
Abstract
Hematopoietic gene therapy has tremendous potential to treat human disease. Nevertheless, for gene therapy to be efficacious, effective gene transfer into target cells must be reached without inducing detrimental effects on their biological properties. This remains a great challenge for the field as high vector doses and prolonged ex vivo culture conditions are still required to reach significant transduction levels of clinically relevant human hematopoietic stem and progenitor cells (HSPCs), while other potential target cells such as primary macrophages can hardly be transduced. The reasons behind poor permissiveness of primary human hematopoietic cells to gene transfer partly reside in the retroviral origin of lentiviral vectors (LVs). In particular, host antiviral factors referred to as restriction factors targeting the retroviral life cycle can hamper LV transduction efficiency. Furthermore, LVs may activate innate immune sensors not only in differentiated hematopoietic cells but also in HSPCs, with potential consequences on transduction efficiency as well as their biological properties. Therefore, better understanding of the vector–host interactions in the context of hematopoietic gene transfer is important for the development of safer and more efficient gene therapy strategies. In this review, we briefly summarize the current knowledge regarding innate immune recognition of lentiviruses in primary human hematopoietic cells as well as discuss its relevance for LV-based ex vivo gene therapy approaches.
Introduction
H
Nevertheless, HSPCs are poorly permissive target cells still imposing the use of multiple hits of high vector doses and prolonged ex vivo culture to reach the high transduction levels observed in some of the recent LV-based clinical trials, 6,7 while other potential therapeutic targets such as primary macrophages can hardly be transduced. Part of the reasons behind the poor permissiveness of primary human hematopoietic cells to gene transfer resides in the retroviral origin of LVs. Extensive research in recent years has revealed the existence of many antiviral factors referred to as restriction factors (RFs) expressed by mammalian cells targeting specific steps of the retroviral life cycle. 8 –12 Some of these RFs are ubiquitously expressed, and others are inducible by specific danger signals such as type I interferon (IFN) or have cell-type-specific expression patterns. LV-mediated gene transfer is likely to be susceptible to these RFs because of some similarities the vector still shares with HIV-1. In particular, LVs rely on the same cellular machinery as HIV-1 to reach the nuclear compartment of target cells and integrate within the host genome. Furthermore, LVs can be recognized by innate sensors expressed by human hematopoietic cells, as discussed in detail below. Engagement of such receptors can result in activation of specific signaling pathways depending on the cell type, leading to distinct outcomes in terms of both transduction efficiency as well as impact on the biological functions of the host cell. Finally, it is emerging that innate immune cues actively regulate human HSPC functions and hematopoietic output (reviewed in ref. 13 ), highlighting the need to better understand the vector–host crosstalk likely occurring during gene transfer. In this review, we will briefly summarize the current knowledge regarding innate immune recognition of lentiviruses in primary human hematopoietic cells as well as discuss its relevance for LV-based ex vivo gene therapy approaches.
