
Abstract
Gene therapy has been applied to cardiovascular disease for over 20 years but it is the application to heart failure that has generated recent interest in clinical trials. There is laboratory and early clinical evidence that delivery of sarcoplasmic reticulum calcium ATPase 2a (SERCA2a) gene therapy is beneficial for heart failure and this therapy could become the first positive inotrope with anti-arrhythmic properties. In this review we will discuss the rationale for SERCA2a gene therapy as a viable strategy in heart failure, review the published data, and discuss the ongoing clinical trials, before concluding with comments on the future challenges and potential for this therapy.
Introduction
T
Background
Heart failure therapy today
Heart failure causes considerable morbidity and mortality in the developed world, affecting over 23 million people. 11 Current pharmacological treatments that improve prognosis in chronic heart failure include β-adrenergic blockers, agents that antagonize the renin–angiotensin–aldosterone system, the selective sinus node inhibitor ivabradine, and recently a new compound that is yet to be licensed LCZ696 (valsartan/sacubitril). 12 Implantable device treatments are also available, including cardiac resynchronization therapy, implantable cardioverter defibrillators, and left ventricular assist devices (LVADs). Many patients with heart failure continue to experience a significant burden of symptoms and poor life expectancy, which these interventions are unable to prevent because of inadequate efficacy, side effects, or issues of cost-effectiveness. There is a need for additional therapeutic options, and gene therapy offers a new approach by allowing us to directly and specifically target the underlying molecular abnormalities seen in the failing cardiomyocyte. Specifically, SERCA2a gene therapy has the potential to be the first heart failure treatment with both positive inotropic and anti-arrhythmic properties. 13
Gene expression in heart failure
There are numerous potential targets for gene therapy in heart failure, as it is recognized that there are hundreds of genes whose expression is disrupted. Microarray profiling of dilated cardiomyopathy (DCM), for example, has reported that over 600 genes are upregulated in DCM and more than 200 genes are downregulated. 14,15 These genes encompass a variety a functional classes, with a tendency for more genes in the categories of protein synthesis and metabolism. In addition, there is a proapoptotic shift in the TNF-alpha pathway 16 and an overall shift back to a fetal gene expression phenotype. Changes in gene expression arise when biomechanical stresses, such as pressure or volume overload, activate signaling pathways, which eventually lead to the activation of transcription factors, coregulators, and microRNAs in the cell nucleus. 17,18
Of the numerous genes whose expression is affected by heart failure, those of particular interest are those that code for proteins known to play a critical role in calcium (Ca2+) handling and the β-adrenergic system. Some of these genes have been studied since the early 1980s and examples include downregulation of gene expression for SERCA2a and β-1 adrenoreceptors. 19,20
Calcium handling in health and disease
The role of Ca2+ in excitation–contraction coupling (Fig. 1) is critically important to understand myocardial dysfunction in heart failure. The cardiac action potential depolarizes the surface membrane, initiating a small amount of Ca2+ entry to the cytoplasm through L-type Ca2+ channels. This triggers a much larger release of Ca2+ from the sarcoplasmic reticulum (SR) store through the ryanodine receptor (RyR). Calcium-induced calcium release activates contraction via the binding of Ca2+ to the troponin C component of the cardiac myofilaments. During diastole there is reuptake of Ca2+ into the SR through the action of SERCA2a and some is extruded from the cell by the Na+-Ca2+ exchanger (NCX). These changes in cytoplasmic Ca2+ concentration occur rapidly in normal myocytes, but it has been recognized for over 25 years that there are significant abnormalities in myocytes from patients with heart failure. 21 In particular, during diastole cytoplasmic Ca2+ concentrations are slow to fall and there is a failure to restore low resting Ca2+ levels reflected in impaired diastolic relaxation. These observations are a result of impaired Ca2+ reuptake into the SR during diastole.

Schematic of a cardiomyocyte showing normal Ca2+ handling during excitation–contraction coupling. (1) Normal Ca2+ cycling begins with the cardiac action potential depolarizing the surface membrane and triggering a small Ca2+ current into the cytoplasm through L-type Ca2+ channels. (2) This triggers a much larger influx of Ca2+ from the sarcoplasmic reticulum (SR) store through the ryanodine receptor (RyR). (3) This calcium-induced calcium release triggers contraction through the binding of Ca2+ to the troponin C component of the cardiac myofilaments. (4) During diastole, Ca2+ is taken back up into the SR through the action of sarcoplasmic (endoplasmic) reticulum Ca2+ ATPase 2a (SERCA2a) and extruded from the cell by the Na+-Ca2+ exchange (NCX) (5). SERCA2a function is regulated by phospholamban (PLN). Ca2+/calmodulin-dependent protein kinase (CaMKII) can modulate excitation–contraction coupling by phosphorylating important regulatory proteins such as RyR, PLN, and L-type Ca2+ channels. Color images available online at
The depleted SR calcium stores mean less is available for systolic release, leading to abnormal systolic contraction. Furthermore, impaired Ca2+ handling predisposes to ventricular arrhythmias 22 : impaired diastolic clearance and increased diastolic levels of Ca2+ in the cytoplasm are partially compensated for by the increased expression of NCX 23 as an alternative mechanism to remove Ca2+. This and the higher opening probability of RyRs in failing hearts 24 increase the Ca2+ leak in diastole, which promotes delayed after depolarizations and triggered activity that can ultimately result in ventricular arrhythmias. 25 Thus, abnormalities of Ca2+ handling in heart failure contribute to systolic dysfunction, diastolic dysfunction, and arrhythmogenesis, identifying Ca2+ handling as an attractive therapeutic target.
