
Abstract
Cytokine-induced killer (CIK) cells consist of a heterogeneous population of polyclonal T lymphocytes displaying NK phenotype and HLA-unrestricted cytotoxic activity against a broad range of tumors. We sought to determine whether transduction of CIK cells with T cell receptor (TCR) genes specific for tumor-associated antigens could generate effector cells endowed with a double mechanism of tumor recognition. HLA-A2-restricted TCR-transduced (TD) CIK directed against the melanoma antigens Mart1 and NY-ESO1 were generated by lentiviral transduction and successfully expanded over a 3–4-week period. TD-CIK cells were both CD3+/CD56− and CD3+/CD56+ (31±8% and 59±9%, respectively), indicating that both major histocompatibility complex (MHC)-restricted T cells and MHC-unrestricted CIK could be targeted by lentiviral transduction. At the end of the culture, the majority of both unmodified and TD-CIK displayed an effector memory phenotype, without considerable expression of replicative senescence and exhaustion markers. Functionally, TD-CIK specifically recognized tumor cells expressing the relevant antigen as well as maintained their MHC-unrestricted tumor activity. The cytotoxic activity of TD-CIK against HLA-A2+ melanoma cell lines was significantly higher than the untransduced counterparts at a low effector:target ratio (cytotoxic activity of TD-CIK was from 1.9- to 4.3-fold higher than untransduced counterparts). TD-CIK were highly proficient in releasing high amount of IFN-γ upon antigen-specific stimulation and were able to recognize primary melanoma targets. In conclusion, we showed that (1) the reproducibility and simplicity of CIK transduction and expansion might solve the problem of obtaining adequate numbers of potent antitumor effector cells for adoptive immunotherapy; (2) the presence of both terminal effectors as well as of less differentiated progenitors might confer them long survival in vivo; and (3) the addition of an MHC-restricted antigen recognition allows not only targeting tumor surface antigens but also a wider range of cytoplasmic or nuclear antigens, involved in tumor proliferation and survival. TD-CIK cells with a double mechanism of tumor recognition are an attractive and alternative tool for the development of efficient cell therapeutic strategies.
Introduction
C
Gene transfer technology is effective in redirecting T cell activity against tumors. Lymphocytes engineered with tumor-specific T cell receptors (TCR) or chimeric-antigen receptors (CAR) are under investigation in clinical trials with encouraging/relevant results. 27 The possibility to apply such gene transfer strategies to CIK cells appears enticing. Indeed, it would be possible to add a tumor-antigen specificity to effector cells already capable of MHC-independent antitumor activity.
Promising preclinical results have been recently reported with CAR-engineered CIK cells 28 against CD19 29 or CD33/CD123 leukemia targets. 30,31 One potential limitation of CAR-based strategies is their exclusive activity against extracellular targets. 27 To overcome this liability, several and alternative strategies are under investigation, taking advantage of tumor-specific TCRs, which are capable of recognizing antigens derived from both intracellular and extracellular molecules. Redirected T cell function has been demonstrated in vitro and in preclinical settings, against a variety of antigens, including MART1, NY-ESO1, gp100, and CEA. 32 Similarly, several in vivo models and initial positive clinical results have been reported in melanoma and soft tissue sarcoma. 32 –34 Although innovative, these strategies have daunting limitations, including the MHC restriction of target antigens, the functional avidity of TCR, and the creation of undesired TCR heterodimers (mispairing). 35 We previously set and validated a lentiviral-mediated TCR transfer strategy to redirect T cell specificity against the melanoma-associated antigen MelanA/Mart1. 36 To avoid the issue of mispairing, we selected a TCR with high intrinsic interchain pairing properties, from a wide panel of Mart1-specific CD8+ clones derived from a vitiligo patient.
TCR transfer strategies have been so far confined to conventional T lymphocytes. Intriguing, but currently poorly explored, it is the possibility of TCR engineering other immune effectors like CIK cells. Such an approach would simultaneously combine the MHC-independent antitumor activity with the MHC-restricted tumor specificity, resulting in functional enhancement of tumor killing ability and better homing of highly efficient effector cells to tumor sites. Furthermore, the great ex vivo expansibility of CIK cells could allow the production of clinical-grade and relevant number of TCR-engineered immune effectors, addressing a main limitation of TCR-engineered T lymphocytes.
We here report preclinical data on CIK cells engineered with exogenous TCR against the tumor antigens Mart1 and NY-ESO1. Phenotypic and functional analyses are provided, supporting the feasibility of this approach, its enhanced efficacy, and the antitumor potential of TCR-engineered CIK cells.
Materials and Methods
Cell lines
The human melanoma cell lines DettMel, WK-Mel, and G4 were obtained from V. Russo (Cancer Gene Therapy Unit, Scientific Institute S. Raffaele, Milan, Italy), while the line Mel-1300 was obtained from M.I. Nishimura (Chicago, IL). They were maintained in RPMI 1640 (Invitrogen Life Technologies) supplemented with heat-inactivated 10% fetal calf serum and penicillin/streptomycin (Gibco Invitrogen). Established 293T is a human embryonic kidney cell line (a gift from L. Naldini, Milan, Italy), which was cultured in Iscove's modified Dulbecco's medium (IMDM; Lonza Verviers) supplemented as described previously.
Primary melanoma cultures M005, M015, M026, and M042 were generated from melanoma patients using the same methodology described by Gammaitoni et al. 3 Cells were maintained in KnockOut Dulbecco's modified Eagle's medium–nutrient mixture F-12 medium (KO DMEM:F12 medium; Gibco Invitrogen) with the addition of penicillin (50 U/ml), streptomycin (50 μg/ml), and Glutamax 100×(all from Gibco Invitrogen). The M005 is HLA-A2−, whereas MO15, MO26, and MO42 are HLA-A2+ primary melanoma cells.
Construction of lentiviral vectors containing Mart1-specific TCR genes and production of recombinant lentiviral particles
The α and β chain genes of a previously described Mart1-specific TCR 36 were cloned in a bidirectional third-generation lentiviral vector. 37 In this vector, the α chain was cloned under the control of the human phosphoglycerate kinase (hPGK) promoter, and the β chain under the control of the minimal cytomegalovirus promoter. Vector stocks were produced by calcium phosphate transient transfection of 293T cells with the following constructs: (1) packaging construct (pMDLg=pRRE), (2) Rev expression plasmid (pRSV-rev), (3) envelope construct (pVSV-G), and (4) a transfer vector encoding the desired TCR α and β chain. The culture medium was replaced 12–16 hr after transfection and the supernatant was collected and filtered 30–48 hr later. Determination of viral p24 antigen concentration was done by HIV-1 p24 core profile ELISA (PerkinElmer).