Innate Sensing of Lentiviral Transduction
Pattern recognition receptors (PRRs) have a central role in innate immune defenses as they recognize evolutionarily conserved structures on pathogens, termed pathogen-associated molecular patterns (PAMPs). Among PRRs, the family of Toll-like receptors (TLRs) has been the most extensively studied. TLRs are membrane-bound receptors, with 10 different TLRs identified in humans. TLR1, 2, 4, 5, 6, and 10 are expressed at the cell surface and mainly recognize hydrophobic molecules unique to microbes and not produced by the host. In contrast, TLR3, 7, 8, and 9 are located almost exclusively in endosomal compartments and are specialized in nucleic acid recognition. 14 Viral recognition is primarily mediated by TLR9 recognizing DNA, as well as by TLRs 7/8, and 3 sensing single-stranded RNA and double-stranded RNA, respectively. 15 –19 In addition, C-type lectin receptors, such as DC-SIGN, dectin-1, and mannose receptor, have emerged as cell surface PRRs that play important roles in induction of immune responses against various pathogens. 20 The retinoid acid-inducible gene (RIG)-like receptors (RLRs), RIG-I and MDA5, are RNA helicases that play a pivotal role in sensing of cytoplasmic RNA. 21,22 Finally, several cytosolic DNA receptors, including DAI, interferon-inducible protein 16 (IFI16), and AIM2, have been recently identified, 23 –25 eliciting great interest also in the field of retrovirology as viral DNA is generated during the viral life cycle. 10
In general, ligand engagement of PRRs leads to activation of signal transduction pathways involving the transcription factors, nuclear factor (NF)-κB, and IFN regulatory factors 3/7 as well as mitogen-activated protein kinase pathways, ultimately resulting in the production of cytokines, chemokines, cell adhesion molecules, and antiviral type I IFN. Recent studies have revealed innate sensors that recognize HIV-1-associated PAMPs. Some of these sensors recognize nucleic acids that are associated with retroviral infection in general, whereas others recognize HIV-1 viral proteins in species-specific manner. 26,27
As opposed to infectious HIV-1, LVs pseudotyped with the VSV-g envelope rely on endocytosis rather than direct fusion at the plasma membrane for entry. For this reason, some of the PRRs that HIV-1 naturally escapes from because of specific intracellular localization, like the endosomal TLR3 and 7, could well be engaged upon LV transduction (Fig. 1). Indeed, although initial studies conducted in a human B cell line showed remarkably low impact of LV transduction on cellular gene expression, 28 recent reports indicate that LVs activate myeloid dendritic cells (DCs) through TLRs 3 and 7. 29,30 Transduction has also been reported to trigger TLR-dependent and TLR-independent innate signaling in murine hepatocytes, leading to decreased transduction. 31 Along these lines, inhibition of TLR signaling in human natural killer cells can lead to improved transduction. 32 In vector particles, the two strands of genomic RNA are entwined within the core as a ribonuclear complex with viral proteins forming a dimeric RNA complex. 33 Thus, higher order dsRNA structures represent potential PAMPs for endosomally located TLR3 or cytosolic RLRs, particularly MDA5. Triggering of RIG-I may be prevented by 5′ capping of viral genomic RNA, making it similar to mRNA of host origin, and precluding its recognition as foreign. 34 Nevertheless, secondary structures present in HIV genomic RNA have been shown to trigger RIG-I signaling in primary human peripheral blood mononuclear cells and macrophages. 35
