
Abstract
mRNA reprogramming results in the generation of genetically stable induced pluripotent stem (iPS) cells while avoiding the risks of genomic integration. Previously published mRNA reprogramming protocols have proven to be inconsistent and time-consuming and mainly restricted to fibroblasts, thereby demonstrating the need for a simple but reproducible protocol applicable to various cell types. So far there have been no published reports using mRNA to reprogram any cell type derived from human blood. Nonmodified synthetic mRNAs are immunogenic and activate cellular defense mechanisms, which can lead to cell death and inhibit mRNA translation upon repetitive transfection. Hence, to overcome RNA-related toxicity we combined nonmodified reprogramming mRNAs (OCT4, SOX2, KLF4, cMYC, NANOG, and LIN28 [OSKMNL]) with immune evasion mRNAs (E3, K3, and B18R [EKB]) from vaccinia virus. Additionally, we included mature, double-stranded microRNAs (miRNAs) from the 302/367 cluster, which are known to enhance the reprogramming process, to develop a robust reprogramming protocol for the generation of stable iPS cell lines from both human fibroblasts and human blood-outgrowth endothelial progenitor cells (EPCs). Our novel combination of RNAs enables the cell to tolerate repetitive transfections for the generation of stable iPS cell colonies from human fibroblasts within 11 days while requiring only four transfections. Moreover, our method resulted in the first known mRNA-vectored reprogramming of human blood-derived EPCs within 10 days while requiring only eight daily transfections.
Introduction
T
While fibroblasts are a common cell type used to generate iPS cells for research, their use in personalized medicine is limited because their isolation is either restricted to neonatal foreskin fibroblasts or to invasive skin biopsies from adults that may not be suitable for young patients or patients with abnormal wound healing (e.g., diabetic patients). Alternatively, peripheral blood provides easy access to adult human somatic cells and there are numerous studies using blood-derived cell types to generate patient and disease-specific iPS cell lines for both research and clinical applications. 19 –25 In late 2012, two groups demonstrated the effective generation of iPS cell lines from blood-outgrowth endothelial progenitor cells (EPCs), which can be clonally isolated from adult peripheral blood 25,26 or cord blood, 27 using viral vectors. A main advantage of EPCs is that they can be derived from fresh as well as frozen mononuclear cells (MNCs) and that the clonally derived primary EPC lines can be cryopreserved. Therefore, peripheral blood MNCs, cord blood MNCs, as well as primary EPC cultures can be either accessed from existing biobanks or stored in such. In contrast to hard-to-transfect lymphocytes, EPCs are adherent and highly proliferative and maintain cell identity for multiple passages, which makes them accessible for repeated transfection with RNA. Nevertheless, to date, no group has been able to demonstrate RNA-based reprogramming of this cell type or any other cell type derived from blood. This limitation has been primarily because of the inability to efficiently and repeatedly deliver mRNA to cells originating from blood without inducing cytotoxicity that is inherent to immunogenic mRNA.
