
Abstract
Current challenges for recombinant adeno-associated virus (rAAV) vector–based cancer treatment include the low efficiency and the lack of specificity in vivo. rAAV serotype 3 (rAAV3) vectors have previously been shown to be ineffective in normal mouse tissues following systemic administration. In the present study, we report that rAAV3 vectors can efficiently target and transduce various human liver cancer cells in vivo. Elimination of specific surface-exposed serine and threonine residues on rAAV3 capsids results in further augmentation in the transduction efficiency of these vectors, without any change in the viral tropism and cellular receptor interactions. In addition, we have identified a potential chemotherapy drug, shikonin, as a multifunctional compound to inhibit liver tumor growth as well as to significantly enhance the efficacy of rAAV vector-based gene therapy in vivo. Furthermore, we also document that suppression of tumorigenesis in a human liver cancer xenograft model can be achieved through systemic administration of the optimized rAAV3 vectors carrying a therapeutic gene, and shikonin at a dose that does not lead to liver damage. Our research provides a novel means to achieve not only targeted delivery but also the potential for gene therapy of human liver cancer.
Introduction
H
Adeno-associated virus (AAV) is a nonpathogenic human parvovirus that has gained attention as a safe vector. Recombinant AAV (rAAV) vectors are currently in use in a number of gene therapy clinical trials and have shown remarkable efficacy in the treatment of Leber's congenital amaurosis, hemophilia B, and aromatic L-amino acid carboxylase deficiency (Mingozzi and High, 2011). Glybera, a rAAV serotype 1 (rAAV1) vector to treat lipoprotein lipase deficiency, is the first gene therapy product in the Western world (Melchiorri et al., 2013). However, although the vast majority (64.4%) of worldwide gene therapy clinical trials to date have addressed cancer (Ginn et al., 2013), little was based on rAAV vectors. Current challenges for rAAV vector-based cancer treatment include the low efficiency and the lack of specificity in vivo. In the early 2000s, conventional, single-stranded (ss) rAAV2 vectors were used by investigators to target HCC in vivo (Peng et al., 2000; Su et al., 2000). Unfortunately, since the transduction efficiency of ssAAV2 vectors is low, no transgene expression was observed in tumors larger than 2 mm through systemic administration. More recently, delivery of specific miRNAs in a mouse endogenous HCC model using rAAV8 vectors was shown to result in inhibition of murine liver tumor proliferation (Hsu et al., 2012). However, rAAV8 vectors have a broad tropism to normal tissues other than the liver in murine models (Gao et al., 2002; Zincarelli et al., 2008) and in nonhuman primates (Nathwani et al., 2006, 2007).
Among all commonly used rAAV serotypes, rAAV3, which fails to efficiently transduce any normal murine tissue in vivo (Zincarelli et al., 2008; Ling et al., 2010; Markakis et al., 2010), was shown in our previous studies to transduce human HCC cells highly efficiently both in vitro (Ling et al., 2011) and in vivo (Cheng et al., 2012). Although rAAV3 vectors also transduce primary human hepatocytes in vitro (Glushakova et al., 2009) and in humanized mice in vivo (Lisowski et al., 2014), the transgene expression could be restricted to malignant cells by using an HCC-specific promoter, alpha-fetoprotein promoter (AFPp). In our subsequent studies, we observed that rAAV3 vectors utilize the human hepatocyte growth factor receptor (hHGFR, also named c-Met) as a cellular coreceptor (Ling et al., 2010), which offered the potential opportunity to utilize rAAV3-based vectors for targeting human liver cancers, since hHGFR is overexpressed in most HCC cells (You et al., 2011). Furthermore, we (Ling et al., 2014a; Wang et al., 2014) and others (Zhang et al., 2010; Mitchell et al., 2013) have recently proposed the idea of using leading compounds isolated from traditional Chinese medicine (TCM) to enhance the efficacy of rAAV vector-based gene therapy. It is worth noticing that most natural drugs are multifunctional (Ling et al., 2014b) and therefore have the potential to play an additive, even synergistic, effect on rAAV vector-based cancer gene therapy. For instance, shikonin, a major component of Lithospermum erythrorhizon, was reported to have various biological activities, including proteasome inhibitor activity (Yang et al., 2009), as well as activities to induce apoptosis in HCC cells through reactive oxygen species (Gong and Li, 2011).
In this report, we demonstrate that (1) rAAV3 vectors transduce various human HCC cells in vivo not only efficiently, but also specifically; (2) a further augmentation of rAAV3 vector-mediated transduction efficiency can be achieved through the elimination of specific surface-exposed tyrosine (Y), serine (S), and threonine (T) residues on the viral capsids, and no observed alteration in cellular receptor interaction in vitro and viral-tropism in vivo was associated with these modifications; (3) shikonin possesses both the ability to inhibit HCC tumor growth and to significantly enhance rAAV vector-mediated transgene expression through its proteasome inhibitory activity; and (4) systemic coadministration of shikonin and a therapeutic rAAV3 vector in vivo results in significant inhibition of tumor growth without inducing toxicity. These findings suggest that the combination of rAAV vector-based gene therapy and multifunctional drug-based chemotherapy has the potential to be developed as an efficient and safe strategy for the treatment of human liver cancers.
Materials and Methods
Chemicals, plasmids, and primers
Grade I-A heparin sodium salt and high-performance liquid chromatography-grade drugs were purchased from Sigma-Aldrich Co. LLC. Recombinant hHGF was purchased from Life Technologies. Plasmid pHelper was purchased from Agilent Technologies. Plasmids pdsAAV-CBAp-EGFP and pAAV-CBAp-Fluc were generously provided by Dr. Xiao Xiao, University of North Carolina at Chapel Hill, Chapel Hill, NC. Plasmid pdsAAV-AFPp-EGFP has been previously described by Glushakova et al. (2009). The TCS gene was synthesized by Life Technologies, based on the published sequence (Trichosanthes kirilowii trichosanthin [TCS] mRNA, complete cds; GenBank: M34858.1), and subcloned into plasmid pdsAAV-AFPp-EGFP using AgeI and HindIII restriction sites. All plasmids were sequenced prior to use.