SERCA2a in heart failure
There are numerous molecules involved in Ca2+ handling but reduced SERCA2a activity appears critical in explaining the abnormalities seen in human heart failure. 26 In humans, three genes (ATP2A1–3) generate multiple isoforms of SERCA through alternative splicing (SERCA1a and b, SERCA2a–c, and SERCA3a–f). SERCA2a is the predominant isoform in cardiomyocytes. In both failing human myocardium and various animal models of heart failure, mRNA levels 27 –29 and activity of SERCA2a are reduced. 30 –32 Further support for the importance of SERCA2a activity in heart failure comes from the following observations: (1) a SERCA2a knock-out mouse develops systolic and diastolic dysfunction in vivo, 33 (2) inhibition of SERCA2a in isolated human myocytes creates a heart failure phenotype, 26 and (3) increasing the activity of SERCA2a with gene therapy improves important cardiac parameters in heart failure (see below).
Recognition of the pivotal role that Ca2+ and SERCA2a play in normal physiology and in the development of heart failure has led to SERCA2a gene therapy being the vanguard for clinical trials of gene therapy in heart failure. The convergence of the acquired phenotype from different etiologies of heart failure suggested that targeting this deficit in calcium handling could correct a final common pathway of disease.
Laboratory Evidence for SERCA2a Gene Therapy
Our research group and others have repeatedly demonstrated that increasing the activity of SERCA2a is beneficial in improving cardiac function and reducing arrhythmias in heart failure. The two main strategies for increasing SERCA2a activity are to either overexpress the SERCA2a gene or to influence the regulators of SERCA2a function. SERCA2a activity is regulated by phospholamban (PLN), which in turn is regulated by protein phosphatase 1 (PP1) and inhibitor-1 (I-1) (Fig. 2). Overexpression of PLN inhibits SERCA2a function resulting in reduced contractility, slow recovery of calcium transients, and reduced Ca2+ stores in the SR, 34 similar to findings observed in heart failure. It is when PLN is unphosphorylated that it inhibits SERCA2a function and it is recognized that the SERCA2a:PLN ratio is reduced in patients with advanced heart failure, with a relative increase in the unphosphorylated PLN fraction. When PLN is phosphorylated by protein kinase A (PKA) or Ca2+/calmodulin-dependent protein kinase II (CaMKII), it forms a pentamer relieving the inhibition of SERCA2a 35 (Fig. 2). PKA also has an indirect mechanism for increasing the phosphorylation of PLN by increasing the activity of I-1, which in turn inhibits PP1 (Fig. 2).
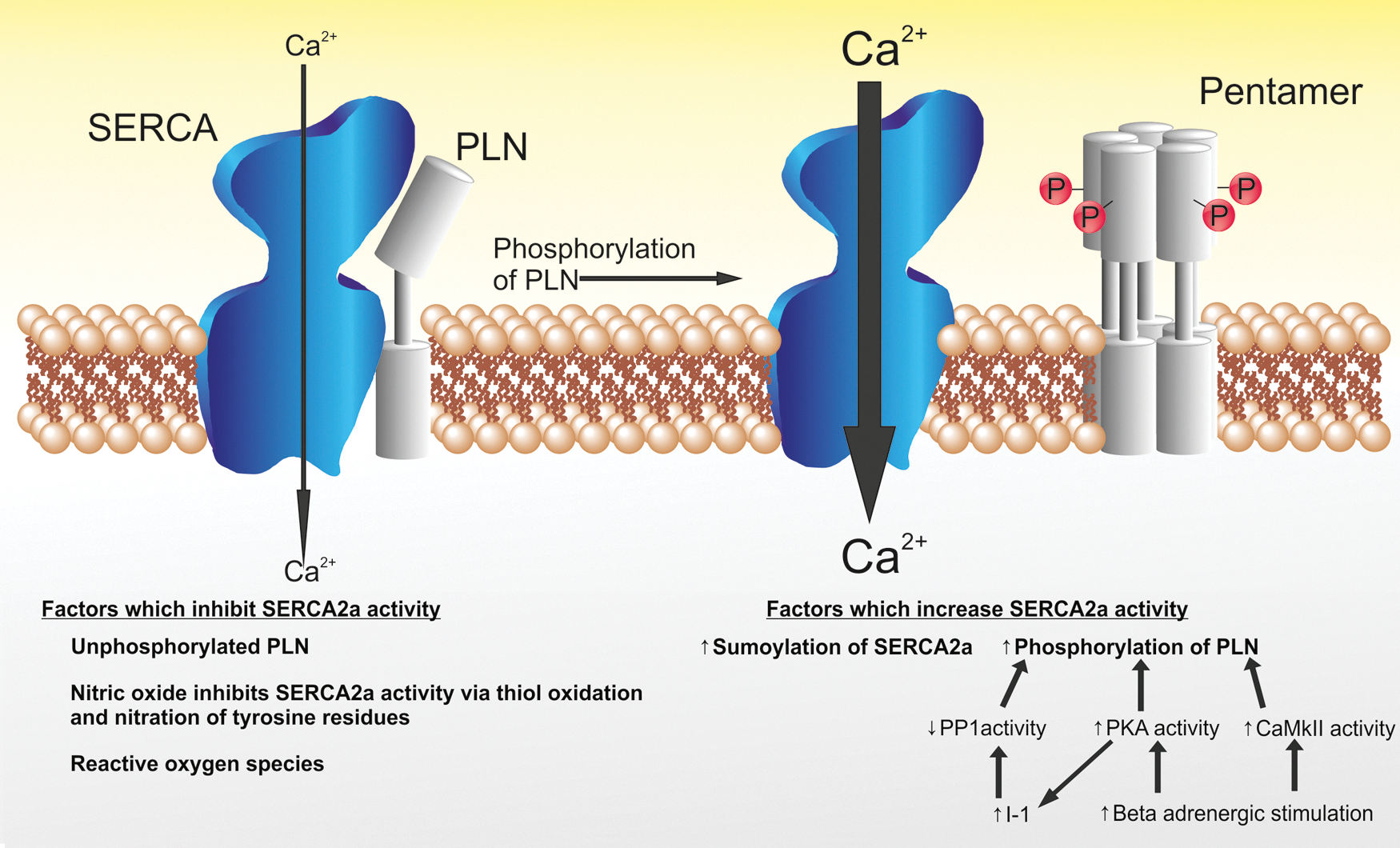