Generation of CIK cells
Human peripheral blood and tumor samples were obtained from healthy donors or metastatic melanoma patients at the Fondazione del Piemonte per l'Oncologia–Institute for Cancer Research and Treatment (FPO-IRCC; Candiolo, Torino, Italy), after providing informed consent under institutional review board-approved protocols. PBMCs were isolated by histopaque density gradient centrifugation (Sigma). To generate CIK cells, PBMCs were cultured at 2×106 cells/ml in RPMI 1640 medium, 10% heat inactivated FBS and penicillin/streptomycin, supplemented with 1000 U/ml of IFN-γ (PeproTech Inc.) were added. After 24 hr of culture, 100 ng/ml of CD3 MoAb (clone OKT3 functional grade; Miltenyi Biotec) and 300 U/ml of IL-2 (Proleukin; Chiron). 38 Half of the culture medium was replaced with a fresh medium with IL-2 every 3 days. Phenotypic analysis was performed weekly, and cytotoxic activity was assessed at the end of 3–4 weeks of culture.
Flow cytometric analysis
Expression of cell surface molecules was determined by flow cytometry using multicolor standard methodology. CIK were labeled with differently conjugated monoclonal antibodies (FITC, PE, PC5, PC7, APC, Pacific Blue, Alexa F750) to detect the following surface markers: CD3, CD8, CD27, CD45RA, CD56, CD57, and CD127 (Beckman Coulter), CD62L, and CD28 (BD Biosciences). Cells were electronically gated according to light-scatter properties and DAPI incorporation to exclude cell debris. Intracellular staining for detection of Mart1 was performed by indirect fluorescence using a Mart1-specific monoclonal antibody (Santa Cruz Biotechnology), and a PE-conjugated goat anti-mouse Ig. Samples were analyzed using FACS Cyan (Cyan ADP; Dako).
Generation of Mart1 TCR-transduced CIK
Human PBMCs from healthy donors or from melanoma patients were cultured with 1000 U/ml of IFN-γ. After 24 hr, they were activated with CD3 MoAb (100 ng/ml) and 300 U/ml of IL-2, and after 48 hours, 1×105 cells were infected with lentiviral nonconcentrated or concentrated particles, using 100 or 200 ng of p24, respectively. All infections were carried out in the absence of Polybrene. The cells were washed in RPMI 1640 medium and 10% heat-inactivated FBS 1 day after infection and were then cultured in a fresh medium with 300 U/ml of IL-2. Half of the culture medium was replaced with a fresh medium with IL-2 every 3 days. Cells were expanded for about 3–4 weeks and stained using an HLA-A2 streptamer labeled with PE or with APCs and containing the Mart1A27L peptide (IBA BioTAGnology), to determine transduction efficiency. Staining with streptamer was performed according to the manufacturer's instruction and analyzed on FACS Cyan (Cyan ADP; Dako).
Generation of NY-ESO1 TCR-transduced CIK
A lentiviral bidirectional construct containing an HLA-A2-restricted, NY-ESO1-specific TCR was provided by the ATTACK consortium (Adoptive engineered T cell Targeting to Activate Cancer Killing) 34 and cloned into a self-inactivating lentiviral vector under the control of the hPGK promoter (Mastaglio et al., article in preparation). Transduction of CIK cells and CD3-CD28-activated PBMCs with the NY-ESO1-specific TCR was carried out using concentrated lentiviral particles using an MOI of 50. For CIK cells, the infection was performed using the same method described above for Mart1-specific TCR. To transduce conventional T cells, PBMCs were isolated from healthy donors and activated with monoclonal antibodies (MoAb) to CD3 and CD28 (used at 200 ng/ml and 1 μg/ml, respectively) and IL2 (50 U/ml) plus IL7 (10 ng/ml). After 72 hr, 1×105 cells were infected with concentrated lentiviral particles using an MOI of 50. CIK cells were expanded for about 3–4 weeks, whereas activated PBMCs were expanded for about 1 week, and a fraction of the cells was stained with an anti-CD8 MoAb together with fluorescein isothiocyanate (FITC)-conjugated Vβ 13.1 MoAb to determine transduction efficiency.
ELISPOT assay
The ELISPOT assay was performed according to the manufacturer's instructions, as already described. Briefly, 96-well flat-bottomed plates were precoated with a 10 μg/ml concentration of primary specific antibody (1-D1K for IFN-γ and GB10 for granzyme; Mabtech, Nacka Strand, Sweden). Effectors were plated at a final concentration of 5×104, 2×104, or 1×104 cells per well in duplicate in the presence of 1×104 HLA-A2+ or HLA-A2− melanoma cell lines. In blocking experiments, melanoma targets were preincubated with 10 μg/ml of the anti-HLA-class I MoAb W632 or with the same amount of an irrelevant isotype control (IgG2a) (both MoAb were from Becton Dickinson). After 24 hr, cells were washed and a biotinylated secondary antibody was added (MoAb 7-B6-1 for IFN-γ and GB11 for granzyme; Mabtech). After 2 hr of incubation, plates were washed and horseradish peroxidase (HRP; Mabtech) was added for 1 hr. After additional washes, the 3-amino-9-ethylcarbazole substrate was added for 20 min (AEC substrate kit; Sigma-Aldrich). The reaction was stopped by the addition of tap water, and the spots were quantified by a computer-assisted ELISPOT reader (AID; Nanogen). ELISPOT assays were carried out in duplicate wells, unless otherwise stated.
CFSE-based cytotoxic activity
CIK's antitumor activity was tested in cytotoxicity assay at the end of cell expansion and tumor cell lines were used as targets. Briefly, all cell lines were harvested and incubated with 10 mM CFSE (Vybrant CFDA SE cell tracer kit; Molecular Probes Invitrogen) at 107/ml for 30 min at 37°C. At the end of the labeling process, the labeled cells were washed twice with sterile PBS and resuspended in RPMI-1640 complete medium.
CIK cells were prepared in fresh medium and cultured with 1×104 target cells for 16 hr at different effector:target (E:T) ratios (20:1, 10:1, 5:1) in 96-well plates. After this time, the co-cultures were harvested and 1 μg/ml of propidium iodide was added for assigning the ratio of cell death; after 15 min at room temperature in dark, the cells were acquired and analyzed with BD FACSCalibur and CellQuest Software (BD Biosciences).
The percentage of specific lysis was calculated as described by Marcusson-Stahl and Cederbrant
39
according to the following equation:
Spontaneous release was obtained by incubating target cells in medium alone, whereas maximal release was obtained after treatment with triton solution.