Construction of optimized rAAV3 capsid plasmids
A two-stage PCR procedure, using Turbo Pfu Polymerase (Stratagene), was performed to introduce site-specific mutations in the rAAV3 capsid, as described previously (Pandya et al., 2013). Briefly, in stage one, two PCR extension reactions were performed in separate tubes for each mutant. One tube contained the forward PCR primer and the other contained the reverse primer. In stage two, the two reactions were mixed and a standard PCR mutagenesis assay was carried out according to the manufacturer's instructions. All mutant plasmids were sequenced before use.
Cell lines and cultures
HeLa, HEK293 and LH86 cells were purchased from ATCC. Huh7 cells (Cheng et al., 2012) and HepG2 cells (Zhu et al., 2012) have been described previously. They were maintained in complete Dulbecco's modified Eagle's medium (C-DMEM; Mediatech, Inc.) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich Co. LLC) and 1% penicillin and streptomycin (P/S; Lonza). A newly established human hepatoblastoma (Hep293TT) cell line was generously provided by Dr. Gail E. Tomlinson (University of Texas Health Science Center at San Antonio, San Antonio, TX) and was maintained in complete RPMI medium 1640 (Life Technologies) supplemented with 15% heat-inactivated FBS (Sigma-Aldrich) and 1% P/S (Lonza). Cells were grown as adherent culture in a humidified atmosphere at 37°C in 5% CO2 and were subcultured after treatment with trypsin–versene mixture (Lonza) for 2–5 min at room temperature, washed, and re-suspended in complete medium.
Cell viability assays
Cells were exposed to different concentrations of drugs for 4 hr and then were cultured in C-DMEM without drugs for additional 20 hr, followed by cell viability assays using Cell Counting Kit-8 (Dojindo Molecular Techonologies, Inc.), as previously described (Dai and Siemann, 2010).
Cell cytotoxicity assays
Cells were exposed to different concentrations of drugs for 4 hr, followed by cell cytotoxicity assays using LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific, Inc.).
RNA isolation, reverse transcription and qRT-PCR assays
Total RNA was extracted with the RNeasy Mini Kit (QIAGEN) and reverse transcribed using the Reverse Transcription System (Promega) as previously described (Gu et al., 2012), with modification. Realtime qRT-PCR amplification was carried out using SYBR Green PCR core reagents kit (Applied Biosystems), as previously described (Lin et al., 2013a).
rAAV vectors production
Highly purified stocks of rAAV vectors were generated by the triple-plasmid transfection protocol as previously described (Liu et al., 2012), with modification. Briefly, HEK293 cells were co-transfected with three plasmids using polyethelenimine (linear, MW 25,000; Polysciences, Inc.), and the medium was replaced 6 hr posttransfection. Cells were harvested 72 hr posttransfection, subjected to 3 rounds of freeze–thaw, and then digested with Benzonase (Life Technology). Viral vectors were purified by iodixanol (Sigma-Aldrich) gradient ultracentrifugation followed by ion exchange chromatography using HiTrap SP/Q HP columns (GE Healthcare), washed with phosphate-buffered saline (PBS; Mediatech, Inc.), and concentrated by centrifugal spin concentrators with 150K molecular-weight cutoff (Life Technologies). The physical genomic titers of recombinant vector stocks were determined by quantitative DNA slot-blot and Southern blot analyses.
rAAV vectors transduction in vitro
Cells were seeded in 96-well plates at 5,000 or 10,000 cells per well in C-DMEM. Twenty-four hours later, cells were mock-treated or treated with indicated chemicals for 2 hr. rAAV infections (MOI: 5×103) were then performed in serum- and antibiotic-free DMEM for 2 hr, with or without indicated chemicals, followed by extensive washes with PBS to remove the vector inoculum. Transgene expression was analyzed by either fluorescence microscopy or flow cytometry 72 hr posttransduction.
hHGF and heparin competition assay in vitro
Huh7 cells were transduced with scAAV3-CBAp-EGFP vectors at an MOI of 5×103 viral genomes (vg)/cell. Vectors were premixed with indicated concentrations of recombinant hHGF or heperin. Cells were analyzed for EGFP expression levels 72 hr posttransduction.
Western blot assay
Western blot assays were performed as previously described (Dai et al., 2011). Briefly, cells were harvested and disrupted in a radio-immunoprecipitation assay lysis buffer. Following normalization for protein concentration, samples were separated using 12% SDS-PAGE electrophoresis, electrotransferred to a nitrocellulose membrane (Bio-Rad), and probed with relevant primary antibodies at 4°C overnight. The membranes were then incubated with horseradish peroxidase–conjugated secondary antibodies (Thermo Scientific), and detected with an enhanced chemiluminescence substrate (Thermo Scientific). All membranes were stripped and re-probed with anti-GAPDH antibodies as loading controls. Antibodies against GAPDH (cat #PA1-988) were purchase from Thermo Scientific. Antibody against ubiquitin (polyclonal) was purchased from Santa Cruz Biotechnology, Inc. Antibodies against Bcl-xL (cat #2764), PARP (cat #9542), Caspase-3 (cat #9662), and β-actin (cat #4970) were purchased from Cell Signaling Technology. Antibody against GAPDH (polyclonal) was purchased from Thermo Scientific. Antibody against ubiquitin (polyclonal) was purchased from Santa Cruz Biotechnology, Inc.