Illustrating the regulation of SERCA2a by PLN. When PLN is unphosphorylated, it acts as an inhibitor to SERCA2a activity (configuration displayed on the left). When PLN is phosphorylated, it forms a pentamer and the inhibition of SERCA2a is relieved. Factors that increase the activity of SERCA2a are displayed. Ca2+, calcium; CaMkII, Ca2+/calmodulin-dependent kinase II; I-1, inhibitor 1; PKA, protein kinase A; PP1, protein phosphatase 1. Color images available online at
The activity of SERCA2a is further influenced during posttranslational modification by small ubiquitin-like modifier type 1 (SUMO1). Small ubiquitin-like modifiers (SUMOs) are a group of proteins that regulate the function of other proteins through posttranslational modification, described as SUMOylation. SUMOylation of SERCA2a increases the stability and activity of SERCA2a, and laboratory studies have demonstrated that SUMOylation of SERCA2a is reduced in heart failure. 36
It can therefore be appreciated that strategies to increase the activity of SERCA2a can include increased expression of SERCA2a; any strategy that reduces the quantity of inhibitory unphosphorylated PLN present (commonly by increasing the phosphorylation of PLN); or increasing SUMO activity.
Some of the beneficial effects of directly increasing SERCA2a activity by transduction of the SERCA2a gene include increased contractility, normalization of LV size, normalization of Ca2+ transients, cardiac microstructure, and reversal of fetal gene expression (Table 1). One of the beneficial effects seen in laboratory studies of particular interest is the effect to reduce ventricular arrhythmias and arrhythmia susceptibility. It had been feared that reloading the SR could exacerbate RyR-mediated leak of Ca2+ and therefore increase arrhythmia. However, the opposite findings have been reported in laboratory studies with SERCA2a gene therapy displaying anti-arrhythmic properties under different experimental conditions: in normal hearts, 37,38 normal hearts subjected to ischemia-reperfusion 39,40 and in heart failure models. 41,42 Up to 50% of patients with heart failure may die from ventricular arrhythmias, and an attractive aspect of SERCA2a gene therapy is the potential for one therapy to be both an anti-arrhythmic agent and positive inotrope. 13 It remains to be seen whether these anti-arrhythmic properties seen in animal studies will translate to humans as there are significant differences in excitation–contraction coupling between species. For example, there are interspecies differences in t-tubule structure, 43 –45 the density of calcium-transporting proteins, 44,46,47 and the relative importance of NCX and SERCA2a for the removal of Ca2+ in diastole.