Immunogenicity of mRNA is caused by the stimulation of antiviral defense mechanisms resulting in apoptosis, cytoskeletal rearrangements, RNA degradation, and shutdown of translation. 28 Synthetic mRNA is thereby recognized by membrane-bound and cytoplasmic sensor molecules that recognize specific molecular patterns that are distinct from regular cellular RNA-like double-stranded RNA contaminants and 5′-triphosphate groups. 29,30 Synthetic mRNA recognition induces an interferon (IFN) response by secreting type I IFNs resulting in profoundly altered gene expression patterns. 30 –32 A central primary IFN response effector and double-stranded RNA sensor is protein kinase R (PKR), which upon activation phosphorylates the eukaryotic initiation factor 2 alpha (eIF2α), which subsequently blocks protein translation. 33 Previous studies achieved cellular reprogramming by reducing immunogenicity of synthetic mRNA with incorporation of modified nucleosides pseudouridine (Ψ) and 5-methylcytidine (5mC) and quenching of residual IFN-secretion with addition of the recombinant B18R protein (rB18R), an IFN decoy receptor from vaccinia virus (VACV) as media supplement. 17,18,34 These cited mRNA reprogramming procedures are labor-intensive, requiring 8–12 daily transfections for neonatal fibroblasts. Aside from inducing cell toxicity and inhibition of translation, activation of innate immunity was shown to increase reprogramming efficiency, 35 indicating that immunogenicity should not be completely removed from RNA vectors. While protocols using nonmodified immunogenic mRNA for reprogramming of fibroblasts into iPS cells have been able to induce pluripotency marker expression, most have failed to robustly and efficiently generate fully reprogrammed iPS cells. 36 –41 Therefore, we aimed to develop an alternative approach that uses a combination of nonmodified synthetic RNAs that decrease cytotoxicity of synthetic mRNA and increase protein translation of transfected reprogramming mRNAs. As B18R had already been introduced into mRNA reprogramming protocols, we hypothesized that VACV PKR inhibitors E3 and K3 should further decrease the IFN response and promote cellular reprogramming by preventing eIF2α phosphorylation and consequently resulting in increased protein translation. 42 –45
In this article, we present novel data demonstrating integration-free RNA reprogramming via cotransfection of synthetic nonmodified reprogramming and VACV immune evasion mRNAs coding E3, K3, and B18R (EKB). When combined with reprogramming-associated microRNAs (miRNAs), this results in an efficient and feeder-free iPS cell generation method that is simple, fast, and cost-effective, while being applicable to cell types from both human skin and blood.
Materials and Methods
Cell culture and reagents
All growth media, supplements, enzymes, growth factors, and solutions were supplied by Life Technologies/Gibco except when stated otherwise. Neonatal human foreskin fibroblasts (HFFs; SBI) were cultivated in minimum essential medium (MEM) containing 15% fetal calf serum (FCS; Invitrogen), penicillin 1 unit/ml, streptomycin 1 μg/ml, 1% nonessential amino acids, 1 mM sodium pyruvate at 37°C, and a CO2 content of 5%. For feeder generation, HFFs were harvested and irradiated with 8000 rad using a gamma irradiator. Human newborn foreskin fibroblasts NuFF (Globalstem) and adult human skin dermal fibroblasts (HDFa; Lifetech) were cultivated in DMEM containing 10% fetal bovine serum (FBS; Atlas). Human umbilical vein endothelial cells (HUVECs; Lonza) and EPCs were cultured at 37°C, 5% CO2, and 21% O2 in EGM-2 MV medium (Lonza) containing 10% embryonic stem cell-grade FBS (Hyclone) instead of the supplied FBS (EPC medium). iPS cells and cells in reprogramming experiments were cultured if not otherwise stated in NutriStem XF/FF hES cell culture medium (Stemgent) at 37°C and a CO2 content of 5%. In case of HFF reprogramming, NutriStem medium was supplemented with 10 ng/ml basic fibroblast growth factor (bFGF) and 0.5 μM Thiazovivin (Stemgent). For passaging of RNA-derived iPS cells, cells were washed once with phosphate buffered saline (PBS) and incubated for 5 min at 37°C with 1 mg/ml collagenase type IV in PBS. Cells were scraped and washed off the plate, centrifuged at 950 rpm for 5 min, resuspended in NutriStem medium, and transferred onto irradiated HFF-Feeder (previously plated 1.4E5 cells/6-well plate). HFF- or NuFF-conditioned NutriStem medium was produced by incubating 2.5E6 irradiated HFF- or NuFF-feeder onto T75 cell culture flask in 20 ml NutriStem medium supplemented with 10 ng/ml bFGF. The medium was collected after 24 hr and plates were replenished with fresh medium. Collection was repeated five times, and the conditioned medium was pooled, sterile-filtered (0.22 μm; Sartorius Stedim), and frozen at −20°C until use. Mature, double-stranded miRNAs 302a–d and 367 were purchased from Thermo Fischer Scientific, mixed in equimolar ratio, and stored at −80°C until use.
