
Abstract
Today, lentiviral vectors are favorable vectors for RNA interference delivery in anti-HIV therapeutic approaches. Nevertheless, problems such as the specific recognition of target cells and uncontrolled expression of the transgene can restrict their use in vivo. Herein we present a new HIV-inducible promoter to express anti-HIV short hairpin RNA (shRNA) by RNA Pol II in mammalian cells. We likewise showed a novel third-generation lentiviral vector system with more safety and a specific tropism to the target cells. The new promoter, CkRhsp, was constructed from the chicken β-actin core promoter with the R region of HIV-1 long terminal repeat fused upstream of minimal hsp70 promoter. This system was induced by HIV-1 Tat, and activates transcription of two shRNAs against two conserved regions of HIV-1 transcripts produced in two steps of the virus life cycle. We also mimicked HIV-1 cell tropism by using the HIV-1 envelope in structure of third-generation lentiviral vector. The new fusion promoter efficiently expressed shRNA in a Tat-inducible manner. HIV-1 replication was inhibited in transient transfection and stable transduction assays. The new viral vector infected only CD4+cells. CkRhsp promoter may be safer than other inducible promoters for shRNA-mediated gene therapies against HIV. The use of the wild envelope in the vector packaging system may provide the specific targeting T lymphocytes and hematopoietic stem cells for anti-HIV-1 therapeutic approaches in vivo.
Introduction
C
Posttranscriptional silencing strategies by RNAi are being used by many studies for the development of new strategies in antiviral therapies. 6,7 RNAi mainly involves the introduction of siRNA, a short, double-stranded RNA molecule with 21–23 nucleotides and 3′ overhang of two nucleotides, modified by the endoribonuclease Dicer. Dicer promotes the activation of the RNA-induced silencing complex that is essential for RNAi. 8,9 The siRNA molecules can be transfected as synthetic double-stranded RNA or can be achieved by expression plasmids as separate sense and antisense strands. Another approach is the use of a short hairpin RNA (shRNA) 21–29 nucleotides long that can be processed to an siRNA molecule. 10 –12
Replication-defective lentiviral vectors derived from HIV-1 have proven to be very potent to transduce both nonreplicating and replicating cells and long-term expression of the therapeutic transgene. These properties suggest the advantages of using these vectors for gene therapy. Today, lentiviral vectors are favorable vectors for RNAi delivery in anti-HIV therapeutic approaches. 13,14 However, problems such as the specific recognition target cells and uncontrolled expression of the transgene can restrict their use in vivo.
To design selective inducible vectors, the HIV long terminal repeat (LTR) has been used as the promoter by several studies, to control transcription of antiviral RNAs and proteins in HIV-infected cells. 15 –19 The LTR promoter is strongly induced by Tat, a protein produced in early steps of HIV-1 life cycle. Tat binds to the transactivation response element (TAR), a stem–loop sequence at the 5′ LTR of viral RNA. 20 Two known chimeric promoters on this approach are the LTR-hsp 18,19 and the CK-TAR. 21,22 The LTR-hsp was constructed from the full HIV-1 LTR and Drosophila minimal heat shock promoter 70 (ΔHSP), and was used to Tat-inducible expression of anti-HIV shRNA and protein. In this promoter, transcription from the ΔHSP element was controlled by the Tat protein attached to TAR in LTR and creates a precise transcriptional start site downstream of the LTR. This property could eliminate non-base-paired sequence in 5′ of shRNA. 18 The CK-TAR promoter was created when Han and colleagues 21 found that LTR sequences other than TAR domain are not required for Tat transactivation. This promoter is composed of the core promoter of the chicken β-actin gene, for the binding of transcription factors, fused upstream of the viral TAR sequence. In comparison with LTR-hsp, the CK-TAR promoter could be safer with regard to vector mobilization and insertional mutagenesis that can occur with the virus LTR. However, the CK-TAR promoter cannot make a favorable transcriptional start site in shRNA expression approaches. It also had a relative basal activity in the absence of Tat, which reduces its competence in selective expression. 22 Hence, both promoters may not be safe or efficient enough in therapeutic approaches.