Animal handling
All animal experiments were approved by the Institutional Animal Care and Use Committee and were performed according to the guidelines for animal care specified by the Animal Care Services at the University of Florida (Gainesville, FL) or the Second Military Medical University (Shanghai, China). Six- to 10-week-old nonobese diabetic/severe-combined immunodeficient, interleukin 2-gamma-deficient (NSG) mice and C57BL/6 mice were purchased from Jackson Laboratory and maintained by the Animal Care Services at the University of Florida College of Medicine (Gainesville, FL).
Human liver cancer xenograft model
Six- to ten-week-old NSG mice received subcutaneous injection of 5×106 Huh7 or Hep293TT cells on the ventral side of the neck between shoulder blades. Animals were kept in sterile cages until the end of the experiment.
In vivo Fluc assays
rAAV vectors were injected intravenously via tail vein, or injected directly into tumors in NSG mice. In vivo Fluc imaging was performed as previously described (Lin et al., 2013b), with modification. Briefly, mice were weighed to calculate the volume of substrate, D-luciferin-K+ salt (Caliper Life Sciences), according to the dose of 4 mg/kg of body weight, and anesthetized. The calculated volume of the 5 mg/ml of stock substrate solution was mixed with 100 μl of PBS and injected intraperitoneally. In vivo bioluminescence images were acquired immediately over a period of 5 min using a Xenogen machine equipped with a cooled couple-charged device camera (Xenogen). Signal intensity was quantified using the camera control program, Living Image software, and presented as photons/second/cm2/steridian.
Statistical analysis
Results are presented as mean±standard deviation (SD). Differences between groups were identified using one-way ANOVA with Sidak's multiple comparison test in GraphPad Prism 6 software.
Results
rAAV3 vectors selectively target human liver tumors in a murine xenograft model in vivo
In the first set of experiments, we compared the transduction efficiency of rAAV3 and rAAV8 vectors in human HCC tumors in a murine xenograft model in vivo. Nonobese, diabetic (NOD), severe-combined immunodeficient (scid), interleukin 2-deficient NSG mice, both male and female (n=4), were injected subcutaneously on the ventral side between shoulder blades with 5×106 human Huh7 cells, which formed tumors. Four weeks later, mice were either mock-injected, or injected via the tail vein with 1×1011 vg/mouse of rAAV3 or rAAV8 vectors carrying the firefly luciferase (Fluc) gene under the control of cytomegalovirus enhancer/chicken β-actin hybrid promoter (CBAp) (Supplementary Fig. S1A; Supplementary Data are available online at

Transduction efficiency of ssAAV3 and ssAAV8 vectors in human HCC tumors in vivo.
To further corroborate these results, in the second set of experiments, we performed direct intratumor injections of rAAV3 or rAAV8 vectors at low (L=1×1010 vg/mouse) or high (H=1×1011 vg/mouse) doses. Whole-body bioluminescence imaging data are shown in Fig. 1C and D. It is evident that, even at a high dose, ectopic expression in the liver in rAAV3 vector-injected mice was minimal, whereas intratumor injection of rAAV8 vectors resulted in strong transgene expression in the liver in a dose- and time-dependent manner. At day 7 after vector injections, mice were sacrificed and the viral genome copy numbers persisting in the liver tissue samples were compared. These results, shown in Fig. 1E, indicated that a large amount of rAAV8 vector genomes were present in the liver, whereas the numbers of rAAV3 vector genomes were minimal, corroborating our previous observations. These studies, together with our previous publications (Glushakova et al., 2009; Ling et al., 2010, 2011; Cheng et al., 2012), provide a clear rationale to further pursue rAAV3 vector-mediated potential gene therapy of human HCC.
Optimized rAAV3 vectors mediate further enhanced transgene expression in human liver tumors in vivo
To further enhance the transduction efficiency of rAAV3 vectors, we performed site-directed mutagenesis of rAAV3 capsids. In addition to our previously described strategy of mutagenesis of surface-exposed tyrosine (Y) to phenylalanine (F) residues (Cheng et al., 2012), various serine (S), threonine (T), and lysine (K) residues were mutagenized to valine (V) and glutamic acid (E) or arginine (R) residues, respectively. Priority was given to the positions that are conserved among various AAV serotypes, and have previously been shown to augment the transduction efficiency of rAAV2 vectors (Zhong et al., 2008; Markusic et al., 2010; Aslanidi et al., 2012, 2013). The wild-type (WT) and all mutant rAAV3 vectors carrying the CBAp-driven enhanced green fluorescence protein (EGFP) reporter gene (Supplementary Fig. S1A) were used to evaluate their transduction efficiencies in a human HCC cell line, Huh7, under identical conditions. A summary of these data is provided in Supplementary Table S1. The seven best mutants as well as the WT rAAV3 vectors carrying the CBAp-driven Fluc reporter gene were then used to transduce Huh7 cells under identical conditions. These results, shown in Fig. 2A, indicated that the transduction efficiency of Y705+731F and S663V+T492V+K533R mutants was increased by ∼10-fold, and that of S663V+T492V mutants was increased by ∼15-fold, compared with the WT rAAV3 vectors. The transduction efficiency of the selected mutants was also evaluated in two different human liver cancer cell lines, HepG2 and Hep293TT (Fig. 2B and C). Thus, the optimized rAAV3 vector may prove useful in the potential gene therapy of human liver cancer.