Plasmids and in vitro transcription
Plasmids used as templates in this study for in vitro transcription are based on pST1-2hBgUTR-A120,
46
which features a tandem repeated 3′-human-β-globin-UTR (hBgUTR) and a template-encoded poly(A)-tail of 120 nucleotides (refer to the schematic in Supplementary Fig. S1; Supplementary Data are available online at
EPC derivation from peripheral blood and cord blood
Human MNCs were obtained from 50 ml of peripheral blood (Research Blood Components) by density gradient centrifugation using Ficoll Paque Plus (GE Healthcare). Cord blood EPCs were derived from 5E7 frozen cord blood MNCs (AllCells). Both peripheral and cord blood MNCs were washed and cultured on rat collagen type I (BD Bioscience)-treated plates at 1.5E6 MNCs/cm2 (up to 1E8 MNCs were seeded on a T75) in EGM-2 MV medium (Lonza) containing 20% embryonic stem cell-grade FBS (Hyclone). Culture medium was changed every other day and EPCs appeared after 8–14 days in culture. EPCs were then passaged onto tissue culture plastic by using Trypsin and maintained in EPC medium containing 10% standard FBS. Once the primary EPCs reached confluency, they were either split for a reprogramming experiment directly, passaged further, or cryopreserved for future experiments.
Transfection of primary fibroblasts and reprogramming
Lipofections of synthetic mRNA were performed in 6-well plates and 1E5 fibroblasts were seeded per well. For reprogramming experiments, plates were previously coated with 0.1% gelatine solution (Millipore) for 20 min at 37°C. Cells were plated in 2.5 ml NutriStem medium without antibiotics. The first lipofection was performed after cells adhered, usually between 4 and 5 hr after plating, and in case of HDFa after about 18 hr. To prepare lipoplexes, 6 μl RNAiMAX was diluted in 250 μl Opti-MEM serum-free medium (both Invitrogen). An amount of 1.4 μg of total RNA was diluted in 250 μl Opti-MEM and combined immediately with the diluted RNAiMAX preparation. In more detail, the reprogramming mRNA mixtures composed of 0.8 μg TF mRNA encoding OCT4, SOX2, KLF4, cMYC, NANOG, and LIN28 (OSKMNL) 1,3 (1:1:1:1:1:1 or 3:1:1:1:1:1) supplemented with 0.2 μg of each EKB (total mRNA = 1.4 μg) and 0.4 μg of miRNAs 302a–d and 367 (0.4 μM each). After 15 min incubation, 500 μl lipoplex solution was added per well to the cells and incubated for 24 hr.
To perform daily lipofections, culture supernatant containing lipoplexes of the previous lipofection was removed and fresh 2.5 ml NutriStem medium without antibiotics were added before lipofection was repeated. If cell growth required splitting during the reprogramming process, cells were split 1:8 onto irradiated HFF feeder previously plated with 1.4E5 cells/6-well plate. In the no-split protocol, the medium was either changed to HFF-conditioned (HFFs) or NuFF-conditioned (NuFFs and HDFa) NutriStem medium on day 5 or cells were kept in nonconditioned NutriStem medium as indicated. Reprogramming efficiency was determined by the total number of pluripotent iPS cell colonies multiplied by 100 and divided by the total number of cells that were seeded.