Given the above explanation, we decided to design a novel lentiviral delivery system to express anti-HIV shRNA against two conserved regions in the HIV-1 transcripts. To increase safety and neutralize vector mobilization, and also to obtain a favorable transcriptional start site in shRNA expression, we devised a Tat-inducible promoter in which chicken β-actin core promoter with the R region of HIV-1 LTR was fused upstream of minimal hsp70 promoter and it was named CkRhsp. We also mimicked HIV-1 cell tropism by using HIV-1 envelope in the structure of lentiviral vectors and investigated its anti-HIV activity in vitro.
Materials and Methods
ShRNA design
In this study, two shRNAs were designed to target two highly conserved sites in the HIV-1 RNA sequence as previously reported. 23 These shRNAs included 28-nucleotide stem and a 9-base loop 11 (Fig. 1A). The first shRNA (T/R) targets Tat/Rev transcripts (early products in HIV-1 life cycle) in position 5547–5568 of the viral genome. The second shRNA (I/V) targets integrase/Vif transcripts (late products) in position 4622–4643 of the viral genome (Fig. 1B).

shRNA information.
Vector constructions
The CkRhsp-shRNA cassettes were synthesized by using sequences of the 278 bp chicken β-actin core promoter (pbetaAct plasmid, ATCC, 37507), the 99 bp R region of HIV-1 LTR (pLenti6.2/GW plasmid; Invitrogen, Carlsbad, CA), the 294 bp minimal hsp70 promoter (pIND plasmid; Invitrogen), shRNA sequences, and minimal polyadenylation (mpolyA), respectively (Fig. 2A). The mpolyA sequence was obtained from Xia et al.'s study. 12 The LTR-I/V cassette was synthesized by using the sequence of HIV-1 LTR (pNL4-3, NIH AIDS Research and Reference Reagent Program) fused to I/V shRNA, upstream of mpolyA.

Construct of the Tat-inducible promoter and the lentiviral packaging system.
The expression vector backbone was a self-inactivating lentiviral vector, pLenti6.2/GW (Invitrogen). Construction and characteristics of the pLenti6.2 vector have been previously described. 24 The pLenti/CkRhsp-shRNA vectors were constructed by insertion of promoter-shRNA cassettes into cut sites of ClaI and MluI restriction enzymes (Fermentas, Sankt Leon-Rot, Germany). The pLenti/LTR-shRNA vector is also constructed by insertion of the LTR-shRNA cassette into cut sites of ClaI and MluI restriction enzymes. All of the described expression cassettes are cloned in reverse orientation (Fig. 2B).
The pLP-Env plasmid is constructed by PCR amplification of the full-length envelope gene of HIV type 1 from HIV-1 provirus plasmid (pNL4-3), using the forward primer AAA
Transfection assay
HEK293 cells (ATCC, CRL1573) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). Twenty-four hours before transfection, the cells were trypsinized and counted, and 5×104 cells were plated in the 48-well format at 80% confluency with 200 μl DMEM and 10% FBS without antibiotics. The next day, cells were cotransfected with 100 ng of pNL4-3 plasmid and 50 ng of shRNA vectors in 0.7 μl lipofectamine according to the manufacturer's protocol (Invitrogen). Six hours after transfection, the medium was replaced with 200 μl medium with antibiotics. Culture supernatants were collected at 24, 48, and 72 hr posttransfection and analyzed for p24 antigen by the enzyme-linked immunosorbent assay (ELISA) kit (Clontech, Seoul, South Korea) and using an ELISA plate reader (BioTek, Winooski, VT) according to the manufacturer's instructions.
