
Abstract
Several translational challenges are currently impeding the therapeutic development of antisense-mediated exon skipping approaches for rare diseases. Some of these are inherent to developing therapies for rare diseases, such as small patient numbers and limited information on natural history and interpretation of appropriate clinical outcome measures. Others are inherent to the antisense oligonucleotide (AON)-mediated exon skipping approach, which employs small modified DNA or RNA molecules to manipulate the splicing process. This is a new approach and only limited information is available on long-term safety and toxicity for most AON chemistries. Furthermore, AONs often act in a mutation-specific manner, in which case multiple AONs have to be developed for a single disease. A workshop focusing on preclinical development, trial design, outcome measures, and different forms of marketing authorization was organized by the regulatory models and biochemical outcome measures working groups of Cooperation of Science and Technology Action: “Networking towards clinical application of antisense-mediated exon skipping for rare diseases.” The workshop included participants from patient organizations, academia, and members of staff from the European Medicine Agency and Medicine Evaluation Board (the Netherlands). This statement article contains the key outcomes of this meeting.
Introduction
A
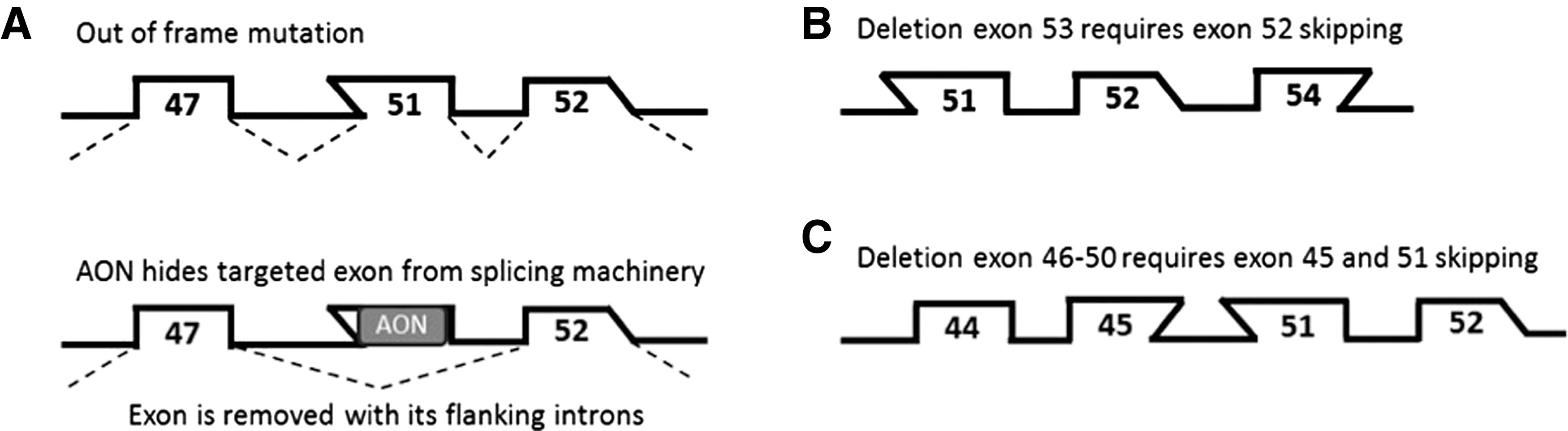
Antisense-mediated exon skipping.
The Example of Duchenne Muscular Dystrophy
These challenges are showcased by efforts to develop therapies for Duchenne muscular dystrophy (DMD) (Aartsma-Rus, 2010), an X-linked disease caused by mutations (generally deletions of one or more exons) that disrupt the reading frame of the dystrophin transcript (Monaco et al., 1988). Dystrophin normally has a function in providing muscle fibers with stability during contraction by connecting the actin cytoskeleton of the fiber to the connective tissue layer surrounding the fiber. Muscle fibers lacking dystrophin are very sensitive to damage during regular exercise. In DMD patients, chronic muscle damage leads to inflammation, replacement of muscle tissue by fibrotic and adipose tissues, and loss of muscle functions (Grounds, 2008).