Statistical analysis
For each variable considered in this study, mean and standard deviation were calculated. Statistical analysis was performed with a two-tailed Student's t-test for paired samples using Prism software, version 5.0 (GraphPad Software). p<0.05 was considered significant.
Results
TCR-redirected CIK cells maintain their phenotype
To induce the expression of a Mart1-specific TCR on CIK cells, PBMCs from six healthy donors were cultured with IFN-γ for 1 day, stimulated with IL-2 and OKT3 for additional 3 days, and transduced with nonconcentrated lentiviral particles (on day 4). Cells were then cultured and expanded for additional 3–4 weeks in the presence of IL-2. With this protocol, Mart1-specific TCR CIK cells were generated that contained a mean of 11±5 positively transduced cells, as determined by staining with a specific HLA-streptamer. TD-CIK expanded overtime similarly to nontransduced CIK (NT-CIK) cells (mean fold expansion 250±62 and 240±71, respectively). To investigate whether TCR transduction might have affected the differentiation of PBMC to CIK cells, we monitored the expression of CD56, CD8, and CD4 on NT-CIK and TD-CIK weekly. As shown in Fig. 1, NT-CIK displayed a pattern of differentiation similar to that of TD-CIK. The percentage of CD3+CD56+ increased from 2±2.45% to 57.1±5.5% in NT-CIK (panel a) and from 2.2±0.24% to 62.2±1.14% in TD-CIK (panel b). As normally observed, 40 the proportion of CD8+ cells increased overtime, while the proportion of CD4+ decreased in both NT-CIK and TD-CIK populations. When the same analysis was performed for Mart1-streptamer− (panel c) and Mart1-streptamer+ (panel d) fractions of TD-CIK, analogous patterns of differentiation were observed.
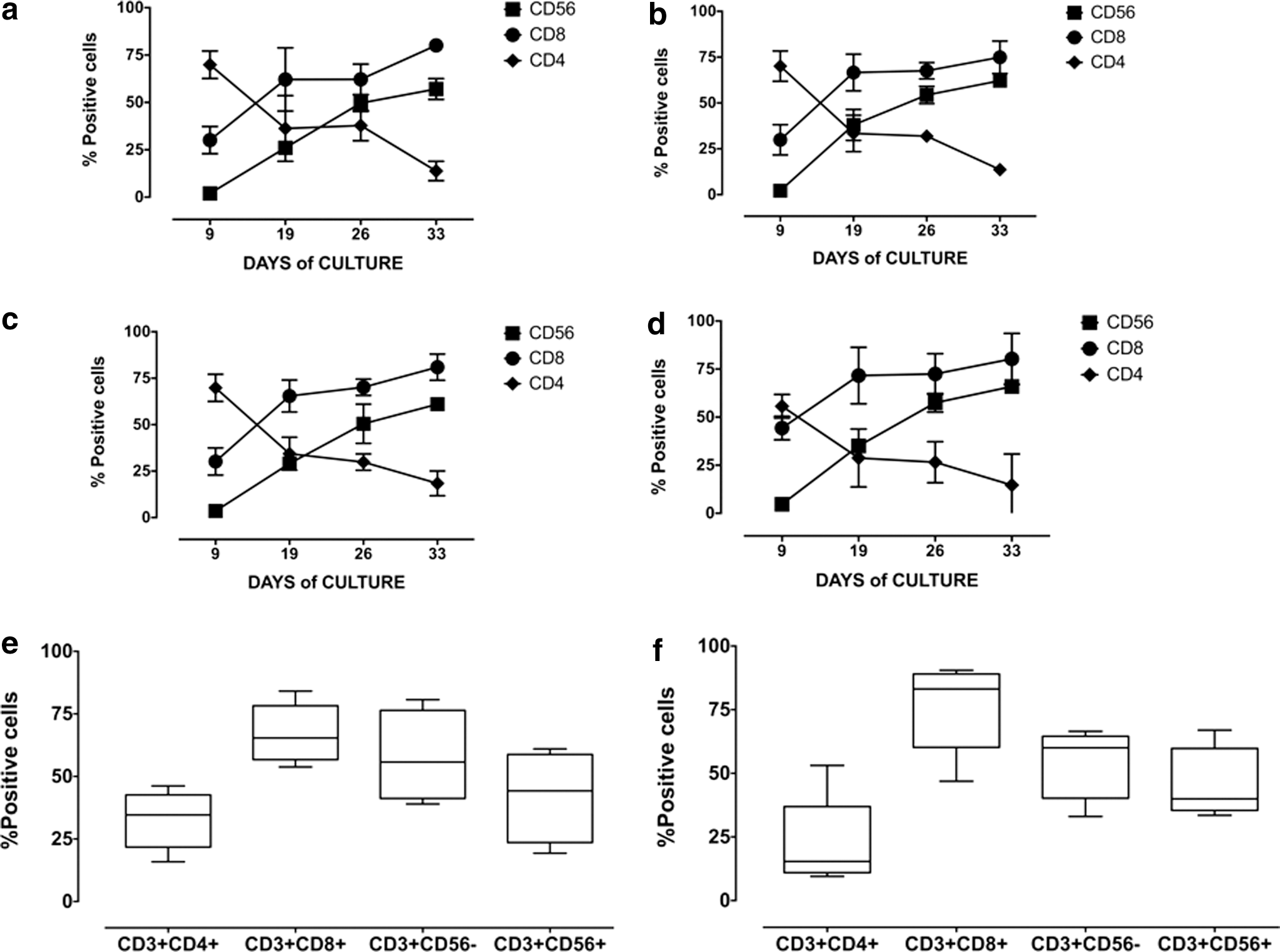
Generation of TCR-transduced CIK. Panels
Next, we analyzed the phenotype of Mart1-streptamer+ CIK cells at the end of cultures, demonstrating that transduced cells had phenotypes similar to that of untransduced cells (Fig. 1e and f). Specifically, Mart1-streptamer+ CIK cells were predominantly CD8+ (76±17%) with a slight increment of CD3+/CD56− (54±14%) over CD3+/CD56+ cells (46±14%). These findings were confirmed even after a prolonged expansion (up to 60 days; data not shown).
Overall, these data demonstrate that CIK cells can be stably transduced with lentiviral constructs encoding a Mart1-specific TCR and that transduction does not affect the phenotype of CIK cells.