ShRNA expression assay
Total RNA was extracted from 107 cells acquired from 24 hr transfection plates by the mirPremier microRNA Isolation Kit (Sigma, Munich, Germany). The first-strand cDNA was synthesized using stem–loop primers and reverse transcription enzyme (Invitrogen) according to manufacturer's protocol. The cDNAs were then quantified in triplicate, using forward and reverse primers and Maxima SYBR Green Master Mix (Fermentas) in Bio-Rad iQ5 Real-Time PCR system (Bio-Rad, Munich, Germany). The shRNA expression was normalized against U6-snRNA endogenous control as previously described. 25 The relative expression was calculated using the 2−ΔCt (ΔCt=Ct of control gene − Ct of target gene).
Lentiviral vector production
A third-generation lentiviral vector system was used to produce transducing vectors as described by Yam et al. 26 HEK293T cells (ATCC, CRL-3216) were cultured to achieve the 90% confluency in a 100 mm culture dish. The cells were cotransfected with 15 μg of transfer vector with the appropriate insert, 15 μg of pLP1(Gag/Pol), 10 μg of pLP2(Rev) (Invitrogen), and 5 μg of pLP-Env (Fig. 2C), by the calcium phosphate precipitation protocol. 27 Eight hours posttransfection, the culture medium was replaced by the fresh medium. After 24 and 48 hr of transfection, the culture supernatants were harvested and pooled. Vector supernatant was collected from the three harvests and cleared by centrifugation at 250 g for 6 min at 4°C, and then filtered through a 0.45 μm filter and concentrated by Lenti-X Concentrator kit (Clontech). The viral titer was determined by p24 level according to the ELISA kit's user guide (Clontech). Lentiviral vector titers ranged from 4×108 to 6×108 infectious units/ml.
Lentiviral vector transduction and HIV-1 challenge
To transduce the human T-cell line CEM (ATCC, CCL-119) and HEK293 cell line (as CD4-negative), 2×105 cells were plated in a 15 ml centrifuge tube containing 1 ml RPMI1640 medium. Lentiviral vectors were added to the tube in a multiplicity of infection (MOI) of 10 without polybrene. Twenty-four hours posttransduction, the cells were centrifuged and cultured in a fresh medium containing 8 μg/ml of antibiotic blasticidin (Invitrogen) every 3 days. After 12 days, 106 live cells were infected with HIV-1 strain NL4-3 at an MOI of 0.001. The cells were then incubated overnight and washed three times with Hanks' balanced salt solution and cultured in the medium with 20% FBS. On days 3–18 after infection, the cell cultures were changed every 3 days, and the supernatant was collected for the HIV-1 p24 assay. Cell viability was also determined by the trypan blue staining. ShRNA expression was analyzed on all time points in cells transduced with the CkRhsp-T/R-I/V vector.
Statistical analysis
The differences between variables were identified by ANOVA test. Data analyses were performed by SPSS software version 17 (SPSS Inc., Chicago, IL). A p-value less than 0.05 was considered statistically significant.
Results
The CkRhsp promoter expressed shRNA in a Tat-inducible manner
In this study, we constructed two types of plasmid vectors in which anti-HIV shRNAs are expressed from two Tat-inducible promoters by RNA polymerase II, the CkRhsp (novel promoter), and HIV-1 LTR (for comparison) (Fig. 2A). To test whether Tat made by HIV-1 could sufficiently induce inhibition from the CkRhsp promoter, the HEK293 cells are cotransfected with shRNA expression plasmids and a HIV-1 infectious clone (pNL4-3). HIV-1 gene expression was then measured by p24 assay on cell supernatants. Results in Fig. 3A show that the virus gene expression significantly decreased when cells were cotransfected with both shRNA vectors and pNL4-3 (plasmid containing Tat gene) (p<0.001). In addition, the inhibition of virus gene expression by pLenti/CkRhsp plasmids was more efficient than that by the pLenti/LTR plasmid (p=0.015) (Fig. 3A).