With good care, most patients become wheelchair dependent in their early teens, need assisted ventilation in their late teens, and die because of respiratory or heart failure in the second to fourth decade of life (Emery, 2002; Bushby et al., 2010a,b). Cognitive problems are seen in one-third of patients, but these are not progressive. Mutations that maintain the reading frame can give rise to dystrophins that are able to connect the actin cytoskeleton to the connective tissue, although with a slightly altered linker. These dystrophins are partially functional, as showcased by the fact that this type of mutation is found in the later onset and more slowly progressive Becker muscular dystrophy (BMD) (Monaco et al., 1988; Emery, 2002). Antisense-mediated exon skipping for DMD aims to restore the disrupted dystrophin reading frame to allow production of a Becker-like dystrophin (Fig. 1) (Aartsma-Rus, 2010). This is achieved by AONs that target a specific exon in the pre-mRNA. Note that different mutations require the skipping of different exons to reframe the transcript (e.g., exon 51 skipping for a deletion of exons 48–50, but exon 52 skipping for a deletion of exon 53), and that skipping of any given exon applies only to a subset of patients (Aartsma-Rus et al., 2009). Since exon 51 skipping applies to the largest group of patients (13%), AONs targeting exon 51 were the first to be developed clinically (Aartsma-Rus, 2014b; Cirak et al., 2011; Goemans et al., 2011; Mendell et al., 2013). Nevertheless, only a minority of DMD patients would benefit from exon 51 skipping. AONs to skip additional exons have to be developed to offer a potential therapy for larger numbers of patients.
While certain groups are reasonably sized (e.g., exon 44, 45, and 53 skipping would apply to 6%, 8%, and 8% of patients), skipping of some of the other exons would apply only to very small groups of patients or even single mutations (Aartsma-Rus et al., 2009). Each AON is considered a new drug, posing a challenge on developing this approach for the majority of patients. Furthermore, for 19% of patients, skipping of a single exon is not sufficient to restore the reading frame and double-exon skipping is required (Fig. 1C).
For exon 51 skipping, two different AON chemistries have been tested in clinical trials (i.e., 2′-O-methyl phosphorothioate [drisapersen] and phosphorodiamidate morpholino oligmer [eteplirsen] AONs) (Cirak et al., 2011; Goemans et al., 2011; Mendell et al., 2013; Aartsma-Rus, 2014b). For both chemistries, systemic dose-finding studies followed by an open-label extension phase showed encouraging results as presented at scientific meetings, that is, stabilization in walking distance measured in the 6 min walk test rather than the decline that would have been anticipated based on natural history for the majority of patients in both trials (Goemans et al., 2013; McDonald et al., 2013a; Pane et al., 2014).
In a small phase 2 trial involving 12 patients, maintenance of the 6 min walk distance was observed for 10 patients for over 2 years. The drisapersen-treated patients outperformed placebo-treated patients in two phase 2 trials involving 5–13-year-old patients (average 7.5) for the primary endpoint (∼30 m difference in the 6 min walk test). However, in a larger phase 3 trial, involving also older, and more severely affected patients (5–16 years, average 8.2 years), the difference between treated and placebo-treated patients was much smaller, that is, a mean difference of ∼10 m in favor of the drisapersen-treated group. Notably, a lot of the natural history data for this disease became available only while phase 2 and 3 trials were already ongoing. Furthermore, the analysis of the open-label extension study following the phase 3 trial suggests that longer treatment (96 weeks) may be required to show benefit in more severely affected patients.
Of course, DMD is just one example and AON-mediated exon skipping is also explored as a therapeutic option for other diseases, such as the genetic skin blistering disorder epidermolysis bullosa. Similarly to DMD, moderation of phenotypic severity has been observed in patients with epidermolysis bullosa through in-frame exon skipping of exons containing nonsense or frame-shift mutations (McGrath et al., 1999). While different genetic diseases pursuing exon skipping face similar challenges, each also faces specific challenges, for example, because of the different tissues that need to be targeted.
Furthermore, other therapeutic approaches are in development for DMD as well. In fact, the week before our workshop took place, it was announced that the committee for human medicinal products (CHMP) of the European Medicine Agency (EMA) gave a positive opinion to the European Commission for conditional approval of Translarna (ataluren) for the treatment of DMD resulting from a nonsense mutation in ambulatory patients aged 5 years and older. The European Commission has since officially granted ataluren conditional marketing authorization (August 4, 2014). The full conditions of the marketing authorization and the related discussion, including the need of a completed confirmatory phase III study, can be found in the European Public Assessment Report and the Annex II of the Summary of product characteristics for Translarna (European Medicines Agency, 2014).