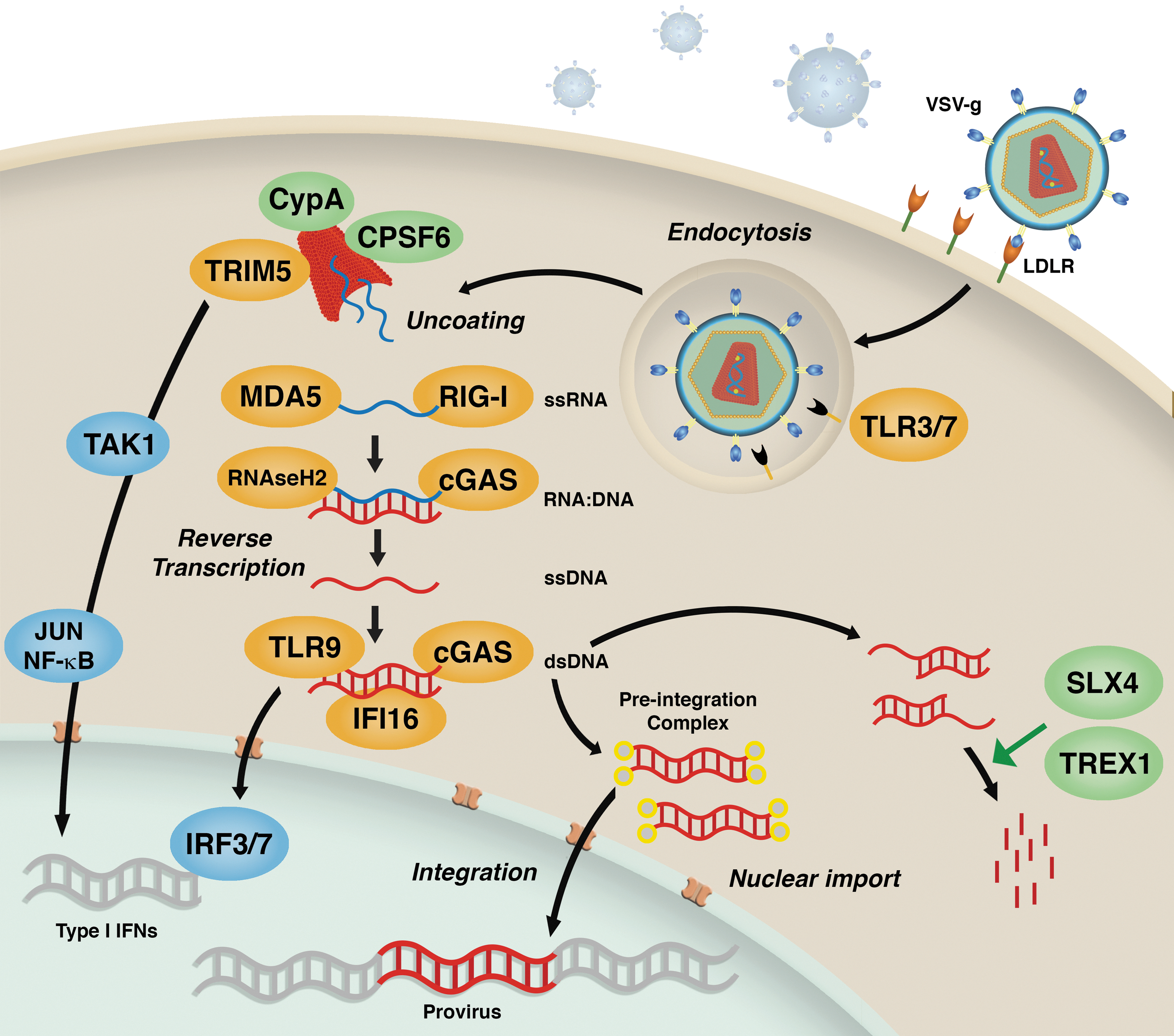
Innate immune sensing of lentiviral transduction. VSV-g envelope-mediated entry of lentiviral vectors (LVs) occurs through the low-density lipid receptor (LDL-R) surface receptor and endocytosis. TLR3/7 potentially recognize viral RNA in endocytic vesicles. During viral uncoating, the capsid lattice can be recognized by TRIM5α and trigger innate signaling through TAK1. Several cytosolic sensors can recognize different viral replication intermediates produced during reverse transcription. MDA-5 and RIG-1 sense single-stranded (ss) RNA, while RNAseH2 and cGAS detect DNA:RNA hybrids. cGAS, TLR9, and IFI16 sense double-stranded (ds) DNA. Ultimately, recognition by these sensors can lead to expression of type I interferons (IFNs) and innate activation of the cell. Innate triggering can be dampened or completely avoided through recognition of the viral capsid by host cofactors CypA and CPSF6. Host nucleases SLX4 and TREX-1 instead are able to clear excess viral DNA products avoiding their recognition by above-mentioned cytosolic sensors.
The HIV-encoded cytosolic DNA produced during reverse transcription does not normally trigger IFN or inflammatory responses in primary targets of infection, namely, activated CD4+ T cells and macrophages. 36,37 HIV-1 achieves this immune evasion through exploitation of one of the most abundant exonuclease in mammalian cells, the DNase TREX1 37 (Fig. 1). TREX1 clears HIV DNA products in the cytosol and maintains them below the threshold of immune activation. When TREX1 is removed, the HIV DNA generated during reverse transcription is sensed by cGAMP synthase (cGAS), which then synthesizes the second messenger dinucleotide cyclic GMP-AMP (cGAMP) that binds to stimulator of interferon genes (STING) to activate downstream IFN signaling. 38 Of note, mutations in TREX1 are associated with Aicardi–Goutières syndrome and chilblain lupus, which may result from accumulation of endogenous retroelements and aberrant stimulation of the cytosolic DNA sensor. 39,40 The cGAS-STING axis has recently been reported to recognize also synthetic RNA:DNA hybrids 41 and could therefore potentially recognize also this type of replication intermediates generated during LV reverse transcription. Another cytosolic ribonuclease, RNase H2, has also been shown to recognize RNA:DNA hybrids 42 and, similarly to TREX1, is implicated in several autoimmune and inflammatory diseases. 43
The cytosolic DNA sensor IFI16 has been recently identified in nonproductively infected “bystander” quiescent CD4+ T cells. 44 In this context, cells harboring HIV DNA products trigger IFI16-mediated IFN signaling and inflammasome response, including activation of caspase-1, secretion of IL-1β, and death of the host cell by pyroptosis. 45 Why TREX1 is not able to counterbalance IFI16-mediated detection of HIV DNA in this setting remains unclear. In the context of LV gene therapy, accurate purification of vector preparations for clinical use 46 could potentially prevent innate triggering, given that plasmid DNA is one of the major contaminant of laboratory-grade vector preparations, potentially activating the cytosolic DNA sensors during gene transfer.