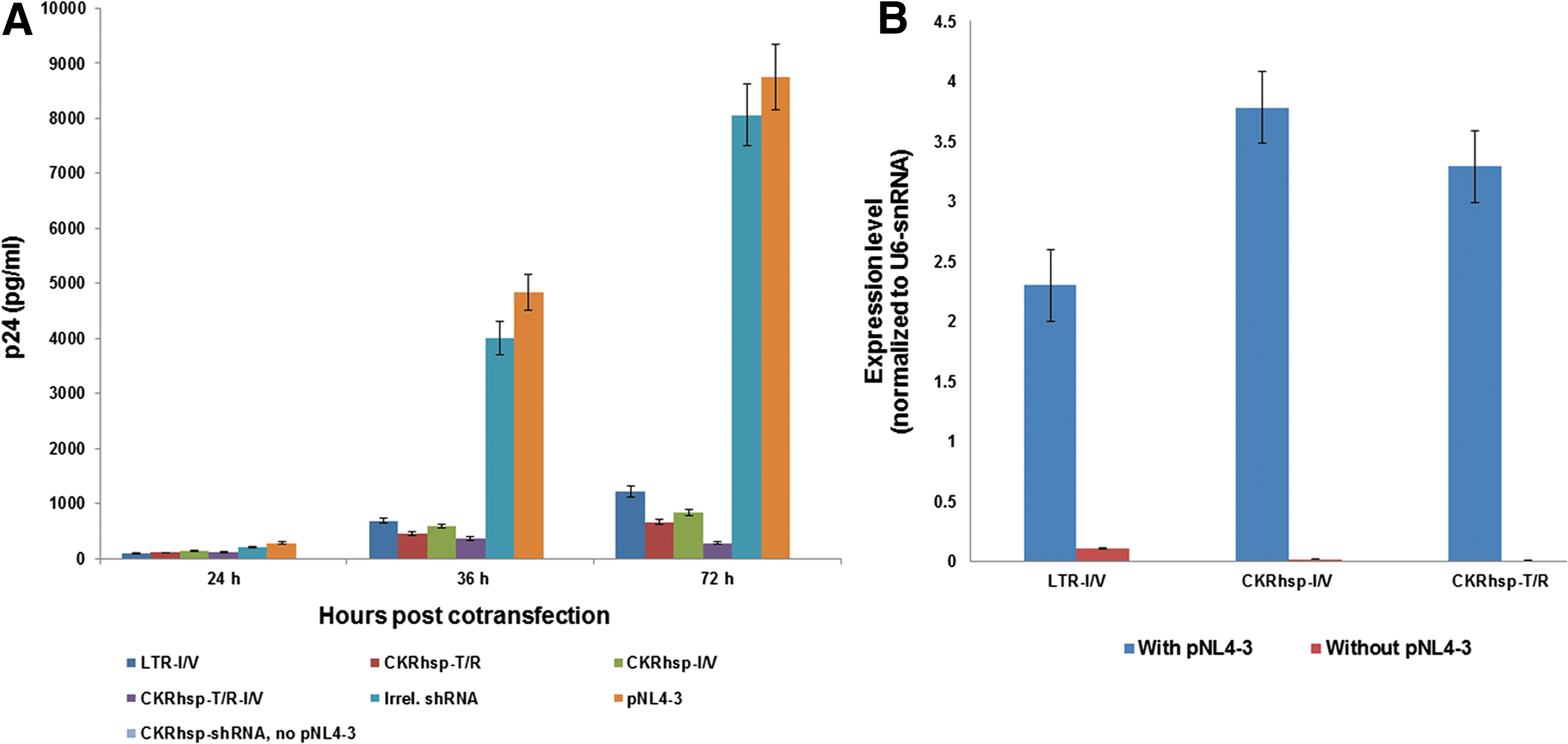
siRNA expression driven by the RNA Pol II promoter inhibits HIV-1 gene expression.