Transfection of primary EPCs and HUVECs for reprogramming
Onto Matrigel (BD Bioscience)-coated plates, 2.5E4 EPCs or HUVECs were seeded per 6-well plate. Lipofection was performed around 18 hr after plating. To prepare lipoplexes, 6 μl RNAiMAX was diluted in 250 μl Opti-MEM serum-free medium (both Invitrogen). An amount of 1.4 or 1.8 μg total RNA was diluted in 250 μl Opti-MEM and combined immediately with the diluted RNAiMAX preparation. In more detail, the reprogramming mRNA mixtures composed of 0.8 or 1.2 μg TF mRNA OSKMNL at stoichiometries of either 1:1:1:1:1:1 or 3:1:1:1:1:1 supplemented with 0.2 μg of each EKB (total mRNA = 1.4 or 1.8 μg) and 0.4 μg of miRNAs 302a–d and 367 (0.4 μM each). After 15 min incubation, 500 μl lipoplex solution was added to each well and incubated for 4 hr on the first 3 days and then switched to 24 hr incubation times for the remaining 3 or 5 days of the transfection schedule. To perform daily lipofections, culture supernatant was removed and fresh 2 ml EPC medium was added before each lipofection. For EPC/HUVEC reprogramming experiments, the medium was supplemented with 20 ng/ml bFGF from day 8 until day 14 and the culture was transitioned to NutriStem medium on day 10. Reprogramming efficiency was calculated by counting the TRA-1-60-positive iPS cell colonies on day 15, multiplying the total number of iPS cell colonies by 100 and dividing it by the total number of cells that were seeded.
Reporter gene assays
For analysis of enhanced green fluorescent protein (GFP) expression, cells were harvested and fixed using 2% paraformaldehyde (PFA; Roth) in PBS. Flow cytometric data were acquired on a FACS Canto II flow cytometer (BD Bioscience) and analyzed by the companion Diva software or FlowJo software (Tree Star). Luciferase expression was analyzed as described 50 using the Bright-Glo Luciferase Assay System (Promega).
Quantitative real-time reverse transcriptase PCR
To quantify transcript levels, total cellular RNA was extracted using the RNeasy Mini or Micro Kit (Qiagen) according the manufacturer's instructions and isolated RNA was quantified by spectroscopy (NanoDrop 2000c; PeqLab). An amount of 500 ng total RNA was reverse transcribed using the Superscript II reverse transcriptase protocol (Invitrogen) with oligo-dT18 primer according to the manufacturer's instructions. Quantitative real-time reverse transcriptase PCR (qRT-PCR) were performed in triplicates using the ABI 7300 real-time PCR System, the companion SDS analysis software (Applied Biosystems), and the QuantiTect SYBR Green PCR Kit (Qiagen). Protocol followed the manufacturer's instruction with 15 min at 95°C, and 40 cycles of 30 sec at 94°C, 30 sec at oligo-specific annealing temperature stated below, and 30 sec at 72°C. Relative expression changes of the target transcript in the probe of interest versus untreated controls was quantified using the 2-ddCT method, normalized to the housekeeping gene HPRT. The following specific primers and annealing temperatures were used: IFNβ, forward: 5′-AAGGCCAAGGAGTACAGTC-3′, reverse: 5′-ATCTTCAGTTTCGGAGGTAA-3′ (60°C); OAS1, forward: 5′-AGGTGGTAAAGGGTG GCTCC-3′, reverse: 5′-GGGTTAGGTTTATAGCCGCC-3′ (60°C); LIN28, forward: 5′-AGGAG ACAGGTGCTACAACTG-3′, reverse: 5′-CCACCCATTGTGGCTCAATTC-3′ (64°C); TERT, forward: 5′-CTGGATTTGCAGGTGAACAGC-3′, reverse: 5′-AGATGACGCGCAGGAAAAATG-3′ (60°C); REX1, forward: 5′-GGTAACAGGGGTTGGAGTGCA-3′, reverse: 5′-GCGCTGACAGGTTCTATT TCC-3′ (58°C); DPPA4, forward: 5′-TGTGTTCACAGGAACAAGGTC-3′, reverse: 5′-TGTAAGAGT CTCTATCTCCAC (58°C); GDF3, forward: 5′-TCCCAGACCAAGGTTTCTTTC-3′, reverse 5′-TTACCT GGCTTAGGGGTGGTC-3′ (60°C); OCT4, forward: 5′-GGCTCGAGAAGGATGTGGTCC-3′, reverse: 5′-CTGTCCCCCATTCCTAGAAGG-3′ (58°C); NANOG, forward: 5′-AGCTACAAACAGG TGAAGACC-3′, reverse: 5′-AGGAGTGGTTGCTCCAGGACT-3′ (60°C); HPRT, forward: 5′-TGACACTGGCAAAACAATGCA-3′, reverse: 5′-GGTCCTTTTCACCAGCAAGCT-3′ (60°C).