Like exon skipping, Translarna (previously PTC124) aims to restore dystrophin, but it achieves this by stimulating read through of premature stop codons (∼15% of DMD patients carry premature stop codons in the dystrophin gene). Translarna is the first drug to receive a conditional market authorization by the European Commission for patients with DMD in the EU. As such, available data on the clinical development of Translarna are useful when discussing DMD exon skipping, for which no marketing authorization has yet been obtained.
Aim of the Workshop
The European Cooperation of Science and Technology (COST) provides funding for networking to overcome scientific and regulatory challenges. In this framework, COST Action BM1207 receives funding to network toward clinical application of antisense-mediated exon skipping for rare diseases (Aartsma-Rus, 2014a). Representatives of the regulatory models working group and biochemical outcome measures working group met in Leiden, the Netherlands, on May 26, 2014, to discuss existing issues and possible ways forward for the development of antisense-mediated exon skipping therapies for rare diseases, focusing on trial design, outcome measures, and market approval models as was done in a larger meeting in 2009 (Muntoni, 2010). Although the scope of the current workshop was to discuss the challenges for all rare diseases, the focus often fell on DMD, because for this disease the most data are available and the approach has been developed the furthest. This statement article contains the key outcomes of this workshop.
Development Plan
AON-mediated exon skipping is generally tested first in patient-derived cell models and animal models to obtain proof of concept of the approach. AONs are mostly species specific, meaning that the AONs tested in patient-derived cells are not identical to those tested in mice. This can be circumvented by developing a humanized mouse model, which allows studying human-specific AONs in vivo (Bremmer-Bout et al., 2004). In contrast to mandatory safety studies, proof-of-concept studies in animal models are not crucial to the process of obtaining marketing authorization, provided that the field has a good understanding of target engagement. However, especially when the phenotype of the model is close to the human disease, animal model studies can be used to inform and guide clinical trial design. Furthermore, the ability to provide proof of concept in relevant animal models is always considered a valuable step in the overall analysis of the totality of available data at the time of marketing authorization.
When the observed clinical benefit is clear from the performed confirmatory trials and the effect size is compelling, the data on proof of concept in animal models may be unnecessary. However, for most rare diseases (as seen for DMD exon skipping in phase 2 and 3 trials), there is the risk that the demonstrated effect size may generally be modest, since trials are small and most therapies aim at slowing down disease progression. In such cases, having supportive preclinical data to demonstrate proof of concept in (humanized) animal models can provide crucial additional justification for the proposed therapeutic approach.
Traditionally, potential drugs are tested first in phase 1 trials in healthy volunteers, followed by dose-finding safety phase 2 trials, and finally larger phase 3 placebo-controlled trials aimed at confirming efficacy and safety and providing the main dataset for the marketing authorization application. For antisense-mediated exon skipping for DMD, the trials in healthy volunteers have been omitted, reasoning that while skipping an exon restores the reading frame for DMD patients, it will disrupt the reading frame in healthy volunteers. Given that the half-life of dystrophin is ∼3 months (Wu et al., 2012; Verhaart et al., 2014), and that the efficiency of exon skipping in vivo with the AON chemistries tested in clinical trial is not that high, it is unlikely that this would lead to DMD in a healthy person. Furthermore, the pharmacodynamics of AONs in healthy volunteers will differ from those of DMD patients, whose muscles are more permeable because of the effect of the disease (Heemskerk et al., 2010). For the AON chemistries currently in trial for DMD (2′-O-methyl phosphorothioate and phosphorodiamidate morpholino oligomer), safety data in human subjects were already available, and therefore performing studies in healthy volunteers would have provided only limited new information, and would have slowed down therapy development.
There are also many new AON chemistries currently in development. Most of these have not yet been tested in humans and for these performing safety tests first in healthy volunteers could be considered. Notably, for dystrophin, exon skipping is unlikely to negatively impact healthy volunteers because of the long protein half-life and because nonfunctional proteins are produced after exon skipping. However, this may not be the case for exon skipping approaches for other diseases, for example, through inducing a dominantly negative effect. Thus, whether or not to test AONs in healthy volunteers will have to be decided on a case-by-case basis.