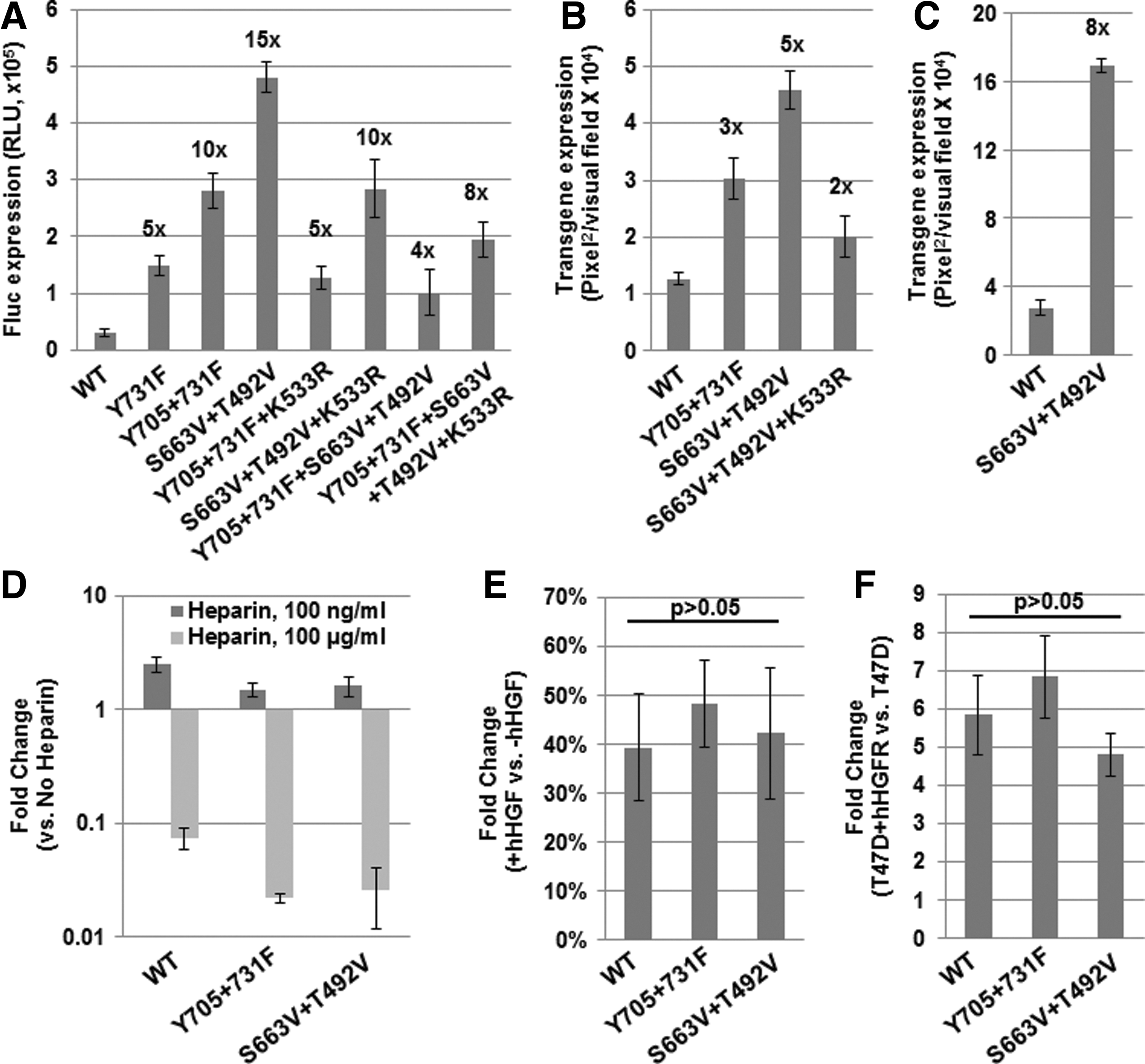
Transduction efficiency of WT and capsid-modified rAAV3 vectors in human liver cancer cells in vitro.
Modifications of specific amino acids on rAAV3 capsids do not alter the virus–cellular receptor interaction
Owing to the uncertainty of viral capsid amino acid(s) responsible for virus–receptor(s) interaction, we wished to examine whether cellular heparan sulfate proteoglycan (HSPG) and human hepatocyte growth factor receptor (hHGFR), previously identified as receptor (Rabinowitz et al., 2004) and co-receptor (Ling et al., 2010) of WT rAAV3 vectors, were involved in our optimized rAAV3 vector transduction. The following three sets of experiments were performed.
First, transduction of Huh7 cells with WT rAAV3 vectors or the two best mutant vectors was performed in the presence of either low (100 ng/ml) or high (100 μg/ml) doses of soluble heparin. These results, shown in Fig. 2D, indicate that both mutants performed in a similar manner as the WT rAAV3 vectors, in which the low dose of heparin enhanced viral vector-mediated transduction efficiency, whereas the high dose dramatically reduced it. Our data are also consistent with a recent report suggesting that fibroblast growth factor receptor is involved in the low-dose heparin-mediated augmentation of WT rAAV3 vector transduction (Messina et al., 2012).
Second, transduction assays were performed in the presence of 5 μg/ml human hepatocyte growth factor (hHGF), which we have previously shown to significantly inhibit the transduction efficiency of WT rAAV3 vectors (Ling et al., 2010). These results, shown in Fig. 2E, demonstrate that the transduction efficiency of the mutant viral vectors was also significantly affected.
Third, the WT and the two mutant vectors were used to transduce a human breast cancer cell line, T47D, that expresses undetectable levels of endogenous hHGFR, as well as T47D cells stably transfected with hHGFR expression plasmids (T47D+hHGFR). These results, shown in Fig. 2F, indicate that both the WT and the mutant vectors transduce T47D+hHGFR cells more efficiently (>5-fold) than the parental T47D cells. Taken together, these data corroborate that the optimized rAAV3 vectors also utilize cellular HSPG and hHGFR as receptor/co-receptor for their transduction.