Different animal models of heart failure have been utilized, which include a volume overload model achieved through chordal rupture of the mitral valve apparatus, a pressure overload model using aortic banding, and a postinfarction model induced by coronary artery occlusion. Adapted with permission from the Japanese Circulation Society: Hayward et al. 72
Targeting PLN as a method to indirectly increase SERCA2a function has also seen success in laboratory studies. Adeno-associated virus (AAV)–mediated overexpression of a mutant form of PLN prevented deterioration in hamsters with cardiomyopathy and in postmyocardial infarction rats. 48,49 Larger animal studies have confirmed these findings in a sheep model of heart failure treated with an inhibitory PLN peptide resulting in improved SERCA2a activity along with improved systolic and diastolic function. 50 Similar findings have been observed in human cardiomyocytes, whereby suppressing levels of PLN improved contraction and relaxation velocities similar to the benefit seen with gene transfer of SERCA2a. 51 RNA interference has also been used in a rat model of heart failure to suppress PLN expression, resulting in increased SERCA2a activity and improved systolic and diastolic cardiac function. 52 These results should be treated cautiously when translating to humans since a naturally occurring mutation of PLN in the human population is not associated with improved cardiac function; conversely, it leads to a severe heart failure phenotype. 53
The inhibitory effect of PLN on SERCA2a activity is maximized when PLN is unphosphorylated. PP1 dephosphorylates PLN and is itself inhibited by I-1 (Fig. 2). As such PP1 indirectly inhibits SERCA2a activity, while I-1 increases SERCA2a activity. Heart failure is associated with elevated PP1 activity in humans. Murine models in which either I-1 is overexpressed in its active form or PP1 is inhibited have been shown to improve cardiac function and protect from heart failure. 54 –56
The final target that has been exploited to increase SERCA2a activity is SUMOylation. Gene therapy with AAV9.SUMO1 to increase SUMO1 expression in an animal model of heart failure led to improved cardiac function, comparable to SERCA2a gene delivery, 36 and was additive when dual AAV9.SERCA and AAV9.SUMO were applied to the failing heart in vivo.
In summary, while only direct SERCA2a gene transduction has been translated into clinical studies to date, this is just one of several potential gene therapy strategies targeting SERCA2a that could prove useful in the future.
Clinical Evidence for Targeting SERCA2a
Clinical gene therapy for heart failure is currently in its infancy. To date, only two clinical trials have published their full findings, one delivering SERCA2a cDNA, which is discussed below, and the other a phase 1 study investigating the safety of SDF-1 DNA injected into the peri-infarct zones of patients with heart failure. 10 The published clinical trial of SERCA2a gene therapy for heart failure is known as the CUPID (Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease) study. 8,57,58 This study involved delivery of AAV serotype 1 (AAV1)-mediated SERCA2a (AAV1.SERCA2a) gene therapy to patients with advanced heart failure. AAV1.SERCA2a consists of an AAV1 capsid containing the human SERCA2a cDNA flanked by inverted terminal repeats and under the regulation of the cytomegalovirus (CMV) promoter. The trial commenced with an open-label phase 1 study in 12 patients to investigate the safety of 4 doses of AAV1.SERCA2a. This was followed by a phase 2 randomized, double-blind, placebo-controlled study in which 39 patients with advanced heart failure were enrolled and randomized to receive one of three doses of intracoronary AAV1.SERCA2a versus placebo. Patients were enrolled with severe heart failure of ischemic and nonischemic etiologies, often on cardiac transplantation waiting lists. Treatment success was determined by examining concordant trends in the following end points: patient's symptoms (New York Heart Association functional class, Minnesota Living With Heart Failure Questionnaire), functional status (6 min walk test, VO2max), N-terminal pro–B-type natriuretic peptide (NT-proBNP) levels, and echocardiographic measures.
The use of concordant trends across multiple end points is not a traditional statistical approach in heart failure. There is typically one primary endpoint, which may be time to first event whether that is death, a cardiovascular event, a hospitalization, or a composite. The approach of using concordant trends in multiple end points has been suggested by the Food and Drug Administration in early trials of cellular therapy in cardiac disease. 59 The chance of concordant changes occurring by chance in this trial was 2.7% (as determined by permutation test). All parameters were required to be numerically superior in the treatment group and at least two of them had to reach a significance level of p<0.2.
Initial results at one year revealed that patients in the “high-dose” treatment group demonstrated improvement or stabilization in symptoms, functional class, NT-proBNP levels, and LV end-systolic volumes. 60 Time to death, LVAD placement, or transplantation was numerically in favor of AAV1.SERCA2a and there was a reduction in cardiovascular hospitalization duration. Importantly, patients in the treatment arm showed no increase in adverse events, disease-related events, or arrhythmias. Recently, three-year follow-up data have been reported, 9 which confirm sustained benefit in the high-dose arm and no safety concerns. At three years there was an 82% risk reduction in recurrent cardiovascular events (myocardial infarctions, heart failure hospitalizations, or worsening heart failure) in the presence of terminal events (death or the requirement for LVAD or cardiac transplantation) in the high-dose group compared with placebo using a joint frailty model analysis.