Host cofactors that bind the viral capsid also modulate sensing of cytosolic HIV DNA (Fig. 1). Mutations of HIV-1 and HIV-2 capsid (CA) proteins that enhance binding to cyclophilinA (CypA), a cofactor involved in viral uncoating, nuclear import, and possibly even integration, 47 have been shown to trigger more robust DNA sensing in monocyte-derived DCs. 48 Similarly, HIV-1 CA mutants N74D and P90A, impaired for interaction with cofactors cleavage and polyadenylation specificity factor subunit 6 (CPSF6) and CypA, respectively, promoted DNA sensing in monocyte-derived macrophages (MDM). 49 It has recently been demonstrated that also the CA lattice itself can trigger innate immunity through the host factor TRIM5α, 50 best known for its direct antiretroviral effect further discussed below. 51 TRIM5α recognition of the capsid lattice stimulates its E3 ubiquitin ligase function, to catalyze synthesis of free K63-ubiquitin chains that activate TAK1 kinase complex. 50 TAK1 activation by TRIM5α promotes AP-1 and NF-κB signaling in DCs. In the absence of capsid recognition, TRIM5α can act downstream of TLR4 signaling to enhance Jun and NF-κB signaling in macrophages and DCs. Because human TRIM5α is capable of only weak restriction of HIV-1, 52 innate sensor activity of TRIM5α against HIV-1 is expected to be limited. Nevertheless, a correlation between levels of endogenous TRIM5α and permissivity to LV transduction was recently reported in rhesus macaque and human CD34+ HSPCs. 53 In the context of VSV-g-pseudotyped LV transduction, in which the endosomal compartment is engaged during entry, TRIM5α could potentially contribute to innate immunity against HIV-1 by acting downstream of TLR3/7-mediated recognition, similarly to the pathway demonstrated for TLR4. 50
In the context of LV transduction for gene transfer, the viral particle load is significantly higher compared with physiological retroviral infection. Therefore, innate regulatory mechanisms that have evolved to naturally containing inflammatory responses against endogenous retroviruses, such as TREX1, may be insufficient or overwhelmed by high vector doses. LVs also lack the viral accessory proteins. 54 It has become increasingly evident that the major role of lentiviral accessory proteins is to counteract host RFs (discussed below) 55 as well as to avoid innate immune activation, as recently discovered for Vpr that promotes escape from innate immune sensing through recruitment of the host SLX4 endonuclease complex. 56
Regarding innate sensing in HSCs, these cells have been thought of generally as dormant and called upon to divide only under specific conditions. However, recent studies suggest that they respond directly and acutely to infections and inflammatory signals, 13 and harbor the capacity to sense foreign nucleic acids as well as PAMPs through several PRRs, linking innate immune signaling with regulation of hematopoiesis. 57 The key mediator of innate immune signaling, IFNα, has recently been shown to impact HSC homeostasis leading to cell proliferation and activation of downstream signaling cascades in mice. 58 Importantly, long-term inflammatory/immune activation of HSCs has been shown to lead to their exhaustion and functional deficits in mice. 13 Furthermore, it is now clear that murine hematopoietic progenitors possess the machinery to directly sense infectious particles. TLRs are present on the surface of hematopoietic progenitors and actively signal upon stimulation leading to proliferation, differentiation, as well as loss of long-term self-renewal capacity upon chronic TLR-ligand exposure. 57,59 Exposure of murine HSPCs to TLR ligands has been also shown to modulate their chemokine receptor expression, 60 suggesting that TLR triggering may regulate their migratory and homing capacities.
Only a handful of studies have addressed innate immune triggering in human HSCs thus far, but this field of research is rapidly expanding. What emerges for now is that TLR triggering in human CD34+ cells can lead to skewed differentiation, mostly toward myeloid lineages, and cause apoptosis depending on which TLR is engaged. 61 –64 Responses may also vary among the CD34+ subpopulations because of differences in the expression levels of PRRs, as have been shown for dectine-1 in the context of murine Lin− HSPCs. 65
Host RFs Against Lentiviruses
After retrovirus entry into target cells, intracellular RFs can provide an additional block to viral replication. Although many of these factors are IFN-induced and thereby potentially induced by the innate sensing mechanisms discussed above, their activity does not necessarily require any virus-triggered signaling or intracellular communication. 11 Therefore, RFs represent an intrinsic mechanism that can act to limit virus replication at many stages of the viral life cycle. To date, several RFs effective against lentiviruses have been identified in mammals, including apolipoprotein B mRNA editing enzyme 3 (APOBEC3G), 66 tripartite motif protein 5α (TRIM5α), 52,67 tetherin, 67,68 SAM domain, and HD domain-containing protein 1 (SAMHD1), 69,70 and most recently Mx2. 71 –73 Those that could potentially be involved in LV–host interactions in the context of ex vivo gene therapy approaches are discussed here (Fig. 2).