Cell viability
Cell viability was determined using the Cell Proliferation Kit II (Roche Applied Science) according to the manufacturer's instructions with the following modification: cells grown in a 6-well plate were washed and covered with 1 ml fresh medium and 500 μl XTT labeling/10 μl electron coupling reagent. After 3 hr incubation at 37°C, 100 μl of the supernatant was transferred three times to a 96-well plate. Absorbance of the triplicates at 450 nm with a reference at 650 nm was measured using an Infinite M200 microplate reader (Tecan). Background absorbance of fresh medium was subtracted.
Alkaline phosphatase staining
To determine alkaline phosphatase (AP) activity, RNA-derived iPS cells grown on 6 wells were washed with 2% FCS/PBS, fixed, and permeabilized for 1 min with Cytoperm/Cytofix solution (BD Bioscience). After 1 min of incubation, cells were washed twice with PermWash solution (BD Bioscience). For staining active AP, the AP Substrate Kit (Vector Red) was used according the manufacturer's instructions. Cells were incubated twice with the AP substrate working solution for 3 min at room temperature (RT) in the dark. Thereafter, AP substrate solution was removed and cells were covered with 96% EtOH for 3 min. Cells were washed twice with 2% FCS/PBS and analyzed by microscopy.
TRA-1-60/TRA-1-81 live staining and immunocytochemistry staining
To stain RNA-derived iPS cells for the hES cell surface markers TRA-1-60 or TRA-1-81, cells were washed and incubated with either TRA-1-60 or TRA-1-81 live staining antibody (Stemgent) at a final concentration of 2.5 ng/ml for 30 min at 37°C. After the incubation, cells were washed again and analyzed by fluorescence microscopy. For intracellular immunocytochemistry (ICC) staining, adherent cells were fixed using 4% PFA, permeabilized using 0.1% Triton X-100 and 20% Animal-Free Blocker (Vector) in PBS, and stained with the appropriate antibodies for overnight at 4°C. EPC-derived iPS cells were stained for pluripotency markers such as NANOG (Stemgent), OCT4 (Stemgent), REX1 (Stemgent), SSEA4 (Stemgent), TRA-1-60 (Stemgent), and TRA-1-81 (Stemgent) using specific antibodies. Primary EPCs were stained using antibodies against CD31 (R&D), CD144 (R&D), ULEX (Vector), or DAPI.
Teratoma formation and immunohistochemistry
EPC-derived iPS cells were cultured on 10 mm plates coated with Matrigel in NutriStem medium. iPS cells were gently dislocated using collagenase. Cell aggregates were then transferred with wide-bore pipet tips from plate/dish into 15 ml tissue culture tubes and washed with PBS with 0.5% bovine serum albumin (BSA). After spinning down gently, supernatant was aspirated. The desirable total volume was around 50 μl. 5E6 cells were injected into the dorsal flank of nude mice. Bulk fibroblast-derived iPS cell stocks were thawed and cultivated onto irradiated HFF feeder for 14 days. Harvested cells were resuspended to 1E6 cells in 100 μl NutriStem medium and mixed with 100 μl Matrigel, and the resulting 200 μl suspension was injected into the dorsal flank of nude mice (Athymic Nude-Foxn1nu; Harlan). Formed teratomas were dissected and, after calculating tumor volume, fixed in 4% PFA and embedded in paraffin. Tissue sections were stained with hematoxylin & eosin (H&E) and/or immunostained by using the following primary antibodies: αDesmin (Abcam), αAFP (Dako) and αNestin (Thermo Fisher Scientific), and secondary antibody PowerVision Poly-HRP (Immunologic). Stainings were performed using the Vector NovaRED Substrate Kit (Vector Laboratories) and analyzed by microscopy.