Heart failure is a chronic condition often associated with recurrent admissions because of decompensation. Traditional statistical analysis commonly compares a treatment group with placebo based on time-to-first event as discussed above. This approach may not take into account a large number of events that the patients experience and as such fail to accurately describe each group. In addition, once a person has died he or she clearly cannot experience any further events. A joint frailty model is able to assess treatment effect with respect to recurrent events in the presence of terminal events. This may be considered a more appropriate methodology for a chronic condition such as heart failure, and indeed it has been shown that some previously published heart failure trials that were negative may become positive if this methodology were applied. 61
While the CUPID trial met its primary outcome, it must be borne in mind that this was a phase 2 study involving a small number of patients. In particular, when one considers the long-term follow-up data, this was a comparison of 9 patients in the high-dose group and 14 in the placebo arm. There is no data available from this study to suggest whether any benefits seen were because of either the positive inotropic effect or anti-arrhythmic effect seen in animal studies. Entry criteria required that patients have an internal cardioverter defibrillator as a safety measure; however, the study was neither powered nor designed to examine arrhythmia incidence as an efficacy end point. An effect on inotropy is difficult to establish in clinical trials even with invasive strategies. Echocardiography did demonstrate a reduction in left ventricular end systolic volume in the high-dose group, but this is more of a measure of cardiac remodeling than it is of inotropy. Should future trials demonstrate significant benefit in important clinical outcomes, then the expectation would be that there will be further trials in which it can be examined whether any benefit seen is because of a reduction in arrhythmias, positive inotropic effects, a combination of these, or perhaps an as-yet-unidentified mechanism.
Practical Considerations for SERCA2a Gene Therapy Trials
Choice of vector for clinical trials of heart failure
All currently active SERCA2a gene therapy trials use AAVs as the vector which are derived from the parvovirus 62 and not known to cause any human disease. Thirteen different AAV serotypes are currently recognized (AAV1–AAV13) each with different tissue tropisms. 63 AAV serotypes 1, 6, 8, and 9 have been shown to transduce skeletal and cardiac muscle efficiently, and it is AAV1 that is the used in the clinical trials in heart failure. Wild-type AAVs are able to integrate into the host genome, and while this often occurs in a site-specific location on chromosome 19, it can occur at sites of DNA damage, making insertional mutagenesis, though rare, still a possibility. However, the recombinant AAVs used in clinical trials are not known to integrate into the host genome but instead persist in the cell nucleus in a concatemeric episomal form, making the risk of insertional mutagenesis theoretically very remote. The lack of integration in the host genome is a problem for cells that divide, as new cells will not contain the transgene. Persistence of the vector transgene is more likely in nondividing cells such as cardiomyocytes. 64 Evidence for longevity of AAV effects has been observed for more than 8 years in a dog model. 65
The main limitation of AAVs is the existence of neutralizing antibodies in human populations, which will be discussed later. Alternative viral vectors that have been studied include adenoviruses, retroviruses, and lentiviruses. Adenoviruses have been used as a vector for gene therapy extensively in cardiovascular disease, but major problems with these vectors are preexisting immunity, as with AAVs, and the immune reaction they generate can limit tolerability and safety for use in clinical trials. Retroviruses integrate into the host genome and as such may theoretically persist for the lifetime of the cell, which is a particularly attractive property when treating a chronic condition such as heart failure. Retroviral vectors based on the Moloney murine leukemia virus were used in a clinical trial to treat X-linked severe combined immunodeficiency. 66 These vectors are problematic as not only did they lead to insertional mutagenesis, 67 but these vectors cannot transduce dividing cells such as cardiomyocytes. However, lentiviral vectors, based on the human immunodeficiency virus type 1, 68 can transduce nondividing cells. The experimental use of such lentiviruses is expanding, and recently two clinical trials have reported safety and benefits of lentiviral-delivered gene therapy targeting hematopoietic stem cells. 69,70
Delivery method in clinical trials of heart failure
SERCA2a gene therapy trials have used intracoronary infusion as the delivery method of choice, but there are other methods that can be considered. The ideal delivery system would be a peripheral intravenous injection with a vector that selectively transduces cardiomyocytes. In humans, this is problematic as the large blood volume causes a dilutional effect and viruses infect organs other than the heart, particularly the liver. Some of the AAV serotypes have high cardiac tropism and conceivably this approach may be feasible in the future. Replacement of the strong CMV promoter currently used in clinical trials of heart failure with a cardiomyocyte-specific one could theoretically improve targeting, but at the expense of increasing viral load to compensate for reduced efficiency. This could raise the likelihood of a T-cell-mediated reaction as the capsid is re-presented on the surface of the infected cell, as observed in a clinical trial of hemophilia. 71
Multiple techniques for delivering gene therapy to the heart have been assessed and include both surgical and percutaneous approaches. These include an antegrade infusion, and antegrade infusion with coronary artery balloon occlusion to reduce immediate washout, a closed loop recirculation method, retrograde infusion through the coronary sinus, pericardial injection, and direct myocardial injection. Each method has advantages and disadvantages, 72 but from a clinical perspective, the most practical route—the one adopted in the clinical trials of SERCA2a gene therapy—is a 10 min antegrade coronary artery infusion without coronary artery occlusion. The various adjuncts to antegrade intracoronary infusion such as coronary artery occlusion or the closed loop recirculation method have not been adopted in humans because of the added complexity of the procedures and the potential damage to the myocardium. One adjunct to intracoronary infusion that is used in clinical trials is intravenous glyceryltrinitrate (GTN) because this has been shown to increase SERCA2a mRNA by more than twofold compared with the control arm receiving vector infusion without GTN. 73 The mechanism of action for this improvement in transduction is unknown but may relate to coronary vasodilatation, changes in vascular permeability, or increasing myocardial perfusion through a reduction in left ventricular end diastolic pressure. Of note, intracoronary GTN did not increase SERCA2a expression, 73 suggesting that the effect may not simply be because of coronary vasodilatation.