Since most rare diseases represent a situation of unmet medical need and are progressive and debilitating, the need for placebo-controlled trials was debated. Using the example of DMD, phase 2 trials for Translarna and drisapersen revealed that the disease progression of the placebo group as measured by the 6 min walk test was very similar to the natural history data (McDonald et al., 2013a; unpublished data). This can be used as an argument to forego the placebo group in confirmatory clinical trials and use the natural history data as a reference. While this may become an acceptable option for the regulators in the future, pending the increased understanding of the disease based on most recent data, for the moment, it seems that some form of placebo treatment will be required to provide the regulators with a more convincing proof that a relevant clinical benefit has been demonstrated.
More specifically for DMD patients, a 30 m improvement over placebo measured by the 6 min walk test was considered a clinically relevant benefit in the Translarna trial. However, the disease progression is relatively slow; for example, DMD patients generally lose ∼30 m in the distance walked in 6 min in 1 year (Goemans et al., 2013; McDonald et al., 2013a; Pane et al., 2014), and it will take time for the AONs to achieve their expected mechanism of action (i.e., accumulation of sufficient amounts of dystrophin to lead to less sensitivity to contraction-induced muscle damage, and a slower disease progression). This implies that it is unlikely that placebo-controlled trials with a duration of less than 1 year will be sensitive enough to show a clinically relevant treatment effect.
For DMD using a 2:1 randomization for treatment (two thirds) and placebo (one third) groups is commonly used (e.g., eteplirsen [phase 2a], drisapersen [two phase 2 and phase 3] and ataluren [phase 2b] trials). Given the heterogeneity of the disease severity and progression, trials should include sufficient numbers in each group, as including small patient numbers can lead to very biased results (Aartsma-Rus and Muntoni, 2013; Goemans et al., 2013).
For DMD, results of natural history studies have been published recently (Goemans et al., 2013; Henricson et al., 2013a,b; Mazzone et al., 2013; McDonald et al., 2013a,b; Pane et al., 2014). This information should be useful to set inclusion criteria and select the group most likely to show clinical benefit over placebo. For instance, it was recently shown that the 6 min walk distance in DMD patients is predictive for the likelihood of losing ambulation in the next year (Mazzone et al., 2013). As such, depending on the design of the trial, patients likely to lose ambulation (or unlikely to lose ambulation) can be included in the study. This information should reduce the number of patients needed to detect a significant difference in the primary endpoint. Notably, there is no prescribed trial setup for exon skipping trials, and regulators are open to an early discussion on innovative trial designs, provided that these are scientifically justified, have internal and external validity, and apply the correct methodology to demonstrate clinically relevant benefit for the patients.
One possible explanation for the phenotypic heterogeneity observed for most rare diseases is the presence of genetic modifiers, which can influence disease progression by modulating the outcome of the mutation; for example, lack of the SMN1 gene causes spinal muscular atrophy, but depending on the number of the homologous SMN2 genes present, patients have a severe early onset disease, or a later onset disease or are even asymptomatic (Taylor et al., 1998). Genetic modifiers that influence disease progression often are single-nucleotide polymorphisms (SNPs); for example, for DMD, SNPs in LTBP4 and SPP1 genes (both encoding proteins involved in the inflammatory response) have been suggested to influence the age of loss of ambulation (Pegoraro et al., 2011; Flanigan et al., 2013). For SPP1, the major allele (allele frequency 0.88) was reported to be protective in a recessive manner, while for LTBP4 the minor haplotype (frequency ∼0.35) was reported to be protective in a recessive manner. Both potential modifiers have been reported to result in a 1-year difference in age of loss of ambulation, which is a significant effect that may influence trial results (Pegoraro et al., 2011; Flanigan et al., 2013). However, because of low patient numbers, recruiting patients for DMD exon skipping trials is challenging to begin with, and excluding patients based on their genotype for a (potential) genetic modifier would not be advisable. Also, stratification will be challenging, because in relatively small trials, it can be anticipated that sometimes only a single patient will be homozygous for, for example, the minor SPP1 allele.
It is also not yet known how the two reported potential modifiers (SPP1 and LTBP4) influence each other. Furthermore, it is very likely that there are additional genetic modifiers that influence disease progression that have not yet been identified. What can be done, however, is a retrospective post-hoc analysis based on genotype to investigate whether there are significant differences (or trends) for the different genotypic groups or to explain outliers who progress more (or less) quickly than other patients because of their genotype.