To investigate efficacy of the new promoter, we measured the level of mature siRNAs resulting from transfection. As shown in Fig. 3B, there is a considerable level of shRNA expression when cells were cotransfected with shRNA plasmids and pNL4-3, but not in cells transfected only with shRNA plasmids. Importantly, low expression of shRNAs in the absence of Tat protein (no pNL4-3) shows a very low basal activity of the CkRhsp promoter compared with the LTR promoter (p<0.001). Moreover, shRNA expression by pLenti/CkRhsp plasmids was little more than that by the pLenti/LTR plasmid (Fig. 3B).
HIV-1 inhibition in transient transfection assay
To test pLenti vectors in a transient expression setting and also to investigate its potency against HIV-1, the HEK293 cells were cotransfected with the shRNA plasmids and pNL4-3. As results in Fig. 3A show, in day 3 posttransfection, there was an approximate 80–97% inhibition of HIV-1 replication by pLenti/CkRhsp-shRNA plasmids relative to pNL4-3 and irrelevant shRNA (p<0.001). The level of inhibition mediated by pLenti/CkRhsp-T/R-I/V was significantly higher than that seen with pLenti/CkRhsp-T/R and pLenti/CkRhsp-I/V (p=0.23).
HIV-1 inhibition in stable transduction assay
The CEM cell line was stably transduced with vectors containing the CkRhsp-shRNA cassettes and wild envelope (Fig. 2B). The transduced cells were then challenged with HIV-1 strain NL4-3. At six time points, culture supernatants were obtained and were analyzed for p24 levels and expressed as a p24 chart. The percentage of live cells was also determined by trypan blue staining and demonstrated as a cell viability chart. As observed in Fig. 4A, our lentiviral vector can potentially infect CEM cells without chemical regents such as polybrene.