TD-CIK cells display a phenotype of effector memory/terminally differentiated cells
To define the differentiation stage of TCR-transduced CIK cells, we checked CD62L and CD45RA expression on CD8+ and CD4+ cells after 3–4 weeks of culture (median time of culture 25 days). According to CD62L and CD45RA expression, CD4 and CD8 T cells were divided into four populations defined as follows: CD62L+/CD45RA+ (naïve, N), CD62L+/CD45RA− (central memory, CM), CD62L−/CD45RA− (effector memory, EM), and CD62L−/CD45RA+ (terminal differentiated RA+, TEMRA). The analysis of the CD8+ subset showed that NT-CIK and TD-CIK were mainly EM cells (31.1±2.7 and 35.4±12.3% respectively) and TEMRA (44.7±14.6 and 33.1±7.9%, respectively). Naïve CIK cells represented ∼20% of the total cells (21±5% in NT-CIK and 28±16% in TD-CIK); meanwhile, CM elements were a minority, in both NT and TD-CIK (<4%; Fig. 2a). Similarly to CD8+ cells, the CD4+ subset did not show substantial differences between NT-CIK and TD-CIK (Fig. 2b), with slightly higher percentage of CD4+ than in CD8+ CM CIK cells, reflecting physiological proportions seen in PBMCs (not shown).
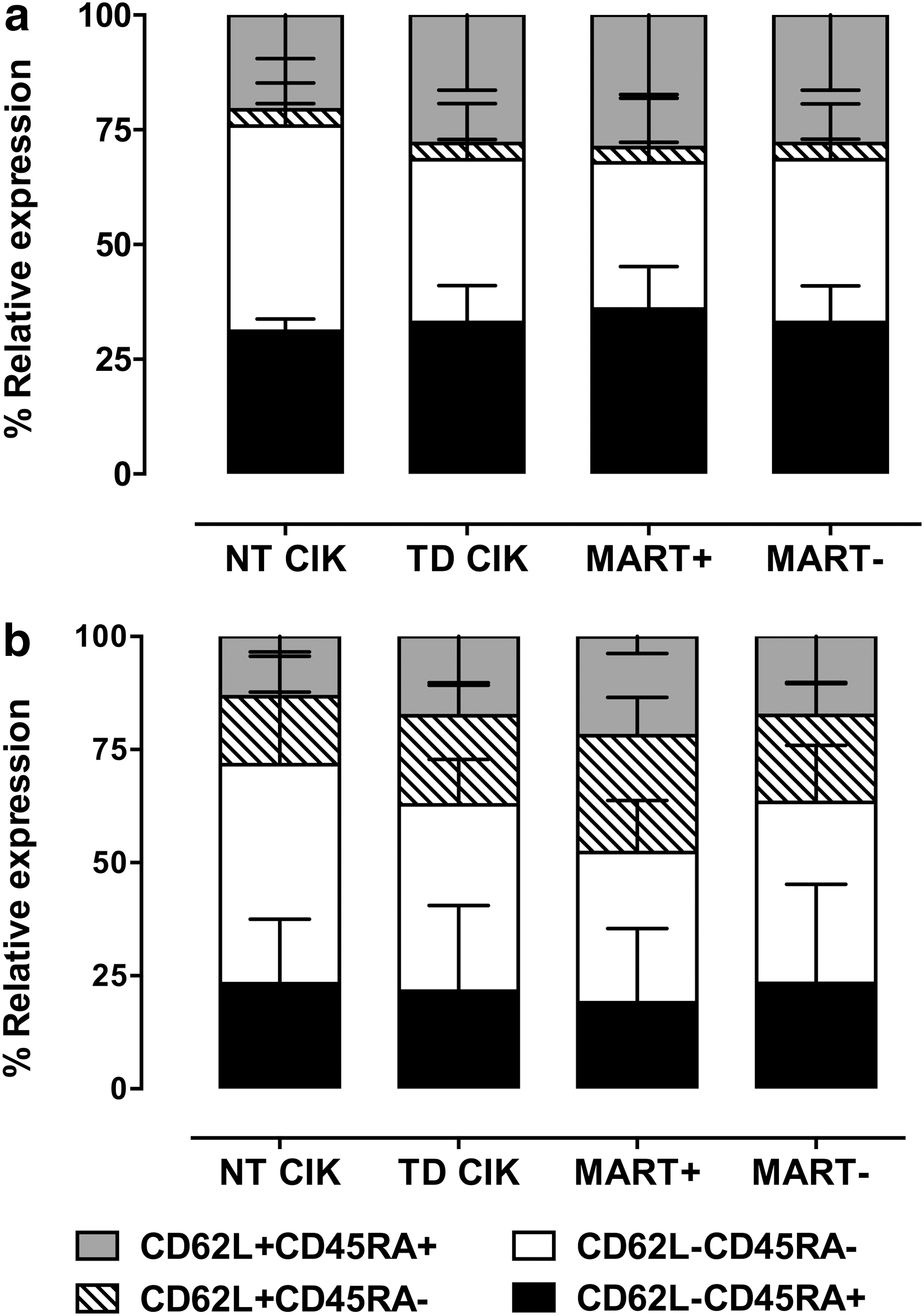
Characterization of naive, central memory, and effector memory compartment of NT-CIK and TD-CIK. Differentiation stage of NT-CIK, TD-CIK, Mart1-streptamer+ fraction, and Mart1-streptamer− fraction of TD-CIK. Panel
TD-CIK CD8+ effector cells do not display evidence of exhaustion
To better define the effector compartment of CD8+ TD-CIK, we analyzed the expression of the co-stimulatory molecules CD28 and CD27. According to the model proposed by Romero et al., 41 EM can be subdivided into four subsets: EM1 (CD28+/CD27+), EM2 (CD28−/CD27+), EM3 (CD28−/CD27−), and EM4 (CD28+/CD27−). Similarly, TEMRA can be subdivided into three subsets: pE1 (CD28+/CD27+), pE2 (CD28−/CD27+), and E (CD28−/CD27−) (Fig. 3a). The flow cytometric analysis of CIK cells showed that CD28 expression was significantly down-modulated compared with starting PBMCs, but the levels of CD27 were persistent in both EM and TEMRA compartments (45.9±12% and 72±10%, respectively), suggesting that mature CIK were mostly belonging to the EM2 and pE2 subtype (Fig. 3a). 42 Similarly to CD28, CCR7 was also down modulated (data not shown).

Lastly, we analyzed the expression of CD57, PD1 (markers of replicative senescence and T cell exhaustion, respectively), and CD127 (IL-7Rα, marker of homeostasis and long-term survival ability), on CD8+ TD-CIK. As shown in Fig. 3b, CD57 and PD1 were expressed in a minute fraction of both EM (10.9±3.53% and 8.2±2.8%) and TEMRA (5.3±2.5% and 3.2±1.6%), whereas CD127 was detectable in a significant fraction of cells (EM: 22±11% and TEMRA: 39±8%, respectively).