Efficacy measures in clinical trials of gene therapy
The goal of any therapy in heart failure is to either improve quality of life or survival. As such, key end points in trials of gene therapy for heart failure will always be mortality, heart failure hospitalizations, quality of life scores, and measures of functional capacity such as the six minute walk distance. It is also important from a scientific and mechanistic point of view to assess the efficiency of gene therapy at each step of the molecular pathway, in particular to measure the quantity of transgene in the target cell and the amount/activity of the therapeutic protein thereby expressed. This is particularly challenging in clinical trials of SERCA2a gene therapy since there is no blood test or imaging investigation that can be used as a surrogate. Cardiac tissue can be analyzed, but this requires either an invasive cardiac biopsy, which has associated risks in patients with advanced heart failure, or for patients to undergo cardiac transplantation, implantation of an LVAD, or to die. Even in these circumstances, the nature of the samples available, such as those reported in the long-term follow-up of the CUPID trial, 9 may only be able to detect the presence of vector DNA and not quantify the efficiency of transgene expression.
This limitation has several important consequences; if clinical trials demonstrate no benefit of SERCA2a gene therapy, it will be impossible to determine at which stage of the process the failure occurred; for example, if no transgene can be detected in the target organ, then the problem could be the delivery method or vector. If benefit is seen, we may assume that we understand the mechanism of action from animal studies, but the benefit could be because of an unknown off-target effect. There are several examples of therapies for heart failure that are only beneficial to subgroups of patients. Without measuring the efficiency of transgene expression, it will be impossible to determine whether “nonresponders” to gene therapy do not respond because there is no expression of the transgene or if expression of the transgene is not beneficial to them. This makes fine-tuning of the therapy in the future challenging as we will not know whether to alter patient selection, delivery method, vector, or dose.
These measures of gene therapy efficacy have been investigated in a porcine model of heart failure. An increase in SERCA2a mRNA and protein levels was observed following AAV1.SERCA2a gene transfer, 73 and gene expression using the reporter gene β-galactosidase on frozen sections was demonstrated. 40 These data are in small numbers of animals and it is unknown whether these findings will translate to human trials.
Ongoing Clinical Trials of SERCA2a Gene Therapy for Heart Failure
The CUPID trial has paved the way for three other clinical trials of SERCA2a gene therapy in patients with heart failure. These trials are all underway and are investigating complementary aspects of the potential benefit of SERCA2a gene therapy; clinical outcomes, gene and protein expression, and the effects on LV remodeling.
Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease 2 (CUPID2—ClinicalTrials.gov id: NCT01643330)
This is the follow-up to the CUPID trial and has now completed recruitment. This is a phase 2, double-blind, placebo-controlled, randomized study evaluating the safety and efficacy of intracoronary administration of AAV1.SERCA2a in subjects with heart failure. Patients were recruited who had chronic heart failure and an ejection fraction of ≤35% with NYHA 3 or 4 class symptoms despite optimal medical therapy. Patients were also required to have either been admitted with decompensated heart failure in the previous 6 months or have a raised NT-proBNP (>1200 ng/liter in sinus rhythm or >1600 ng/liter in atrial fibrillation). Patients who met these criteria were prescreened for the presence of neutralizing antibodies to AAV1. If the antibody was present at a titer of >1:2, then they were excluded from the trial. Two hundred and fifty patients were recruited across more than 50 sites around the world, including our own center. Patients were randomized 1:1 to receive a single 10 min intracoronary infusion of placebo or the high dose of AAV1.SERCA2a (1×1013 DNAse resistant particles). GTN was coadministered intravenously during investigational product infusion.
The primary end point of the trial is time-to-recurrent heart failure-related hospitalizations in the presence of terminal events (all-cause death, heart transplantation, LVAD implantation) analyzed using the joint frailty model. The secondary efficacy endpoint is the time-to-terminal event (all-cause death, heart transplantation, LVAD implantation). A press release from the trial's sponsor (Celladon Corporation) in April 2015 reported the disappointing news that the study failed to meet both primary and secondary endpoints. There had been hope that this iteration of SERCA2a gene therapy (the investigational product is also known as MYDICAR) would lead to significant improvements in outcomes for those with the most advanced heart failure. This optimism is reflected in the U.S. Food and Drug Administration granting MYDICAR “breakthrough” and “fast-track” status. A detailed analysis of the data is clearly required to unravel the reasons why this trial was negative and it is likely that the complete data will be published later in 2015.