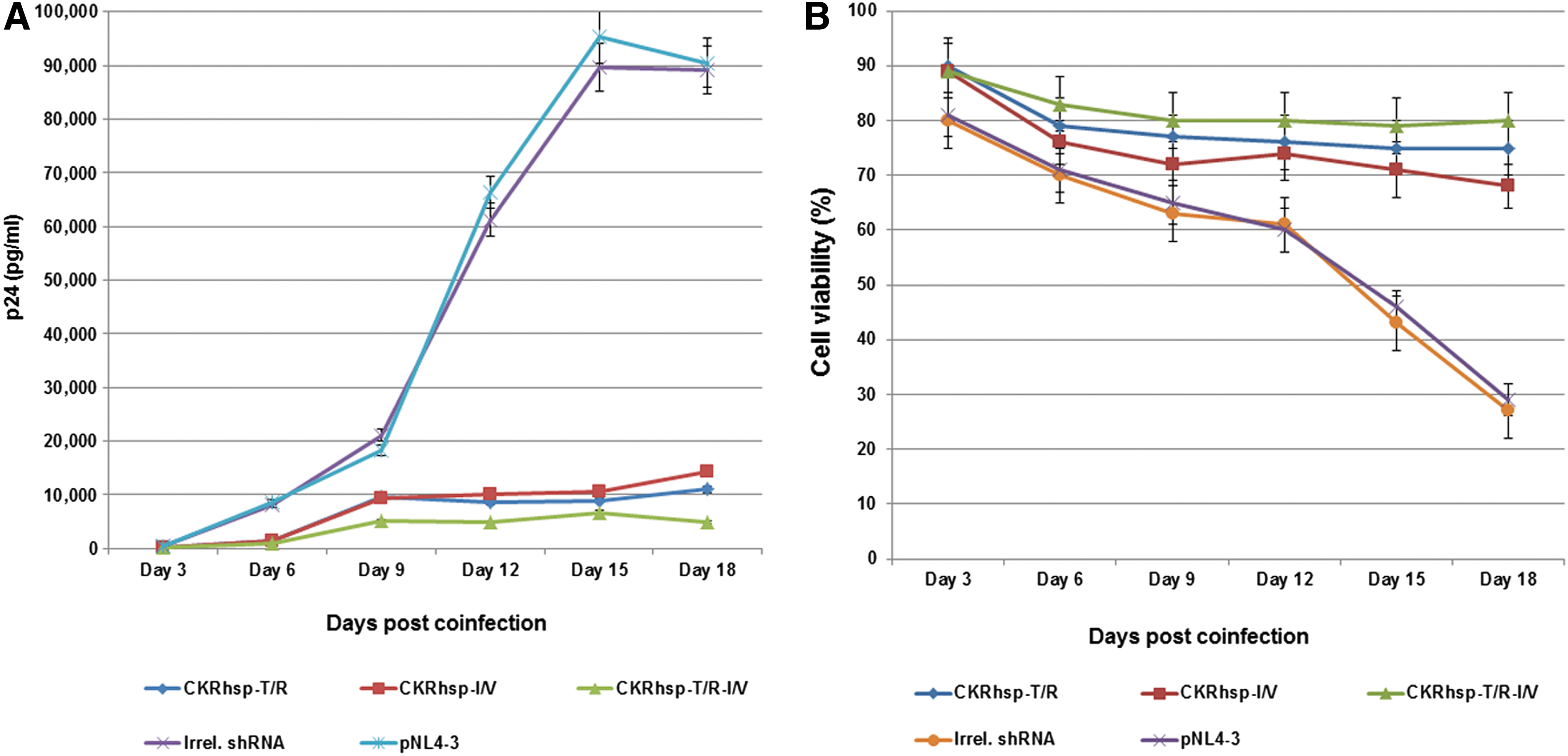
HIV-1 challenge assay.
Our results in Fig. 4A show that a strong virus inhibition was observed in cells transduced with shRNA vectors relative to NL4-3 strains (p<0.001). At 15–18 days postinfection, cells transduced with single-shRNA vectors showed viral replication as an increase in p24 level, whereas the dual-shRNA vectors decreased virus replication and increased cell viability percentage (p=0.021) (Fig. 4).
Lentiviral vectors with wild envelope have a specific tropism to CD4+cells
To investigate efficacy of novel vector in specific tropism to CD4+cells, we have analyzed shRNA expression level in CEM (as CD4+) and HEK293 (as CD4−) cells transduced with CkRhsp-shRNA vectors. Our results in Fig. 5 show that shRNA was considerably expressed only in CEM cells and no expression was detected in HEK293 cells.
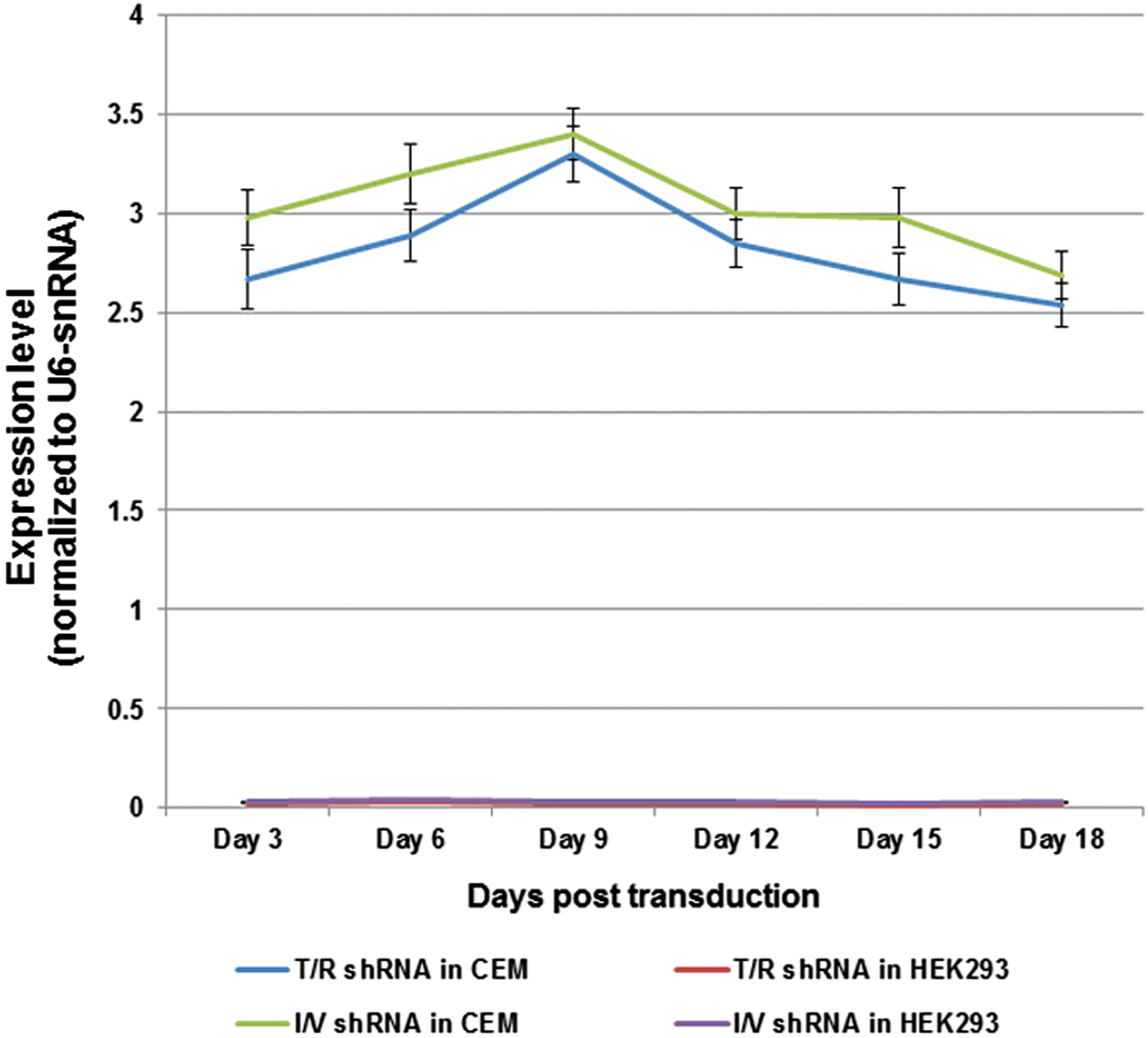
Investigation of vector-specific tropism to CD4+cells via siRNA expression analysis in transduced cell lines. siRNAs extracted from CEM and HEK293 cells were transduced with the CkRhsp-T/R-I/V vector and expression levels were determined by real-time PCR. All the data are from the three independent experiments. Color images available online at
Discussion
To avoid the side effects of chemotherapy and development of drug-resistant HIV-1 strains in the treatment of HIV-infected patients, there is a need to develop alternative therapeutic approaches such as gene therapy. 