Altogether, these findings demonstrate that the effector compartment of transduced CIK cells is not exhausted and it has not reached terminal differentiation.
Dual activity of TCR-redirected CIK cells against Mart1+ melanoma targets
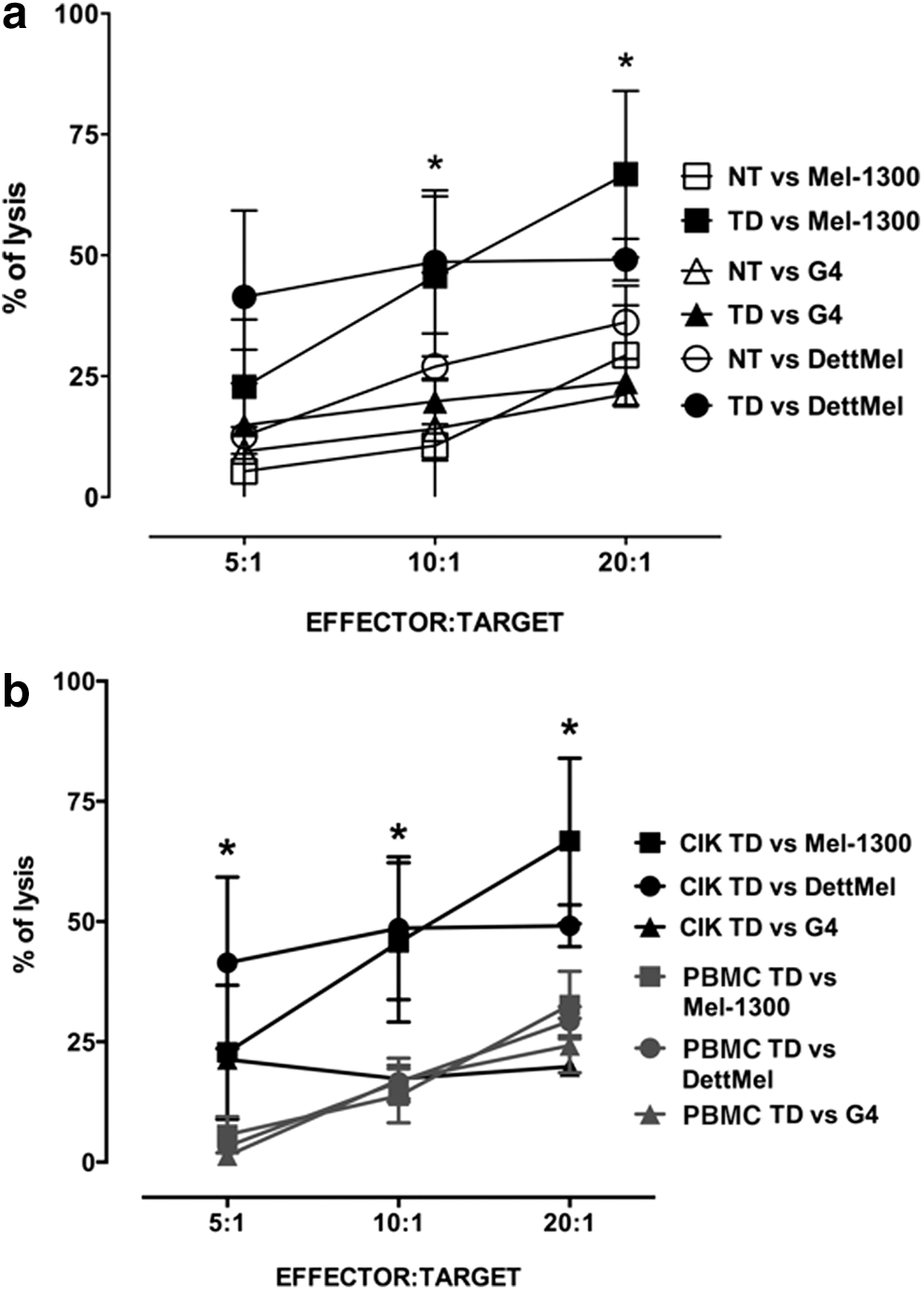
Functional activity of TD-CIK was tested by cytotoxicity and by ELISPOT against four melanoma cell lines; two of them were HLA-A2+ (Mel-1300 and DettMel) and the other two were HLA-A2− (G4 and WK-Mel). Figure 4a shows the expression of Mart1 by the melanoma cell lines as determined by intracytoplasmic staining with a MoAb: Mel-1300, DettMel, and G4 are Mart1 positive, while WK-Mel is Mart1 negative. To show that the antitumor ability of CIK cells was enhanced after TCR transduction, we performed a series of cytotoxicity assays. For these experiments, we used CIK obtained from 6 donors; 3 of them were the same used also for phenotypic analysis, while the other 3 were different donors used only for functional assay. Concentrated lentiviral particles were used in 3/6 cases, allowing for higher transduction efficiency (19%, 22%, and 26.5%; average 22.5%) than nonconcentrated particles (4.6%, 8%, and 8%; average 6.9%). Altogether, the transduction efficiency of this group was 14.7±9.9 (n=6) (data not shown). For cytotoxicity, CIK cells were cultured at different E:T ratios (5:1, 10:1, and 20:1) with the three Mart1+ cell lines (G4, DettMel, Mel-1300). Target cells were initially labeled with CFSE, and cytotoxicity was measured by flow cytometry after an overnight incubation with effector CIK cells (see Materials and Methods). Notably, TD-CIK cells killed Mart1+/HLA-A2+ melanoma cells more efficiently than NT-CIK (Fig. 4b). In particular, we detected an increase of 3.8- and 3.3-fold for the Mel-1300 and DettMel cell lines (E:T ratio 5:1); meanwhile, NT-CIK and TD-CIK showed a basal and similar antitumor activities against the Mart1+/HLA-A2− G4 melanoma cell line (Fig. 4b). Supplemental Fig. 1 shows how the cytotoxic activity of TD-CIK increases along with transduction efficiency.

Mart1 expression by melanoma cell lines and enhanced recognition by redirected CIK.
We then assessed the functional profile of TD-CIK by IFN-γ ELISPOT assay using melanoma cell lines as stimulators. In this assay, the IFN-γ-secreting ability of both NT-CIK and TD-CIK was similar when CIK cells were challenged with the Mart1+/HLA-A2− G4 cell line, suggesting that the level of MHC unrestricted activity was not different between the two subsets (Fig. 4c). On the contrary (and accordingly to their cytotoxic activity), TD-CIK released higher amount of IFN-γ when stimulated via Mart1+/HLA-A2+ melanoma cells (Mel-1300 and DettMel), whereas NT-CIK (47±18) failed to respond in the same conditions (Fig 4c). These results show that TD-CIK acquired the ability to specifically kill Mart1+/HLA-A2+ melanoma cell lines and to efficiently respond producing high levels of cytokines.