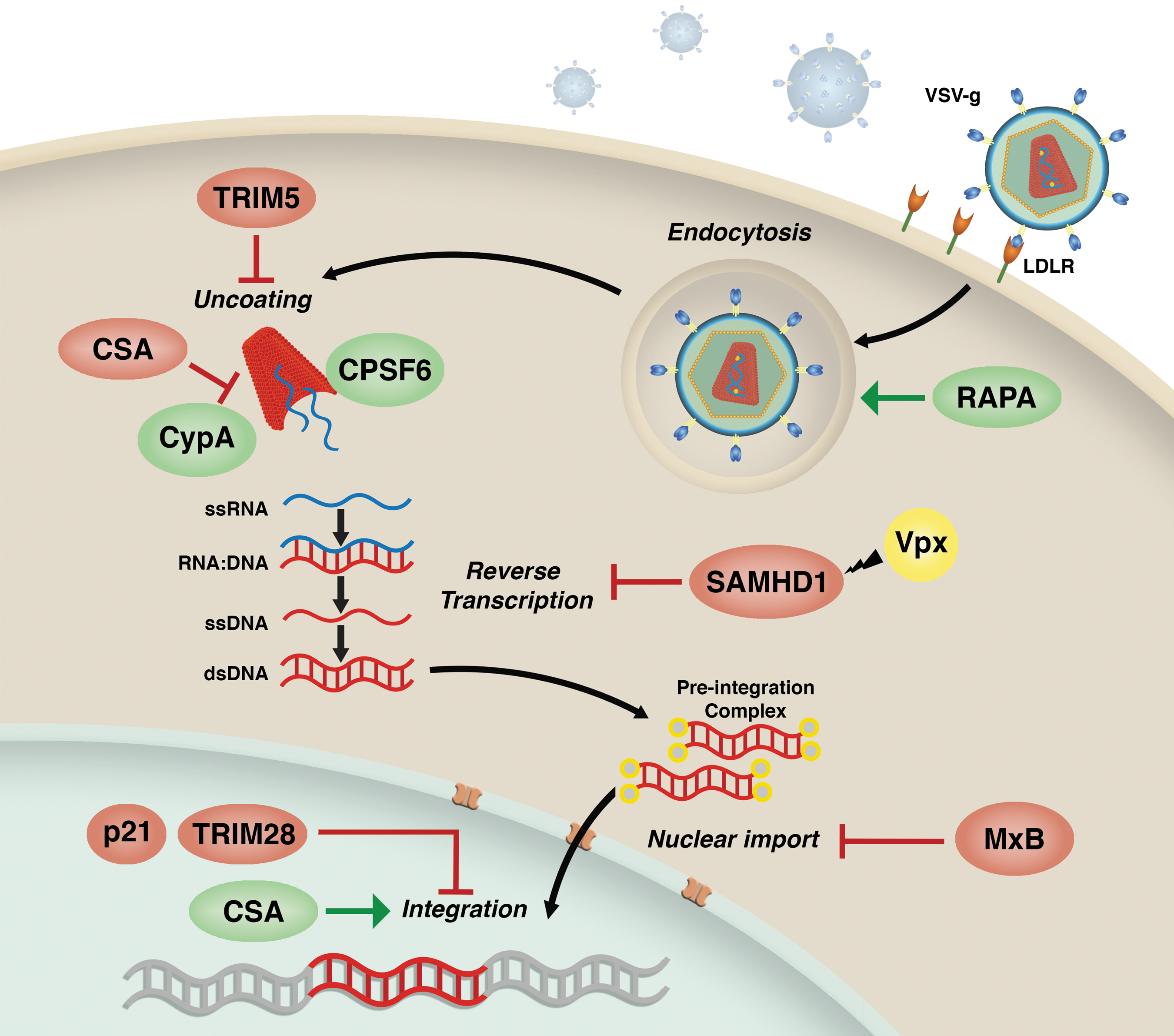
Factors potentially impacting lentiviral transduction efficiency. Levels of LDL-R surface expression have been suggested to influence LV transduction efficiency at the entry step. Rapamycin has been shown to improve LV transduction in human and murine hematopoietic stem and progenitor cells (HSPCs) by enhancing postbinding endocytic events. In Old World monkey cells, TRIM5α potently restricts LVs through premature degradation of the viral capsid. CsA disrupts the capsid-CypA interactions negatively affecting the uncoating process. In human cells of myeloid origin, SAMHD1 potently inhibits the process of reverse transcription but can be efficiently counteracted by the SIV-derived accessory protein Vpx. The IFN-inducible GTPase Mx2 inhibits LV transduction potentially acting at the level of nuclear import. In the nucleus, TRIM28 and p21 have been reported to impair the LV integration process. Finally, CsA is able to increase LV integration in human and murine HSPCs.