Interestingly, Celladon Corporation has published the details of an additional trial using MYDICAR in advanced heart failure with a study start date of April 2015 (
The publication of these topline results means that the future of MYDICAR, as well as indeed SERCA2a gene therapy for heart failure, is uncertain, but the full details of the trial need to be explored before one should speculate any further.
SERCA2a Gene Therapy in LVAD Patients (SERCA-LVAD—ClinicalTrials.gov id: NCT00534703)
This trial has commenced recruitment at our institution and is recruiting patients with advanced heart failure who have received an LVAD for chronic heart failure. Patients receive an intracoronary infusion of either AAV1.SERCA2a or placebo. Myocardial tissue samples are taken at the time of LVAD implantation, before gene therapy treatment, and follow-up samples are taken either at the time of transplantation or via percutaneous biopsy. This trial will assess the potential benefit of SERCA2a gene therapy in end-stage heart failure. There are three unique aspects of this trial. First is the opportunity to study the hypothesis that mechanical unloading may potentiate the functional response to transduction. Second, by taking tissue biopsies the investigators will be able to correlate changes in clinical outcome with the expression of SERCA2a and correction of the underlying calcium handling abnormalities of heart failure. Third, both neutralizing-antibody-positive and neutralizing-antibody-negative patients will be enrolled, to evaluate the effect of antibody status on gene expression efficacy and safety, and whether the presence of neutralizing antibodies is a justifiable exclusion criterion.
Along with the publication of the full data from CUPID2, the biopsy data from SERCA-LVAD may help to better understand why CUPID2 was negative.
AAV1.SERCA2a Gene Therapy Trial in Heart Failure (AGENT-HF—ClinicalTrials.gov id: NCT01966887)
This trial is a phase 2, single-center, double-blind, randomized, placebo-controlled study that has started recruitment at the Pitié-Salpêtrière Hospital Institute of Cardiology in Paris, France. The primary objective of this study is to investigate the impact of AAV1.SERCA2a on cardiac remodeling in patients with severe heart failure. Patients with ischemic and nonischemic cardiomyopathy are currently being recruited. The primary outcome will be the effect of gene therapy on left ventricular end-systolic volume (measured with a 256-slice CT-scan) 6 months after treatment. Secondary endpoints will include changes in the ejection fraction, diastolic volumes, VO2max, echocardiographic remodeling, BNP, and biological safety profile.
Patient selection for gene therapy trials in heart failure
Patients selected for current clinical trials are those with advanced systolic heart failure. They generally have severely reduced ejection fraction, heart failure symptoms, and other markers of severe disease and poor prognosis such as heart failure hospitalizations or high levels of natriuretic peptide. This is despite optimally tolerated guideline-driven heart failure therapy. If the clinical trials report benefit in this group of patients, then it is likely that the first approved therapy will be an add-on to current therapy in those with advanced heart failure, akin to the use of cardiac resynchronization therapy pacemakers in those not improving despite optimal evidence-based therapy. One can envisage at this stage that clinical trials may be designed in those with less severe heart failure to assess whether adding gene therapy to optimal current medical therapy is beneficial or an alternative direction clinical trials may take is to identify patient factors that predict those most likely to benefit from gene therapy; for example, it is conceivable that those who will derive most benefit will be those with a greater proportion of surviving cardiomyocytes and so those with a DCM may theoretically benefit more than those with large full-thickness myocardial infarctions. If the ongoing trials of SERCA2a gene therapy prove positive, then there is a whole range of directions opened up that future research could take.
Future Challenges
Should future trial evidence suggest that SERCA2a gene therapy is safe and efficacious, then a major obstacle to the widespread application of this iteration of gene therapy is the prevalence of neutralizing antibodies to AAV vectors. 74 There are 12 serotypes of the wild-type virus and it is a recombinant version of the AAV serotype 1 that is being used in clinical trials. Exposure to wild-type AAVs results in neutralizing antibodies that can block transduction of future AAVs and therapeutic gene expression. 75 People who are AAV neutralizing antibody positive are not generally eligible for enrolment in clinical trials and are likely to be excluded from treatment should the therapy become licensed. The prevalence of neutralizing antibodies in the population is dependent on a number of factors, which include the AAV serotype investigated, 76 –78 the country studied, 76 the age, 79,80 and possibly gender 79 of the subjects investigated. Globally, there is a wide variation in the prevalence of neutralizing antibodies to AAV1, ranging from 13% to 79%. 76,77,79,81,82
The exact effect of neutralizing antibodies on the success of AAV-delivered gene therapy is largely unknown. Our group is currently undertaking a study that will hopefully address the effect of neutralizing antibodies on transduction in humans (SERCA-LVAD-
Another strategy involves removing neutralizing antibodies from the patient using plasmapheresis 87 before delivery of gene therapy. Not only is the issue of neutralizing antibodies relevant to those people who have been exposed to the wild-type virus, but also it is expected that people who receive the recombinant AAV-delivered gene therapy will develop neutralizing antibodies, thus making repeated administrations of the same treatment problematic. The longevity of this treatment is as yet unknown, but it would be desirable with a chronic condition such as heart failure to have the capacity for repeated administrations.