Outcome Measures
Every positive opinion for granting a marketing authorization adopted by the CHMP is based on the conclusion of a positive benefit/risk ratio for the drug in question. The demonstration that the observed effect leads to a clinically relevant benefit for the patient plays a major part of this positive benefit/risk conclusion. What clinical benefit entails will obviously vary for different diseases, depending on their symptoms and rate of disease progression. For DMD, a 30 m difference in the 6 min walking distance between placebo-treated and drug-treated patients was proposed as clinically relevant benefit (Henricson et al., 2013a), and used in the recent Translarna CHMP positive opinion. As mentioned earlier, the 6 min walk test is predictive for when patients may lose ambulation (Mazzone et al., 2013), and it is known from treatment with corticosteroids that prolonged ambulation is associated with prolonged independent breathing and survival, suggesting that the 30 m difference in the 6 min walk test may reflect long-term impact on disease progression. However, the 6 min walk test cannot be performed on nonambulatory DMD patients (who represent the majority); for that reason, efforts to develop additional functional scales, such as the “performance of upper limb function,” are ongoing to allow designing clinical trials in nonambulatory patients (Mayhew et al., 2013). These scales may become acceptable to use as primary outcome measures in clinical trials if the regulators are convinced that they are indeed valid measures of a true clinical benefit.
Ideally, the results demonstrated for the primary endpoint would be expected to be supported by the data from other endpoints. Upon validation and acceptance of any newly developed outcome measures, and given that the majority of DMD patients are nonambulant, we may see future trials performed in both ambulatory and nonambulatory patients, even using different primary endpoints based on the baseline disease status of the patients. In fact, pulmonary function tests have been used as a primary outcome measure for a phase 3 trial testing Idebenone (an antioxidant) in nonambulant DMD patients. However, for pulmonary function, more insight in the rate of decline and effect of disease stage in the contemporaneous, modified natural history of DMD is needed to define clinical benefit as measured by respiratory outcome measures.
It has become clear that while beneficial effects measured through appropriate functional outcome measures are an indispensable part of a successful marketing authorization, the use of validated and qualified biomarkers may provide useful additional information. In the regulatory sense, the term “biomarker” is not restricted to measures used to evaluate and examine normal or pathogenic biological processes, or pharmacologic responses to a therapeutic intervention, but includes also innovative methods used specifically in the context of research and development of new pharmaceuticals. These methodologies can be used in a variety of different ways, for example, to enrich a patient population for likely responders, to show a pharmacodynamic confirmation of activity (e.g., dystrophin restoration for DMD exon skipping), or, if properly validated, to be used as primary endpoints in the sense of surrogate endpoints.
Regardless of the intended purpose, these methodologies need to undergo a regulatory qualification process, which involves the analysis of data supporting the intended context of use (i.e., for a biomarker to be used in a clinical trial as a surrogate endpoint, data will need to be provided on the correlation between the biomarker and outcome measure detecting the expected clinical benefit). If the data are considered sufficient, the methodology (biomarker) will be qualified and become publically available for the intended context of use.
For DMD treatments aiming at dystrophin restoration, dystrophin has historically been proposed as the logical primary (surrogate) endpoint, based on the fact that the lack of the protein is the primary cause of the disease (lacking in DMD patients and present in reduced levels in BMD patients) and arguing that even its partial restoration is bound to bring beneficial effects to the patients. However, in interventional studies aiming at dystrophin restoration, data studying a possible correlation between dystrophin levels and clinical outcomes are currently unavailable (Aartsma-Rus, 2014b); thus, the confirmation that an increase in dystrophin levels indeed leads to a clinical benefit is currently lacking. One also has to bear in mind that while in BMD patients low levels of dystrophin are associated with a slower disease course, these patients express dystrophin since birth. By contrast, DMD patients express no or only very low levels of dystrophin before they are treated, and consequently their muscle quality will deteriorate with age. This is an additional reason why the assumption that “any dystrophin increase in DMD patients is bound to be clinically beneficial” first needs to be proven.