Modifications of specific amino acids on rAAV3 capsids further enhance viral transduction efficiency in vivo following systemic administration
We next wished to evaluate the transduction efficiency of the two optimized rAAV3 vectors in murine xenograft models in vivo. To this end, human Huh7 or Hep293TT tumor-bearing NSG mice were used (n=4), and a relatively low dose (5×1010 vg/mouse) of rAAV3−Y705+731F or rAAV3−S663V+T492V vectors was delivered via the tail vein. Whole-body bioluminescent imaging was performed 3 days after vector injections. From the results shown in Fig. 3A, it is clear that both Huh7 and Hep293TT tumors were efficiently targeted by the optimized rAAV3 vectors, and that transgene expression in both types of tumors was significantly enhanced by rAAV3−S663V+T492V vectors (Fig. 3B), compared with rAAV3−Y705+731F vectors, which we have previously shown to be significantly more efficient than the WT rAAV3 vectors in vivo (Cheng et al., 2012). Three days after vector injections, mice were sacrificed, and liver and tumor tissues were harvested. Total DNA samples were isolated from both liver and tumor tissues, and subjected to qPCR analyses to determine the vector genome copy numbers.

Transduction efficiency of optimized rAAV3 vectors in vivo.
From the data shown in Fig. 3C, it is evident that the persisting vector genomes of rAAV3−S663V+T492V mutant in the tumor were significantly higher than those of rAAV3−Y705+731F mutant (upper panel), which also correlates well with the Fluc transgene expression (Fig. 3A and B). No significant difference in the persisting vector genomes of the two vectors in the liver was observed, which is also comparable to that reported by others (Zincarelli et al., 2008). It is noteworthy that the studied tumor grew progressively in mice after establishment, which may account for a roughly fivefold lower vector genome copy numbers in the tumor than that in the liver. Nevertheless, despite the presence of vector genomes in the liver, little transgene expression was detected with either of the two optimized rAAV3 vectors. Most importantly, the rAAV3−S663V+T492V mutant vectors showed similar ability to transduce various human HCC tumors through systemic administration in vivo (Fig. 3D).
Systemic administration of optimized therapeutic rAAV3 vector suppresses liver tumor growth in vivo
Previous studies identified a new therapeutic gene that encodes trichosanthin (TCS), a ribosome-inactivating protein, isolated from a traditional Chinese medicinal herb, T. kirilowii (Sha et al., 2013). Although the nucleotide sequence of the TCS gene was determined more than 20 years ago, the delivery of the gene into mammalian cells has never been pursued. We synthesized TCS gene-expressing cassettes (Supplementary Fig. S1B) under the control of the AFPp, whose intracellular expression significantly inhibited the growth of human HCC cell lines in vitro and HCC tumors in vivo (Zhang et al., 2014).
Next, we generated rAAV3−S663V+T492V mutant vectors carrying this novel therapeutic gene. In addition, to allow initiating the treatment at an early time point, before the tumors are palpable, we also generated a genetically modified human HCC cell line, Huh7-Fluc, in which the Fluc gene under the control of the CBAp promoter is stably transfected, which also allowed for monitoring the tumor growth by whole-body bioluminescent imaging. Four weeks postxenografts, NSG mice were divided into 2 groups (n=5), and 5×1010 vg/mouse of rAAV3−S663V+T492V−AFPp−TCS vectors were injected via the tail vein in the first group (day 0). The second group of mice was injected with 5×1010 vg/mouse of rAAV3−S663V+T492V-AFPp-EGFP vectors to serve as appropriate controls. Whole-body bioluminescent imaging of mice was performed at days 0, 3, 8, and 11 after vector administration. These results, shown in Supplementary Fig. S2A, document that whereas Huh7-Fluc tumors grew progressively in mice injected with negative control vectors, tumor growth in mice injected with therapeutic vectors was significantly inhibited until day 11 (p<0.05), with maximal growth inhibition at day 8 (p<0.01). Whole-body bioluminescent images of mice performed at day 8 after vector administrations are shown in Supplementary Fig. S2B. Moreover, on day 11, all mice were sacrificed, and serum levels of aspartate transaminase (AST) and alanine transaminase (ALT) were determined. No significant differences were observed between two groups, suggesting little liver injury in mice (Supplementary Fig. S2C).
Shikonin significantly enhances rAAV vector-mediated transgene expression
So far, seven leading compounds isolated from TCM have been reported to possess HCC growth inhibitor activity. It was of interest to investigate their ability to enhance rAAV vector-based transgene expression. To this end, assays were initially performed using HeLa cells infected with rAAV2 vectors, the most extensively studied rAAV serotype. Interestingly, shikonin possesses the best ability in these assays, ∼10-fold increase for ssAAV2 and ∼5-fold for scAAV2 vectors (Fig. 4A and B; Supplementary Fig. S3A and B). Flow cytometry analysis revealed that not only the percentage of EGFP-positive cells but also the EGFP mean value in each positive cell was significantly enhanced in the presence of shikonin, compared with DMSO control (Fig. 4C). A dose-dependent effect was also observed with various rAAV vectors, including the capsid-modified rAAV3 vectors in human liver cancer cells (Fig. 4D and 4E; Supplementary Fig. S3C). On the other hand, no significant effect was observed when HeLa cells were treated with shikonin 48 hr after vector transduction (Supplementary Fig. S3D), indicating that shikonin plays a role in enhancing rAAV vector-mediated transgene expression during the early steps of viral infection.