4 The unique features of HIV-1 virus and the various mechanisms of pathogenesis involved in disease progression and ultimately death pose considerable problems in the design of effective gene therapeutic setting. Stable integration, high mutability of the viral genome, the tendency of virus to latency, and its accumulation in immunologically privileged cells such as brain are some of these features. 28 Therefore, it is more appropriate that different stages of virus life cycle and multiple products be targeted for more effective therapy.
During the past decade, RNAi gene silencing has proven to be an efficient tool for disrupting specific genes' function and is considered as a potential antiviral agent in gene therapy strategies. Successful application of this novel technology in vivo requires vector development in the fields of safety and specific transducing target cells. An advantage of the RNAi-based therapies compared with protein-based approaches is lack of immunogenicity of the RNAi molecules. 29,30 For the effective inhibition of HIV-1 replication with an RNAi-based gene therapy, the use of multiple shRNAs simultaneously and preferably targeting highly conserved sequences have been proposed. 31 –34 In most previous studies, the targets of siRNAs or shRNAs in HIV-1 transcripts are not in fact highly conserved, and only a few studies focused on targeting conserved HIV-1 sequences. 23,35 –38 As previously reported 39,40 and our results at 15–18 days postinfection show (Fig. 4A), HIV-1 can escape from inhibition by mutations in sequences targeted by a single shRNA. We therefore decided to design two shRNAs for targeting two highly conserved regions in the HIV-1 genome in a dual-shRNA expression from a novel RNA polymerase II promoter.