Although studies in animal models can offer predictive values, only data obtained from clinical trials will be able to reveal exactly what amounts of dystrophin are needed to lead to clinical benefit, whether dystrophin restoration always leads to clinical benefit, whether the amount of dystrophin needed to achieve clinical benefit differs depending on the muscle quality at the time of intervention, and whether there is a “point of no return” when muscle quality has deteriorated to such an extent that dystrophin restoration does not lead to clinical benefit. Considering the above, dystrophin levels cannot be regarded as a surrogate endpoint for clinical efficacy in DMD patients. In cases where the observed clinical efficacy is compelling and the clinical benefit has been clearly demonstrated, data on dystrophin production may not be required for approval, but when available, such data and their correlation with clinical efficacy outcomes would increase the robustness of the results. However, dystrophin restoration can in all cases be used as a pharmacodynamic marker, providing confirmation that the applied therapeutic approach worked as predicted (Aartsma-Rus, 2014b).
It is anticipated that patients with early stage disease will benefit more from AON-mediated exon skipping than patients with poorer muscle quality, since exon skipping targets dystrophin transcripts and these are expressed by muscle tissue (and not by fibrotic or adipose tissues), and since dystrophin restoration is expected to slow down disease progression, but will not restore lost muscle tissue. Thus, methods to measure muscle quality (e.g., magnetic resonance imaging) could be developed to be used for selecting patients who are anticipated to be the best responders to a certain therapeutic approach, increasing the power of clinical trials. Furthermore, patients with deletions bordering certain exons (e.g., exon 44) or in certain areas of the gene (e.g., before exon 8) have been found to have a more benign disease course, probably because a subset of these patients can produce very low amounts of dystrophin because of spontaneous exon skipping or the use of alternative initiation sites (Flanigan et al., 2009).
Indeed, patients involved in an exon 44 skipping trial showed higher dystrophin levels at baseline than patients involved in exon 51 skipping trials (Beekman et al., 2013). Hence, a baseline dystrophin analysis could also be a possibility to stratify patients. Once it is known from currently ongoing trials which patients respond best to treatment (e.g., based on muscle quality, baseline dystrophin levels, disease trajectory, and genetic modifiers), it may be possible to select the most appropriate patient population for future trials, allowing for smaller and/or shorter trials without compromising the quality of the outcome data.
However, stepping away from the specific example of DMD and BMD, when developing drugs in rare diseases, there is always a risk that testing a drug in very specific subgroups may make extrapolation to the whole disease population challenging. Currently, for a number of conditions, efficacy data cannot be extrapolated from one disease stage to another, as information is lacking that the demonstrated clinical benefit in the narrow subpopulation will also be relevant and bring about a positive benefit/risk ratio in a population with a more progressed disease stage. Therefore, it might be important that additional efficacy data be provided for the whole population, so that the benefit/risk ratio for all potential subgroups of patients can be assessed by the regulators and/or reimbursement bodies.
Regulatory Models for Marketing Approval
In EU various regulatory pathways are available for achieving a centralized EU marketing authorization, which include the full marketing authorization application, but also the conditional marketing authorization and the marketing authorization under exceptional circumstances. Full marketing authorization requires a comprehensive data package on clinical benefit and safety to be present at the time of approval, which can often be difficult in rare diseases because of limited patient numbers and limited trial duration. Conditional marketing authorization is a mechanism that allows approval for drugs for severe and/or rare life-threatening diseases where there is an unmet medical need. Here, data allowing the regulators to conclude on a positive benefit/risk ratio at the time of granting of the approval have to be provided as well, but additional data that are crucial to the benefit/risk of the drug need to be collected in the future in the form of postauthorization commitments. The granted marketing authorization in this case is conditional and will be re-evaluated on a yearly basis. Once sufficient data have been collected, a company can apply and convert this into a full marketing authorization.
Conditional authorization is a useful tool for rare diseases, since it can provide earlier access to drugs for patients. However, since authorization is conditional, drugs can technically be taken off the market if at any point during the evaluation of the provided postapproval data, the benefit/risk ratio turns negative, or the marketing authorization holder fails to satisfactorily fulfill the postapproval commitments (i.e., fails for whatever reason to provide the data required in the “condition” of the marketing authorization).