Since these findings were generated using normal donors as source of CIK cells, we next explored the possibility of generating functional TD-CIK from PBMCs from melanoma patients. TD-CIK from two metastatic melanoma patients were tested for cytotoxicity and IFN-γ release, generating data analogous to those seen with normal healthy donor CIK cells (Fig. 4d and e).
The activity of Mart1-redirected CIK cells is MHC dependent
To test whether the increased IFN-γ production and cytotoxic ability of TD-CIK was HLA dependent, we incubated with an anti-HLA-class I MoAb (W6/32) or with an isotype control the melanoma cell lines and measured CIK activity by IFN-γ and GranzymeB ELISPOT assays. As shown in Fig. 5a, the treatment with W6/32 MoAb abrogated IFN-γ production by TD-CIK cells when these cells were co-cultured with Mart1+/HLA-A2+ melanoma cells (Mel-1300 from 378 to 20 spots and DettMel from 268 to 7 spots, p<0.05). GranzymeB analysis (Fig 5b) revealed a similar pattern, with a higher production when TD-CIK cells were challenged by Mart1+/HLA-A2+ melanoma cells (251 spots for Mel-1300 and 206 for DettMel) compared with Mart1+/HLA-A2− melanoma cells (145 spots for G4 and 125 for WK-Mel). When blocking of class I was performed, the number of granzyme spots produced by TD-CIK in response to Mart1+/HLA-A2+ melanoma cells was reduced to the levels detected in NT-CIK-based assays (p<0.05 between TD-CIK and TD-CIK blocked with anti-HLA-class I).
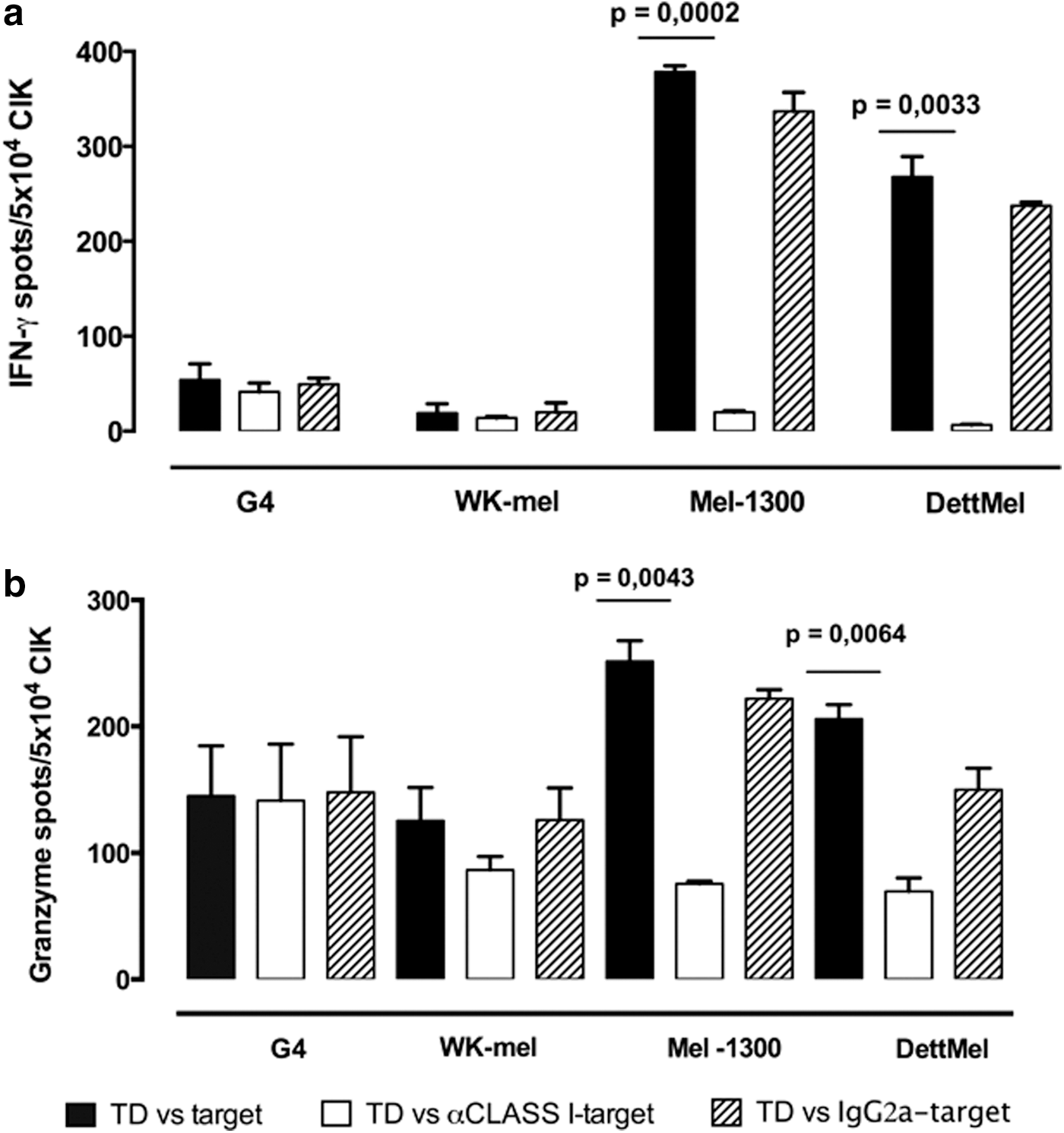
IFN-γ and GranzymeB release: TCR-related and major histocompatibility complex-unrestricted properties of TD-CIK. IFN-γ
Dual activity of TCR-redirected CIK cells against NY-ESO1 melanoma targets
We next explored whether this strategy could be applied more widely, using a different HLA-A2-restricted TCR specific for the cancer testis antigen NY-ESO1. 34 CIK cells were transduced using concentrated lentiviral particles containing an NY-ESO1-specific TCR, with the same protocol used for Mart1-redirected CIK. Transduction efficiency was 38±11 (n=3), as determined by staining with a specific Vbeta 13.1 MoAb. TD-CIK cells were tested against the three melanoma cell lines previously described. All lines (Mel-1300, DettMel, and G4) were found to express NY-ESO1 by RT-PCR (data not shown), confirming published data on Mel-1300. 43 Results obtained are shown in Fig. 6a: TD-CIK cells killed NY-ESO1+/HLA-A2+ melanoma cells more efficiently than NT-CIK. In particular, we detected an increase of 4.3- and of 1.9-fold against Mel-1300 and DettMel cell lines, respectively (E:T ratio 5:1), while NT-CIK and TD-CIK showed a basal and similar antitumor activities against the NY-ESO1+/HLA-A2− G4 melanoma cell line.
