University of Helsinki, Marie Curie FP7-PEOPLE-IRG-2008, K. Albin Johanssons Stiftelse, ASCO Foundation, HUCH Research Funds (EVO), Sigrid Juselius Foundation, Academy of Finland, Biocentrum Helsinki, Biocenter Finland, and Finnish Cancer Organizations. M.H. was supported by Finnish Cultural Foundation, Emil Aaltonen Foundation, and Waldemar von Frenckell Foundation.
Author Disclosure Statement
A.H. is a shareholder in Oncos Therapeutics, Ltd. A.H. is an employee and shareholder in TILT Biotherapeutics Ltd. All other coauthors declare no potential conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.