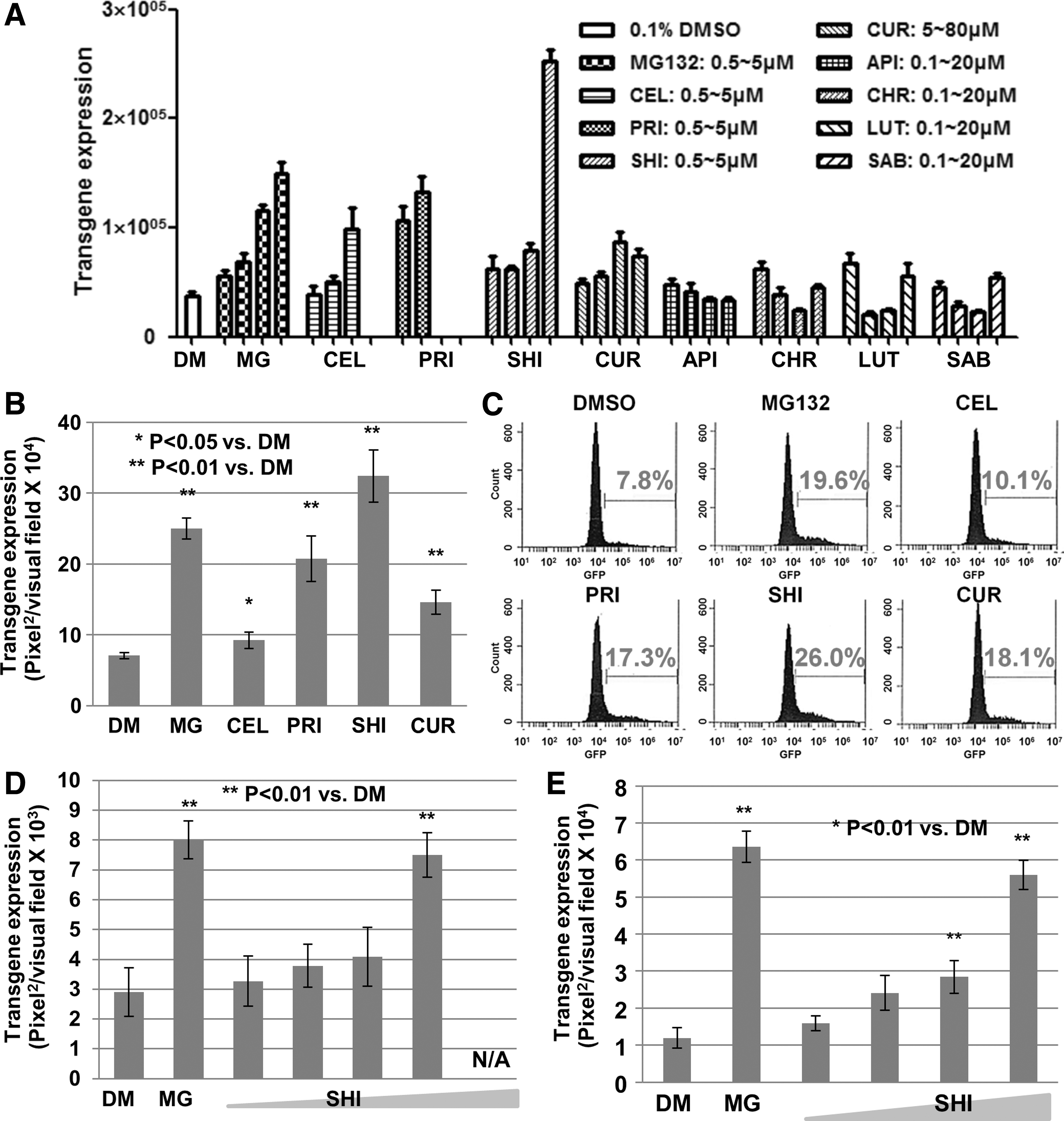
Shikonin significantly enhances rAAV vector-mediated transgene expression.
Shikonin affects rAAV transduction through its proteasome inhibitor activity
To further address the potential mechanism that shikonin significantly enhances rAAV vector-mediated transgene expression, we conducted the following experiments. First, total RNAs were extracted from cells at 24 hr postinfection with rAAV2 vectors, co-treated with various drugs, and were subjected to qRT-PCR assays. The increase in mRNA copy number in the presence of drugs correlates well with the increase in the transgene expression (Fig. 5A), suggesting minimal effect of these drugs on protein translation. Second, the intracellular biodistribution assay indicated that the percentage of viral genomes in nucleus 24 hr after viral infection was drastically affected by the shikonin treatment (Fig. 5B). Third, as shown in Fig. 5C, no further increase was observed when cells were co-treated with shikonin and MG132, a well-known proteasome inhibitor. As a control, co-treatment of 1-Azakenpaullone, a known GSK-3 inhibitor (Kunick et al., 2004), has an additive effect over either shikonin treatment or MG132 treatment. Fourth, we generated Cy3-labeled rAAV2 particles, which could be visualized under a Leica TCS SP5 confocal system (Leica Microsystems). Although the Cy3-modification reduced the viral vector-mediated transduction efficiency (Fig. 5D), it is evident that the transgene expression still could be further augmented by shikonin treatment (Fig. 5E). Most importantly, confocal microscopy results indicated that shikonin behaved the same as MG132, regarding the nuclear translocation of intact viral particles (Fig. 5F). Fifth, upon shikonin treatment, the levels of polyubiquitinated total cellular proteins were accumulated in the cells in a dose-dependent manner, similar to MG132 treatment (Fig. 5G).