Activity of TCR-redirected CIK cells against NY-ESO1 melanoma targets.
We then compared the potency of TD-CIK with that of conventional T cells. PBMCs from the same donors used for CIK generation were stimulated with MoAb to CD3 and CD28 and transduced with concentrated lentiviral particles. One week after transduction, activated T cells were tested for their cytotoxic activity against the previously described melanoma cell lines and their activity was compared with that of TD-CIK cells. Transduction efficiency of activated T cells was 43±9, similar to that of TD-CIK (38±11), as determined by staining with a specific Vbeta 13.1 MoAb. Cytotoxic activity of TD-CIK was superior to that of activated PBMCs and this difference reached statistical significance for both Mel-1300 and DettMel at 10:1 and 20:1 E:T ratio, while at 5:1 E:T ratio the difference was significant only for DettMel (Fig. 6b).
TCR-redirected CIK cells are active against primary melanoma targets
To strengthen the clinical efficacy and applicability of redirected CIK, we finally checked their activity against primary tumor cell culture targets that were recently established in vitro. Mart1 and NY-ESO1 TD-CIK were tested against four melanoma primary cells: M005 (HLA-A2−, Mart1−, NY-ESO1+), M015 (HLA-A2+, Mart1±, NY-ESO1−), M0026 (HLA-A2+, Mart1+, NY-ESO1±), and M0042 (HLA-A2+, Mart1+, NY-ESO1+). We show in Fig. 7 that there was a significant increase of cytotoxic activity over NT-CIK when Mart1 TD-CIK were tested against Mart1+/HLA-A2+ targets (M026 and M042) and when NY-ESO1 TD-CIK were tested against NY-ESO1+/HLA-A2+ cells (M042, panel d). TD and NT-CIK displayed the same activity when tested against an HLA-A2-negative primary target (M005) or when they were tested against primary cells not expressing adequate levels of relevant antigen (M015 for Mart1, M015, and M026 for NY-ESO1) (Fig. 7a–d). The levels of Mart1 expression in the four primary cultures were determined by intracytoplasmic staining with an MoAb and are reported in Fig. 7e. Expression of NY-ESO1 was determined by RT-PCR (data not shown)
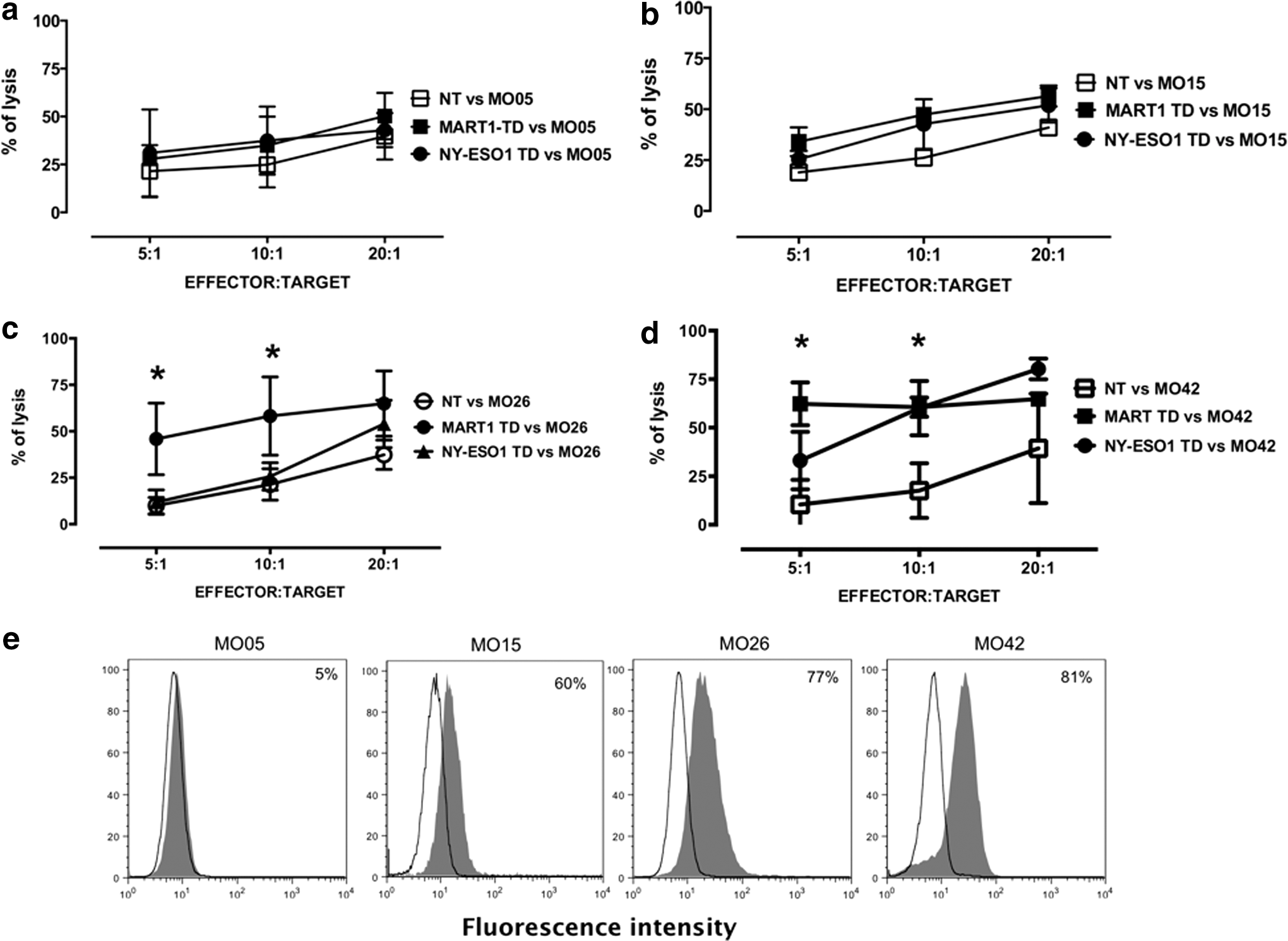
Activity of TCR-redirected CIK against primary melanoma targets. Cytotoxic activity of Mart1 and NY-ESO1 TD-CIK was tested against four melanoma primary cultures:
Discussion
Our data demonstrate that the stimulation of PBMCs with IFN-γ, CD3, and IL-2 followed by gene transfer of a tumor-specific TCR resulted in the generation of gene-modified CIK cells endowed with a double mechanism of tumor recognition. The core message is that the tumoricidal potential of CIK cells can be strongly enhanced by TCR transfer, making CIK cells capable of MHC-restricted and MHC-unrestricted recognition of tumor targets at the same time. Notably, our lentiviral-mediated gene transfer did not affect the differentiation of PBMCs to CIK cells, resulting in the efficient and reproducible expansion of large numbers of tumor-specific effector cells, equally divided into CD3+/CD56− and CD3+/CD56+ elements. Normally, untransduced CD3+/CD56+ CIK cells have phenotypic characteristics of terminally differentiated CD8+ effector memory T cells, and they are able to mediate a potent non-MHC-restricted cytotoxicity, but display a limited proliferative capacity. On the contrary, CD3+CD56− CIK cells have a reduced cytotoxicity but higher proliferating potential. 23,44 Thus, we hypothesize that, in our experimental setting, transduced CD3+/CD56+ cells have a double mechanism of tumor recognition, while CD3+/CD56− cells preferentially mediate MHC-restricted recognition and maintain a higher proliferative capacity.