Potential Future Directions for SERCA Gene Therapy
SERCA is present in all types of muscle in its various isoforms, which raises the possibility that SERCA gene therapy could be applied to other diseases. Impaired Ca2+ handling is present in various myocytes contributing to diseases such as asthma, pulmonary arterial hypertension (PAH), and coronary artery disease.
Airway smooth muscle cells from patients with asthma display increased contractile activity 88 –90 that contributes to airway narrowing. Analogous to the importance of Ca2+ handling in the cardiomyocytes, it was suggested over 30 years ago that alterations in Ca2+ handling in the airway smooth muscle may be critical to explain the pathophysiology of asthma. 91 –93 Impaired Ca2+ handling leads not only to impaired relaxation of the airway smooth muscle cells, but also to enhanced proliferation and secretion of proinflammatory chemokines that interact to produce the asthma phenotype. The main isoform of SERCA2 in airway smooth muscle cells is SERCA2b and protein expression is reduced in proportion to disease severity. 94 In addition, suppression of SERCA2 by small interfering RNA in normal airway smooth muscle cells results in an asthma phenotype, 94 supporting the causal link between SERCA function and asthma. This raises the hypothesis that SERCA gene therapy may be a future treatment for asthma, and this would potentially be amenable to an inhaled delivery route as has been used in cystic fibrosis. 87
Analogous to SERCA2 downregulation in asthma contributing to abnormal cell proliferation and impaired smooth muscle relaxation, there is also evidence of changes in SERCA expression in the pulmonary circulation in PAH. SERCA2 expression is reduced in remodeled pulmonary arteries from patients with PAH and in a rodent model of PAH. 95 Stretch of pulmonary smooth muscles leads to calcium influx, which is exaggerated in animal models of PAH. 96 SERCA2a gene transfer in vitro reduced proliferation and migration of human pulmonary artery smooth muscle cells. In vivo SERCA2a gene transfer using inhaled AAV1 containing the human SERCA2a gene decreased pulmonary artery pressure, vascular remodeling, right ventricular hypertrophy, and fibrosis in a rodent model of PAH. 95 This study has moved on to large animal models and the hope is that this may then translate to clinical trials as PAH can be a highly morbid disease with a high mortality rate and few effective treatments.
Vascular smooth muscle cells express two SERCA genes (ATP2A2 and ATP2A3) simultaneously. The major SERCA2 isoform that determines Ca2+ handling is either SERCA2a or SERCA2b depending on the cell phenotype (proliferating/migratory vs. contractile/quiescent, for example). Mature vessels are mostly of the contractile/quiescent phenotype and express SERCA2a. In response to injury, however, the phenotype can switch to a proliferating/migratory one such as in atherosclerosis. This switch leads to the predominant SERCA isoform being the slowly pumping SERCA2b isoform that allows Ca2+ levels to remain high for longer and this activates proliferation-related transcription factor nuclear factor of activated T-cells (NFAT). 97 In vitro SERCA2a gene transfer inhibits proliferation and migration of these cells secondary to inhibition of NFAT. 98 –100 It is plausible that coronary artery disease may be another future target for SERCA gene therapy. Furthermore, AV fistulas created for renal dialysis commonly fail and vascular smooth muscle cell proliferation can contribute to this. There are current plans for a clinical trial investigating the efficacy of AAV1.SERCA2a in the prevention of failure of AV fistulas.
Conclusions
Gene therapy as a treatment for patients with heart failure has been in development for over 20 years, but it is only more recently that it has translated into clinical trials. On the foundation of the first published trial of SERCA2a gene therapy, there are now three ongoing trials that will recruit over 300 heart failure patients between them. Laboratory studies have shown that SERCA2a gene therapy can improve contractility and reduce ventricular arrhythmias in animal models of heart failure in an energetically favorable manner. This could potentially mean that SERCA2a gene therapy is the first positive inotrope that reduces the risk of arrhythmias. However, the largest of the three ongoing clinical trials recently published the topline results that reported failure to achieve both primary and secondary endpoints. We eagerly anticipate the full report of this study and of the other clinical trials, but it remains to be seen whether the failing human heart can be successfully transduced with therapeutic DNA in sufficient levels to impact upon the underlying failing myocardial substrate and initiate clinically meaningful reverse remodeling.
Footnotes
Acknowledgments
C.H. is supported by the NIHR Cardiovascular Biomedical Research Unit. A.M.-S., A.R.L., and S.E.H. are supported by the British Heart Foundation.
Author Disclosure Statement
All authors declare that they have no competing interests.