An alternative approach for rare, debilitating, and life-threatening diseases is presented by EMA's pilot on adaptive licensing. This is a process where a drug is authorized early for a restricted group of patients (e.g., certain age range) based on strong trends in the absence of safety issues, followed by adaptive authorization for a wider group based on additional evidence gathered (e.g., in younger or older patients). This would be an attractive procedure for exon skipping therapies. Finally, marketing authorization under exceptional circumstances is another mechanism that is specifically designed for situations where it is impossible to acquire sufficient data in clinical trials (e.g., because of very limited patient populations). Like with conditional marketing authorization, a yearly evaluation of the benefit/risk ratio will take place; however, for exceptional marketing authorization, the additional information obtained is not expected to ever lead to a full marketing authorization. Exceptional marketing authorization may be a possibility for AONs that apply only to very small patient groups, but probably only once marketing authorization has been obtained for other AONs, thus confirming the therapeutic principle.
It is also possible for patients to gain early access to a drug that has not yet obtained marketing authorization through compassionate use programs. This applies only to drugs that are in the development stage or subject of a marketing authorization procedure. The regulations for compassionate use differ per country; for example, the responsibility can lie with the treating physician or the license holder, reimbursement can be from the company, and the individual requesting compassionate use or through a national system (e.g., temporary authorizations for use in France) and reporting can include adverse events and/or efficacy or only serious adverse events. Compassionate use can be provided only during the development of a drug if there is a reason to believe that it would also be beneficial for a group of patients not included in a clinical trial. However, once marketing authorization is obtained in EU countries, compassionate use is no longer an option for these countries.
Currently, each AON is considered a new drug by the regulators (both when the AON target is identical but the chemical modifications of the nucleotides differ, and when the chemistry is identical but content and order of the nucleotides differ). For the AON chemistries currently in clinical trials for DMD, it is known that the pharmacokinetic, pharmacodynamics, and safety profiles depend on the chemistry rather than the nucleotide order and composition of the AON. Therefore, academics have proposed class approval for a certain AON chemistry, which would avoid small trials in which only a few patients could be included because of the rarity of their mutations (e.g., exon 38 skipping would apply to only 0.2% of DMD patients). However, this is unlikely to be obtained for AONs in the near future, as class approval is generally given when this is justifiable by a significant amount of long-term data on safety and efficacy for a certain class of drugs, while AONs are a relatively recent development and only limited data are available for long-term safety and efficacy. Nevertheless, preclinical data obtained for a chemistry can of course be used for future AONs of the same chemistry. Furthermore, it is probably possible to extrapolate data on pharmacokinetics, pharmacodynamics, and safety (being) obtained in past and ongoing clinical trials. As such, it is likely that future AONs can be developed quicker.
The discussion on the possible ways to extrapolate data can happen only when AONs have obtained marketing authorization (which is currently not the case), and there are robust data on at least one or two medicinal products, allowing the regulators to conclude convincingly on their efficacy and safety. Even then, extrapolation may be accepted only in cases where the AONs have identical chemistries and are used to treat the same disease. For each AON sequence, specific data (e.g., on off-target effects) will also have to be provided.
Extrapolation between diseases will be more difficult. This is because clinical benefit is obviously not transferable from one condition to another; for example, a 30 m difference in 6 min walk distance is considered a clinically significant benefit for DMD patients, but this may not be the case for less severely progressive muscular dystrophies. Furthermore, while the safety profile is anticipated to be similar for a certain AON chemistry regardless of the disease phenotype, its effect on the different populations may lead to a different level of risk. For example, side effects such as mild nephrotoxicity may create an acceptable risk in view of the potential benefit for DMD patients (who in general do not have disease-induced kidney damage), while at the same time be completely unacceptable for a different disease with pronounced disease-related kidney involvement. So for each disease the balance between benefit and risk will have to be evaluated, requiring a certain amount of data that can be obtained only from clinical trials.
For DMD and other rare diseases, exon skipping sometimes would apply to only single patients. It may be possible that small foundations or even individuals will develop these AONs for individual patients. However, it is unlikely that these AONs will be developed for marketing authorization, and reimbursement will be challenging in most countries. Since compassionate use is not possible for AONs that are not in a clinical trial process, compassionate use is not a mechanism to treat patients with new AONs applicable only to small groups of patients that are not in clinical development.