TRIM5α is a prototypic RF that targets the incoming lentiviral capsids to proteasomal degradation. This leads to a block in reverse transcription and a process that dismantles the virus. 51,52 Although human TRIM5α has little or no antiviral activity against HIV-1, as discussed above, its newly identified innate sensor functions could contribute to sensing and potentially restricting LV transduction in human hematopoietic cells. However, TRIM5α potently blocks HIV-1 infection in Old World monkey cells, 52 an aspect that should be kept in mind when eventually choosing primate models for preclinical testing of LV-based gene therapy approaches. Indeed, modifications in the HIV-1 capsid that alter its recognition by TRIM5α can partially overcome lentiviral restriction in nonhuman primate epithelial cells, but not in hematopoietic cells. 74
The dNTP triphosphohydrolase SAMHD1 is expressed widely in myeloid cell lineages and resting T cells. 70,75 Restriction by SAMHD1 is mediated by reduction of nucleotide pools to levels where reverse transcription cannot proceed, although recent investigations suggest that more complex mechanisms are likely involved. 76,77 Many simian immunodeficiency viruses (SIVs) are not susceptible to SAMHD1 because they encode the accessory protein Vpx that antagonizes SAMHD1 by recruiting a ubiquitin ligase complex that leads to its proteasomal degradation. 55 HIV-1 does not encode a Vpx homolog and is thus sensitive to SAMHD1. This sensitivity has an important impact on HIV-1 tropism, with evidence that SAMHD1 prevents HIV-1 infection of DCs and resting T cells. 70,75 Although HIV-1 can replicate in macrophages, addition of Vpx significantly improves infectivity, confirming a functional role for SAMHD1 also in these cells.
The most recently identified HIV-1 RF is the GTPase Mx2. 71 –73 Mx2 suppresses HIV-1 infectivity after reverse transcription, possibly at the level of nuclear entry (Fig. 2). Curiously, several HIV-1 capsid mutants, including the CypA-binding mutants P90A and A88T as well as the CPSF6-binding mutant N74D, have been shown to be insensitive to Mx2 restriction, suggesting direct interactions between the capsid and Mx2 as well as involvement of CypA in its antiviral mechanism of action. 73 Interestingly, we have observed that the A88T CA mutant performs better than wild-type LVs in presence of CsA, a compound that normally inhibits HIV-1 infection by disrupting the CA-CypA interaction 78,79 but that we have found to significantly increase LV transduction in human and murine HSPCs. 80 Nevertheless, involvement of Mx2 in this mechanism remains unclear as its activity may not depend solely on the CA-CypA interaction, 81 and Mx2-resistant CA mutants have been shown to remain susceptible to IFN treatment, suggesting the existence of additional IFN-induced host factors capable of restricting HIV-1 and LVs. 82
Host factors directly impairing the HIV-1 integration process, such as TRIM28 83 or the cyclin-dependent kinase inhibitor CDKN1A (p21), 84 have also been described (Fig. 2). Although TRIM28 is ubiquitously expressed in many cell types, it was originally discovered as a strong transcriptional repressor of retroviruses in embryonic cells 85 and was later shown to control the expression of endogenous retroviruses in mouse embryonic stem cells. 86 In the context of HIV-1 infection, TRIM28 was shown to interact with acetylated HIV-1 integrase (IN) and to form a complex with histone deacetylase 1, leading to IN deacetylation and reduced integration efficiency. 83 CDKN1A (p21) instead was shown to restrict HIV-1 replication in MDM at the stage of reverse transcription. 87 However, this effect was recently suggested to be most likely related to p21-mediated regulation of SAMHD1 phosphorylation that in turn will influence viral reverse transcription in MDM. 88 Interestingly, prior work performed in primary human bone-marrow-derived CD34+ HSPCs suggests that in these cells instead endogenous p21 blocks HIV-1 by complexing with the viral IN and aborting chromosomal integration. 84 We have shown that CsA, as opposed to its well-documented inhibitory effect in differentiated hematopoietic cells, increases LV transduction in human and murine HSPCs at the level of the viral integration process 80 (Fig. 2). It will be of interest to understand whether TRIM28, p21, or other yet-to-be-discovered host factors could be counteracted by CsA in HSPCs.