There are potent advantages to utilizing RNA polymerase II in shRNA expression, including inducible transcription and tissue-specific promoters. 12 Several studies have focused on the use of HIV LTR for HIV induction of anti-HIV genes' expression. 41,42 In the present study, we demonstrated an HIV-1-inducible shRNA promoter in which the chicken β-actin core promoter with the R region of HIV-1 LTR was fused upstream of the Drosophila minimal hsp70 promoter (Fig. 2A). It is already known that the full LTR other than TAR element is not necessary for Tat transactivation. 21 In addition, the use of HIV LTR may increase the risks of vector mobilization and insertional mutagenesis, which may not be appropriate in vivo approaches. Therefore, we exchanged the U3 region in HIV LTR with a safer alternative promoter such as the chicken β-actin promoter for transcription of the TAR sequence, which may be more favorable for in vivo applications. The U5 region of HIV LTR is also removed. The U5 sequence contains sequences such as primer binding sites that are involved in reverse transcription. 43 With regard to this point, adding these sequences among the two LTRs in the vector genome (triple U5) may interfere with the efficient functioning of vectors in the target cells. As previously reported by Xia et al., 12 the hsp70 promoter and HIV-1 LTR show considerable similarities in transcription initiation. Both promoters are arrested by promoter-proximal pausing and are released by the Tat element (Fig. 2A). Hence, we decided to create a Tat-inducible promoter by fusing the minimal hsp70 promoter to downstream of the chicken β-actin core promoter and TAR element (Fig. 2A), in contrast to Unwalla et al.'s study, 18 in which the hsp70 promoter had been attached to full-length LTR. In the CkRhsp promoter, transcription initiation from the hsp70 promoter is dependent on binding Tat to TAR element in its upstream. Therefore, the shRNA expression occurs only in HIV-1-infected cells containing the Tat protein. As is understood from the results of Fig. 3, the new chimeric promoter can efficiently help to minimize no-Tat basal activity and elimination of non-base-paired sequences of 5′ end in shRNA expression. These results demonstrated that, unlike HIV LTR in the CkRhsp promoter, transcription initiating from the hsp70 can be more efficiently processed into an siRNA. Transcripts resulting from the HIV LTR would have an additional sequence (TAR and U5) at the 5′ end of the sense strand that would reduce recognition and processing to mature siRNAs. 12,18,19
Compared with the previous studies that used VSVG-pseudotyped vector to cell transduction, a remarkable change in this work is to deliver anti-HIV shRNA constructs via a lentiviral vector with the HIV-1 envelope. This modification mimicked specific tropism to the target cells without using chemicals such as polybrene, which raises the chance of using these vectors for in vivo approaches. As siRNA expression analysis in Fig. 5 shows, only CD4+ cells (CEM) were transduced by our new vector.
In summary, we have provided a novel third-generation lentiviral vector system to inhibit HIV-1 replication with specific features, including more safety, HIV-inducible, and specific targeting, one step closer to a treatment of the HIV-infected individuals. These experiments indicated that RNAi can be expressed by a Tat-inducible promoter other than the full HIV-1 LTR. The chimeric CKRhsp promoter, as well as LTR, can efficiently be induced by Tat, but with more safety, lower risk of LTR mobilization, and favorable start site in shRNA expression. RNAi is a potent antiviral agent in a gene therapy approach, specially expressing multiple shRNAs to target high conserved sequences from a single viral vector. The use of wild envelope in the vector packaging system may provide the specific targeting T lymphocytes and hematopoietic stem cells for anti-HIV-1 therapeutic approaches in vivo.
Footnotes
Acknowledgment
This study was supported by the Research and Technology Council of Golestan University of Medical Sciences (Grant No. 241756).
Author Disclosure
No competing financial interests exist.