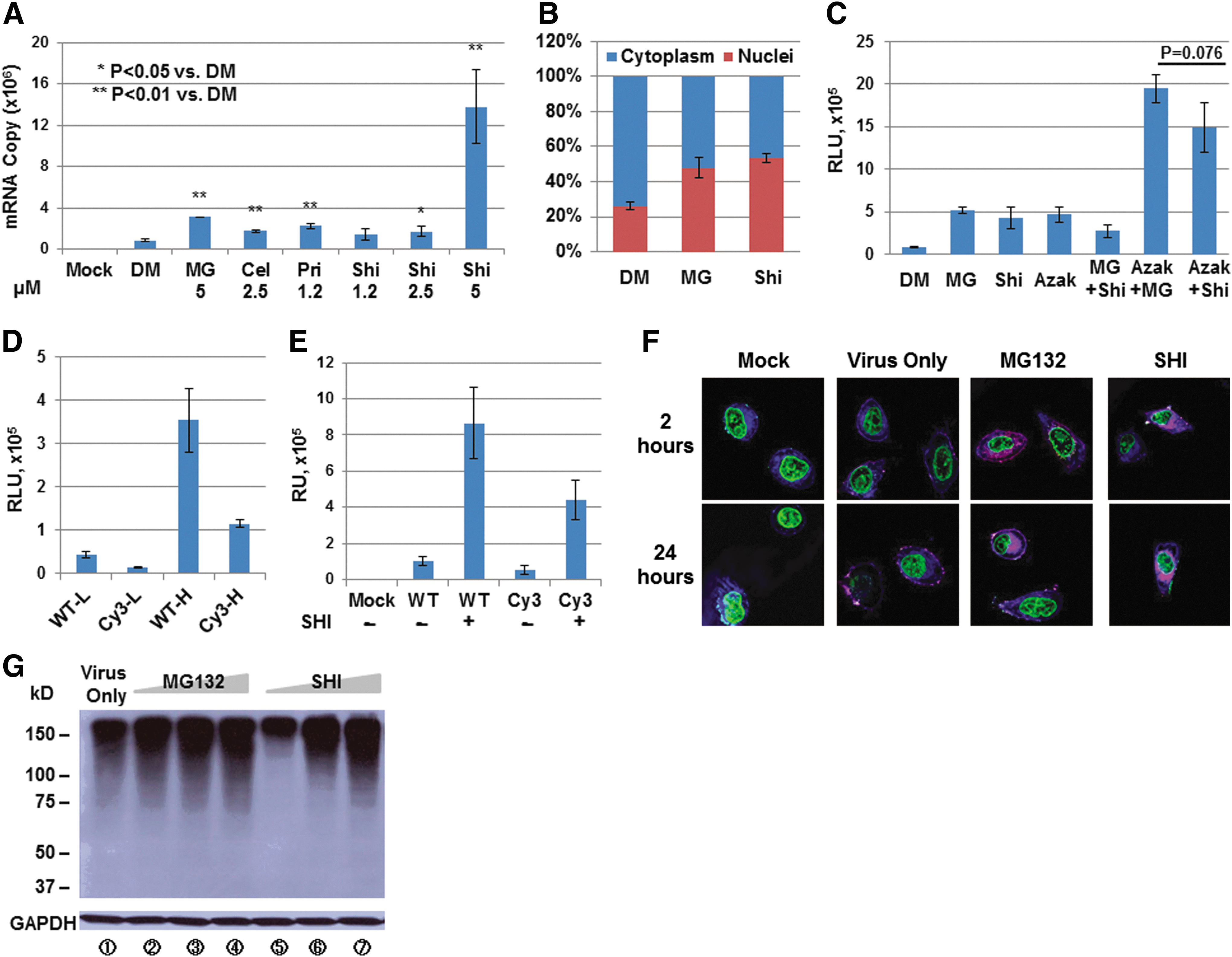
The mechanism of shikonin's effect on enhancing rAAV vector-mediated transgene expression.
Shikonin inhibits liver cancer cell growth both in vitro and in vivo with little hepatic cytotoxicity
Next, we wished to investigate shikonin's effect on liver cancer cell growth. During the TCM drugs screen assay, we observed that the IC50 of shikonin to inhibit Huh7 cell growth in vitro was similar to that of pristimerin, and that shikonin was more effective than all other drugs tested (Supplementary Fig. S4A). LDH assays suggested that an increase in cell death was accompanied by increased dose of shikonin (Supplementary Fig. S4B). In addition, intraperitoneal injection with shikonin at 1 mg/kg/day for 14 consecutive days significantly reduced Huh7-Fluc tumor size (Supplementary Fig. S4C and D), although the tumor growth was restored quickly once the shikonin administration was stopped. To address the potential hepatic cytotoxicity of shikonin treatment, we compared its effect on various cultured cells in vitro. Interestingly, the IC50 of shikonin to inhibit various liver cancer cell growth was significantly lower than that to inhibit cultured primary human hepatocytes (Supplementary Fig. S5A). Furthermore, we intraperitoneally injected normal male C57BL/6 mice with either DMSO control or shikonin at different doses for 10 consecutive days, and then mice were sacrificed and serum levels of AST, ALT, and ALP were determined. No significant differences were observed between control mice and mice treated with shikonin at 1 mg/kg/day, suggesting that little liver injury occurred at the therapeutic dose (Supplementary Fig. S5B).
A combination of the optimized therapeutic rAAV3 vector and shikonin further suppresses tumor growth in a mouse xenograft model
Finally, the Huh7-Fluc tumor-bearing male NSG mice were generated and the following treatment strategies were compared side by side: (1) mock treatment; (2) rAAV3−S663V+T492V−EGFP vectors at 5×1010 vg/mouse; (3) rAAV3−S663V+T492V-EGFP vectors at 5×1010 vg/mouse and 5 consecutive days of shikonin at 1 mg/kg/day; (4) rAAV3−S663V+T492V-TCS vectors at 5×1010 vg/mouse; and (5) rAAV3−S663V+T492V-TCS vectors at 5×1010 vg/mouse and 5 consecutive days of shikonin at 1 mg/kg/day. Both transgenes were driven by AFPp, and all rAAV3 vectors and drugs were administrated by tail-vein and intraperitoneal injection, respectively. Tumor growth was monitored either by whole-body bioluminescence in vivo imaging of Fluc expression over 20 days after stopping the shikonin treatment (Fig. 6A), or by tumor volume measurement at the end of the experiment (Fig. 6B). Although it was not possible to evaluate the long-term efficacy of this approach because of restrictions imposed by the Institutional Animal Care and Use Committee (IACUC) with reference to the extent of tumor growth in animals in the control, untreated group, these results clearly indicate that both rAAV3-based gene therapy and shikonin-based chemotherapy have a significant inhibitory effect on tumor growth, and that the combination of both strategies is highly effective in further preventing tumor growth. In addition, there is little alteration in the serum AST, ALP, and ALT level from different mice groups, suggesting minimal cytotoxicity on normal hepatocytes induced by the combination therapy (Fig. 6C).