Potential Strategies to Overcome Innate Barriers to Transduction
One of the best examples of how knowledge on host RFs and their counteraction by viral protein can allow the development of improved vector platforms, tailored to reach higher transduction efficiency in a specific target cell type, is the SAMHD1-Vpx axis.
As sentinels of the immune system, DCs play an essential role in regulating cellular immune responses. They are attractive targets for many therapeutic approaches that aim to harness the potential of the patient's immune system to prevent or treat human diseases, such as cancer immunotherapy, tolerogenic therapies, and vaccine delivery. 89 –91 Macrophages, like DCs, are professional antigen-presenting cells constituting a first line of defense against invading pathogens. They also regulate tissue growth, homeostasis, repair, and remodeling via their expression of numerous cytokines, chemokines, growth factors, proteolytic enzymes, and scavenger receptors. 92,93 As such, macrophages play a central role in developmental processes, such as tissue morphogenesis and vascular and neuronal patterning, but also in pathophysiological responses, like inflammation and organ healing/regeneration. 94,95 Macrophages are also a major cellular component of murine and human tumors, where they are commonly termed tumor-associated macrophages. In this context they represent an attractive cellular target for anticancer therapies. 96
One of the major drawbacks for the developments of myeloid cell-based gene therapies has been their natural resistance to lentiviral infection. The capacity of the SIV-encoded Vpx to degrade SAMHD1 and to dramatically increase LV transduction efficiency in human myeloid cells has been proposed by many to be exploited in the context of gene transfer applications. 97 –99 Target cells can either be preexposed to virus-like particles that contain Vpx (Vpx-VLPs) 100 or Vpx can be directly incorporated into the LVs during vector production through the use of a modified packaging plasmid engineered to contain an SIV-derived Vpx-interacting sequence. 97,101 We have successfully taken advantage of the Vpx-VLPs to efficiently transduce primary human monocytes with an LV encoding for human IFNα under the control of the myeloid-specific TIE2 promoter in the context of targeted antitumor strategies 102 as well as to test myeloid-specific LV encoding gp91phox in primary human MDM for the treatment of chronic granulomatous disease. 103 It should, however, be pointed out that although murine SAMHD1 has been recently shown to restrict LV replication, 104 Vpx is not able to degrade it. 105 Therefore, the Vpx vector platforms cannot be investigated in the context of murine models of disease, rendering their potential preclinical validation challenging and costly.
It is also important to note that it has been observed that, when SAMHD1 restriction is experimentally bypassed through the use of Vpx in human DCs, the cells are able to detect the virus, become activated, and secrete large amounts of type I IFN. An initial study suggested that the mechanism behind this innate immune activation would be postintegration sensing of de novo synthesized viral Gag 106 and thus of no concern for single-round LV transduction. However, work from the same group recently reported that it is the cytoplasmic DNA sensor cGAS that detects HIV-1 reverse transcription products in this setting, leading to activation of innate signaling cascades and the maturation of the DCs, 48 similarly to what has been suggested to occur in monocyte-derived macrophages when specific capsid–host factor interactions are altered. 49 Based on these observations, potential innate immune triggering should be taken into consideration and carefully assessed when considering Vpx, LV capsid mutants, or any treatment that can alter the vector–host interactions for clinical application. Nevertheless, it is also possible that the extent of innate activation triggered by modifications of the vector–host interaction may also depend on the target cell type and is likely to be more pronounced in professional antigen-presenting cells. Indeed, we did not observe any type I IFN secretion in human CD34+ HSPCs upon transduction in presence of CsA or with an LV harboring the A88T capsid mutant, 80 both impairing vector interaction with the host cofactor CypA.