This hypothesis is supported by our functional data, which showed that the MHC-dependent engagement of tumor-specific TCR antigens on CIK cells strongly enhanced their tumoricidal activity, synergizing with the conventional MHC-unrestricted activity typical of CIK cells. The clinical efficacy and applicability of this approach was supported by the observation that TD CIK cells can be easily generated also from melanoma patients and, most notably, that TD-CIK displayed enhanced cytotoxic activity also against primary melanoma targets. Clinical feasibility was also strengthened by the demonstration that TCR with different antigen specificities can be easily introduced into CIK cells with equal efficacy.
The enhanced tumor killing features of TD-CIK were detected even when a low effector/target ratio (5:1) was tested, demonstrating the impressive properties of these engineered CIK cells. Moreover, TD-CIK cells produced high amounts of IFN-γ after antigen-specific stimulation. T cell antigen activation triggers intracellular pathways resulting in cytotoxic activity and/or immunomodulatory cytokine production (i.e., IFN-γ). 45 Production of IFN-γ is typically stimulated by antigen encounter, which suggests that the increased IFN-γ production by TD-CIK cells should be ascribed to the MHC-dependent TCR activation. The increased production of IFN-γ by transduced CIK cells might be relevant in controlling tumor growth. Since CIK cells can infiltrate melanoma xenografts tumor masses in mice, 3 we hypothesize that TD-CIK might home to tumor site, kill tumor cells, and modulate the microenvironment through IFN-γ production.
In the balance between immune response and tumor growth, transduction of CIK cells could counteract some of the immune-escape mechanisms engendered by tumors. For instance, TCR-mediated recognition might overcome the inhibition of NKG2D-mediated death because of soluble MICA products released by tumor cells. 46
As expected, 47 TD-CIK cells showed an effector memory phenotype, although a small subset displayed a putative naive profile (CD62L+/CD45RA+) (approximately 28% of CD8+ and 15% of CD4+ CIK). Interestingly, TD-CIK frequently expressed the co-stimulatory molecule CD27, which is usually expressed by naïve, memory, and early effector T cells, 48 while it is downregulated in late effector T cells. The CD27 molecule is a relevant partner for TCR signaling, providing high level of activation and proliferation after interaction with its ligand, the CD70 molecule. Thus, we anticipate that CD27 expression in both CD56+ and CD56− cells might promote TCR triggering and antigen-dependent expansion of effector cells in vivo. To this regard, Huang and colleagues 49 have demonstrated that, in melanoma patients treated with TIL infusion, the number of CD27+ CD8+ T cells within TIL cells significantly correlated with tumor regression following adoptive transfer.
Finally, the analysis of the effector compartment revealed that CD8+ TD-CIK do not express CD28 (Fig. 4) and CCR7 (not shown) antigens, indicating that EM and TEMRA TD-CIK are at an intermediate rather than late stage of differentiation (namely, EM2 and pE2 in according to the model proposed by Romero et al. 41 and Rufer et al. 42 ), and that they may have the ability of further proliferation rounds, as also suggested by previous data generated in a different CIK model. 50
Regardless of the brisk and prolonged cell expansion in vitro that implies multiple rounds of cell divisions, TD-CIK lacked the senescence marker CD57 and the exhaustion marker PD1, suggesting that they could undergo further replication. Altogether, these features diversify TD-CIK cells from late effector cells with high lytic activity but poor replicative capacity. 51 These intermediate effector cells might drive in vivo an immediate and very potent killing of tumor cells, while the presence of CD62L+CD45RA+ naïve cells bearing an antitumor-specific TCR might drive a more persistent MHC-restricted memory response.
In conclusion, our study demonstrates that the TCR gene transfer technology of CIK cells is a powerful and novel strategy. TCR-engineered CIK might solve the stumbling problems of generating adequate numbers T cells targeting tumor-associated antigens. Lastly, CIK cultures have the advantage of being heterogeneous, comprising both differentiated effectors as well as proliferating progenitors. These features make the transduction of CIK cells an attractive tool for the development of efficient cell therapy strategies against both solid and hematological malignancies, which could be readily transferred to the clinical arena.
Footnotes
Acknowledgments
This work was supported by the Italian Association for Cancer Research (AIRC) through the Special Program in Clinical Molecular Oncology, Milan (5×1000 No. 10007), through My First Airc Grant Program (MFAG 2014 N.15731) and through AIRC IG 2013-14299 Regione Piemonte (ONCOPROT, CIPE 25/2005). The work was also supported by ImmOnc (Innovative approaches to boost the immune responses, Programma Operativo Regionale, Piattaforme Innovative BIO F.E.S.R. 2007/13, Asse 1 ‘Ricerca e innovazione’ della LR 34/2004), by Ricerca Finalizzata-Giovani Ricercatori (GR-2011-02349197), by the Oncology Program of Compagnia di San Paolo, Torino and by EU grants SUPERSIST and ATTACK. The authors wish to thank Stefania Rapelli, Vincenzo Russo and Luigi Naldini for helpful contributions. The authors also are grateful to healthy donors and melanoma patients for participating in this study.
Author Disclosure Statement
Chiara Bonini has a research contract with MolMed s.p.a. No competing financial interests exist for the remaining coauthors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.