Another complication for DMD exon skipping is that 19% of patients require the skipping of two exons to restore the reading frame; for example, 0.3% would in theory benefit from the skipping of exon 44 and exon 45, and 1.1% from the skipping of exon 45 and exon 51. AONs to skip these exons are in clinical development with potential plans to apply for a marketing authorization for the combination in the future. However, when this is done, additional tests will be needed to provide data that patients treated with the combination of AONs are not subjected to additional safety risks. Furthermore, most probably, additional preclinical tests would have to be conducted to optimize dosing. An option currently being investigated is the possibility to reduce the dose of each individual AON and still obtain double-exon skipping. However, since both AONs will have to target the same transcript in order to restore the reading frame, it is also conceivable that the dose of each individual AON will have to be higher than for single-exon skipping.
If preclinical data suggest that double-exon skipping is possible and safe, a postmarketing study where two AONs are combined may be a possible way to assess the safety of the combination. However, such speculations are too early at present, and in the case of a combined AON therapy, things are not as straightforward as just prescribing the two individual AONs in patients. It is good to bear in mind that double-exon skipping is an option for DMD because the single-exon skipping by-products will not lead to a protein. For other diseases, for example, epidermolysis bullosa, the single-exon skipping by-products may actually be detrimental. Thus, it will have to be assessed on a case-by-case basis whether double-exon skipping is an option.
Multiexon skipping (skipping of more than two exons) has been proposed as a method to reduce the mutation specificity of DMD exon skipping (Aartsma-Rus et al., 2004; Aoki et al., 2013). Using a combination of 11 AONs targeting 11 exons, the skipping of exons 45–55 should be induced. This would apply to ∼40% of all patients with deletions or small mutations between exons 45 and 55 (Aartsma-Rus et al., 2009).
An added benefit is that Becker patients with an exon 45–55 deletion have a very mild phenotype (Beroud et al., 2007). However, from a regulatory perspective, this multiexon skipping would not be straightforward. First, there would be safety concerns for combining any number of AONs (individual AONs can be toxic and/or a combination of two or more can be even more toxic). Second, multiexon skipping will lead not only to the generation of exon 45–55 deleted dystrophins, but also to many other in-frame and out-of-frame versions (e.g., for a patient with an exon 48–50 deletion, multiexon skipping can lead to dystrophins lacking exons 45–51, exons 45–55, exons 47–51, exons 47–55, exons 48–51, exons 47–53, exons 48–53, exons 48–55, or combined deletions of exons 45–46 plus exons 48–51, exons 48–53 or exons 48–55, or exons 54–55 plus exons 47–51 or exons 48–51, in addition to an almost infinite list of out-of-frame possibilities). Thus, multiexon skipping will dilute the effect of AONs (it will generate more out-of-frame transcripts than in-frame transcripts) and it will give rise to many dystrophins that may not all be functional (this is especially likely for the nonconsecutive in-frame deletions [e.g., exons 45–46 and exons 48–51]). This underlines that like double-exon skipping, multiexon skipping is also not as straightforward as developing an “AON cocktail.” However, it is possible that with new technical developments or AON chemistries, multiexon skipping will be possible in the future.
Looking into the Future
AON-mediated exon skipping is a new type of therapeutic approach that would be applicable to rare diseases. For DMD, there has been a steep learning curve once AON-mediated exon skipping, Translarna, and other therapies were tested in clinical trials. Outcome measures were developed in parallel with the trials and natural history data became available only during or after phase 2 and 3 trials were conducted. Obviously, this was far from ideal, and so it is advisable that groups developing exon skipping for other rare diseases take outcome measures and natural history into account from early phases of drug development. Biomarkers to be used as pharmacodynamics markers should be taken into account from an early stage as well.
Notably, at the moment no marketing authorization has been granted to AON-mediated exon skipping for any disease. Nevertheless, it is anticipated that obtaining this for the first AON will probably facilitate the development for additional exon skipping AONs.
Footnotes
Acknowledgments
All authors are members of COST Action BM1207, which is gratefully acknowledged for funding the workshop.
Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties.
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Medicines Evaluation Board of the Netherlands.
Author Disclosure Statement
A.A.-R. discloses being employed by LUMC, which has patent applications on exon skipping. As coinventor on some of these patents, she is entitled to a share of potential royalties. N.G. discloses being a principal investigator for AON (Prosensa Therapeutics) and Translarna (PTC therapeutics) clinical trials in DMD patients. A.F. is principal investigator for AON (Prosensa Therapeutics) clinical trials in DMD. Other authors have nothing to declare.