Given the pivotal role the viral capsid plays both in evading and in getting recognized by the innate immune factors, several efforts to engineer it or modulate its interactions with host factors have been made with the aim of improving lentiviral transduction efficiency in human HSCs. 74,107,108 Unfortunately, none of the thus-far-tested capsid variants, including a panel of LV capsid mutants generated by us, 80 have given a significant advantage compared with unmodified LVs in human HSPCs in absence of any additional stimulation. These findings underscore the importance of the interaction between the viral capsid and host cofactors for optimal transduction efficiency. Interestingly, however, the A88T capsid mutant outperformed LVs harboring WT capsid in human HSPCs in presence of CsA, 80 indicating that the capsid–host factor interactions may also be involved in LV restriction in HSPCs.
Transient exposure of HSPCs to compounds potentially affecting vector–host interactions has also been explored as a strategy to improve LV transduction efficiency. In particular, proteasomal inhibition has been shown to significantly improve LV transduction in both murine and human HSPCs, 107,109 although the precise mechanisms behind this observation remain elusive. We and others have observed that rapamycin acts early during the LV life cycle to improve transduction in human and murine HSPCs, independently of the viral capsid and autophagy 80,110,111 (Fig. 2). Moreover, we found that rapamycin not only improved LV transduction efficiency but also lead to better engraftment of human HSPCs in NSG mice. 80 This could be of further benefit in clinical settings in which engraftment kinetics are crucial for successful gene therapy outcome. 112
Finally, it has recently been suggested that the VSV-g envelope currently used in all LV-based clinical trials may not be the optimal choice in certain cell types or under specific experimental conditions because of variable levels of the low-density lipid receptor (LDL-R) expression, 113 previously identified to be the cellular receptor for VSV. 114 LVs pseudotyped with a baboon retroviral envelope glycoprotein 115 or Measles virus glycoprotein 116 that do not rely on LDL-R for entry have been suggested to outperform the VSV-g-pseudotyped one in T and B cells as well as in quiescent CD34+ HSPCs. Targeting LVs toward more primitive subsets of HSPCs by exploiting the subset-specific expression of surface markers such as CD105 and CD133 has also shown promising results regarding preferential transduction of long-term repopulating HSCs. 117,118
Conclusions and Future Considerations
We have discussed here how LV transduction could potentially trigger innate immune responses not only in differentiated hematopoietic target cells, such as DCs or macrophages, but also in HSPCs, with potential consequences in terms of both efficiency of gene transfer as well as biological integrity of the target cells. Having said this, the relatively short ex vivo exposure of cells to LVs might not be sufficient to alter their biological properties in a clinically relevant manner, as comparison of colony-forming and engraftment capacities of human HSPCs showed no differences between transduced and untransduced cells in preclinical safety and efficacy studies for the treatment of Wiskott–Aldrich syndrome. 119 Along these lines, LV transduction was recently shown not to have any long-term impact on the transcriptional profiles and telomere length of rhesus macaque HSPCs in vivo 10 years posttransplantation, 120 and unaltered long-term human hematopoietic reconstitutions after LV gene therapy have been documented in recent clinical trials. 4,6,7 Nevertheless, some of these aspects may turn out more relevant in specific disease contexts in which the patient background can potentially be more prone to aberrant immune activation such as autoimmune syndromes, or if specific features affecting vector–host interactions are modified to improve transduction efficiency.
Overall, efforts to improve LV transduction efficiency are still needed, as even relatively small improvements in cell permissivity to transduction could greatly impact on sustainability of vector production as well as contribute to avoid triggering of innate sensors and effectors. Finally, despite the increasing evidence regarding the mechanisms infectious HIV-1 has evolved to counteract innate immunity, it is clear that the virus is unable to antagonize all of the innate barriers it encounters in the presence of type I IFN. 82,121 This suggests that the repertoire of innate effector proteins includes yet-unidentified RFs with anti-HIV-1 activity 10 that could potentially be expressed or induced also in primitive HSPCs. This leaves plenty of room for investigation of these innate barriers also in the context of LV gene therapy. Insights from molecular retrovirology and innate immune sensing could inspire the development of innovative vector platforms and promote understanding of how these treatments could potentially impact the therapeutic outcomes long-term.
Footnotes
Acknowledgments
Our research discussed here was supported by grants from the Italian Ministry of Health and the Italian Telethon Foundation to A.K.-R. (Bando Giovani Ricercatori GR-2009-1471693, TGT14-D04) and from the Italian Telethon Foundation and the European Research Council (TARGETINGGENETHERAPY) to L.N. The authors wish to thank Dr. Andrea Di Pietro for kindly creating the figures for this review.
Author Disclosure Statement
No competing financial interests exist.