Therapeutic effects of the shikonin-based chemotherapy and capsid-optimized rAAV3 vector-based gene therapy in vivo. Huh7-Fluc tumor-bearing male NSG mice were used for various treatments.
Discussion
As stated above, it is readily clear that the development of novel therapeutic options for HCC is sorely needed (Zhang et al., 2014), and the combination of rAAV vector-based gene therapy and shikonin-based chemotherapy is a promising option. Indeed, recombinant AAV vector-mediated gene therapy of HCC has been attempted. Most of the work was focused on rAAV2 previously (Peng et al., 2000; Su et al., 2000) and rAAV8 vectors more recently (Hsu et al., 2012). However, rAAV2 vectors were generally inefficient through systemic injection (Zincarelli et al., 2008) and rAAV8 vectors have a broad tropism to normal tissues of mice other than the liver (Gao et al., 2002; Wang et al., 2005; Zincarelli et al., 2008). In addition, most cocktail strategies to combine gene therapy and chemotherapy demonstrated only additive therapeutic effects. Only chemical drugs acting on specific molecular targets were utilized (Liu et al., 2014). Our studies, on the other hand, report a synergistic effect by combining multifunctional drug-based chemotherapy and rAAV vector-based targeted cancer gene therapy.
In our studies described here, we have shown, for the first time, that the remarkable tropism of rAAV3 vectors for human liver cancer cells in vitro can also be exploited to achieve targeted delivery of these vectors to various human liver tumors in a xenograft mouse model in vivo. In addition, site-directed mutagenesis of specific amino acid residues on the rAAV3 capsid can further augment the transduction efficiency of rAAV3 vectors. Furthermore, the optimized rAAV3 vectors expressing a novel therapeutic gene can be used to suppress human liver tumorigenesis in a murine xenograft model.
While our studies were in progress, Lisowski et al. (2014) reported the development of a shuffled rAAV vector, designated LK-03, using a chimeric human–mouse liver model. It is noteworthy that LK-03 shares 97.7% homology at the DNA level, and 98.9% homology at the amino acids level, with rAAV3. However, a number of distinct differences are also readily apparent. For example, Lisowski et al. (2014) used NOD/SCID mice to establish human Huh7 xenografts, and injected 1×1011 vg/mouse of rAAV3 or LK-03 vectors, but did not detect any transgene expression in tumors transduced with rAAV3 vectors. In our studies, on the other hand, using NSG mice and 5×1010 vg/mouse of rAAV3−S663V+T492 vectors, we were readily able to not only efficiently transduce the tumors, but also suppress tumor growth. These differences notwithstanding, the fact that S663 and T492 residues are conserved in LK-03 as well, it would be of interest to now introduce the S663V+T492V mutation in LK-03. Thus, regardless of which vector would prove to be more efficacious, our studies as well as those described by Lisowski et al. (2014) have significant implications in the further development of rAAV3 vectors for their optimal use in the potential gene therapy of human liver cancers. Interestingly, our data indicate that the rAAV3 vector-mediated transgene expression in HCC tumors in vivo is not significantly affected by gender, at least in mice. Although the incidence of HCC in the female population is lower than that in males, the total patient number is not insignificant (Chen et al., 2013).
For hundreds of years, natural products, mostly herbs, have become a commonly used treatment for liver diseases (Dong and Su, 2014), including HCC (Song et al., 2013), in China because of its unique advantages, such as being less expensive, controlling symptoms, improving quality of life, and preventing disease recurrence. Shikonin, a natural naphthoquinone isolated from the herb Lithospermum erythrorhizon, has been found to inhibit tumor growth and induce cell cycle arrest and apoptosis of various cancer cells, including HCC, through reactive oxygen species (Gong and Li, 2011). Our data presented here show that shikonin also has the ability to enhance rAAV vector-mediated transgene expression through its proteasome inhibitor activity. As a multifunctional drug, safety instead of efficacy is yet another challenging issue. Fortunately, there was no significant difference in serum AST, ALP, and ALT levels between control mice and those treated with shikonin at a therapeutic dose (1 mg/kg/day), suggesting that it may be a safe and effective antitumor as well as rAAV enhancer agent.
However, neither rAAV3-based gene therapy nor shikonin-based chemotherapy is ideal; the former requires a large dose for systemic administration (Supplementary Fig. S2), and the latter requires repeated injections at a therapeutic dose without inducing liver injury (Supplementary Fig. S4). These observations, along with those that shikonin enhances rAAV vector-mediated transgene expression, prompted us to combine these strategies to achieve the maximum therapeutic effect. Our data indicate that tail-vein injection of rAAV3 at 5×1010 vg/mouse, and a 5-day intraperitoneal injection with shikonin at 1 mg/kg/day, resulted in no further tumor growth until the day when mock-treated mice had to be sacrificed because of the large tumor volume. Next, it is of interest to perform additional experiments with primary human liver tumor xenografts, especially in liver microenvironment and those with large animal models. In sum, our studies indicate that the combination of shikonin-based chemotherapy and rAAV3 vector-based gene therapy might prove useful in targeting and treating human liver cancers.
Footnotes
Acknowledgments
This work was supported in part by the Alex's Lemonade Foundation, and Bankhead-Coley Cancer Research Program, Florida Department of Health (to C.L.); Public Health Service Grants R01 HL-097088 (to A.S.), and R21 EB-015684 (to A.S. and G.G.) from the National Institutes of Health; and institutional grants from the Children's Miracle Network (to A.S., C.L., and G.A.) and the National Natural Science Foundation of China grant No. 81273881 (to CQL) and No. 81303112 (to LNW); E-institutes of Shanghai Municipal Education Commission, Project Number: E-03008 (to CQL).
Author Disclosure Statement
None of the authors have any competing financial interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.