Intracellular flow cytometry analysis
Cells were washed with PBS, trypsinized, pelleted, and fixed and permeabilized using BD Cytoperm/Cytofix solution for 20 min at RT. Cells were stained by using specific antibodies for OCT4, SOX2, and NANOG or the respective isotype controls of the Human Pluripotent Stem Cell Transcription Factor Analysis Kit (BD Bioscience) and following the instructions of the manufacturer. Flow cytometric data were acquired and analyzed as described above for GFP. Percentage of OCT4/SOX2/NANOG-expressing and living cells and mean fluorescence intensity were determined.
Transfection efficiency analysis
To measure transfection efficiency, 2.5E4 EPCs were seeded in a 12-well format and cultured at 37°C and 5% CO2. The next day, the cells were washed once with PBS, and transfected with 0.7 μg of nonmodified GFP mRNA. After 18–24 hr of transfection, the EPCs were incubated with propidium iodide (PI) and analyzed by flow cytometry for GFP expression and cell viability.
DNA fingerprinting and karyotyping
Testing for intraspecies contamination and/or misidentification of a human cell line was done by DNA fingerprinting using short tandem repeat (STR) analysis. This was performed by Idexx RADIL in this study using the Promega CELL ID System (8 STR markers and amelogenin) and was used to verify that the genetic profile of the sample matches the known profile of the cell line (parent primary line vs. resulting iPS cell lines). Karyotyping was performed by Cell Line Genetics. The analysis can detect microscopic aberrations at a >5 Mb resolution and can detect chromosome abnormalities such as aneuploidy, inversions, duplications/deletions, and translocations.
Statistics
Data are shown as mean ± SEM and analyzed by the unpaired Student's t-test on raw data using Graph Pad Prism (Graph Pad Software).
Results
Cotransfection of E3, K3, and B18R encoding mRNAs increases mRNA translation and counteracts immunogenicity of synthetic mRNA
To investigate whether cotransfection of EKB-encoding mRNAs increases translation and combats immunogenicity of synthetic mRNA, we co-lipofected HFFs with nonmodified synthetic mRNAs coding for EKB and enhanced GFP. The synthetic mRNAs as well as all other synthetic mRNAs used in this study were thereby designed as shown in Supplementary Fig S1. As media supplementation with rB18R was shown to counteract IFN response during reprogramming with modified mRNAs, 17 B18R mRNA was substituted by rB18R in the control samples (EKrB). We observed a three-fold higher GFP mRNA translation when cells were cotransfected with E3 and K3 mRNAs, which was elevated to a 4.6-fold induction by adding B18R mRNA or rB18R (Fig. 1A). Co-lipofection with either B18R mRNA or treatment with rB18R alone did not notably enhance mRNA translation. To confirm these data and assess the kinetics of mRNA translation, we repeated the experiment with luciferase (Luc) mRNA. In all samples, translation peaked after 24 hr and was again clearly increased by EKB mRNAs or EKrB (Fig. 1B). Importantly, EKB addition massively extended Luc mRNA translation. Seventy-two hours after transfection, Luc expression was higher in samples with EKB compared with the highest expression in control samples (Fig. 1B). To measure the effect of EKB on IFN-response, we quantified transcript levels of IFNβ and 2'-5′-oligoadenylate synthetase 1 (OAS1), a prototypic IFN response gene. Induction of IFNβ and OAS1 transcripts in response to lipofection of immunogenic nonmodified mRNA was thereby used as a reference and set to 100% (Fig. 1C). We found that cotransfection with E3 and K3 mRNAs reduced IFNβ transcript levels by 80%, which was further reduced by addition of B18R mRNA or rB18R. B18R alone did not reduce IFNβ induction independently of its source. With respect to OAS1 transcripts, rB18R alone but not B18R mRNA alone inhibited upregulation. However, in combination with E3 and K3 mRNAs, B18R mRNA was as potently inhibiting OAS1 induction as rB18R (Fig. 1C). We concluded that the combination of EKB mRNAs abrogated IFN response and that, in this combination, B18R mRNA could replace rB18R. rB18R was therefore excluded from following experiments.
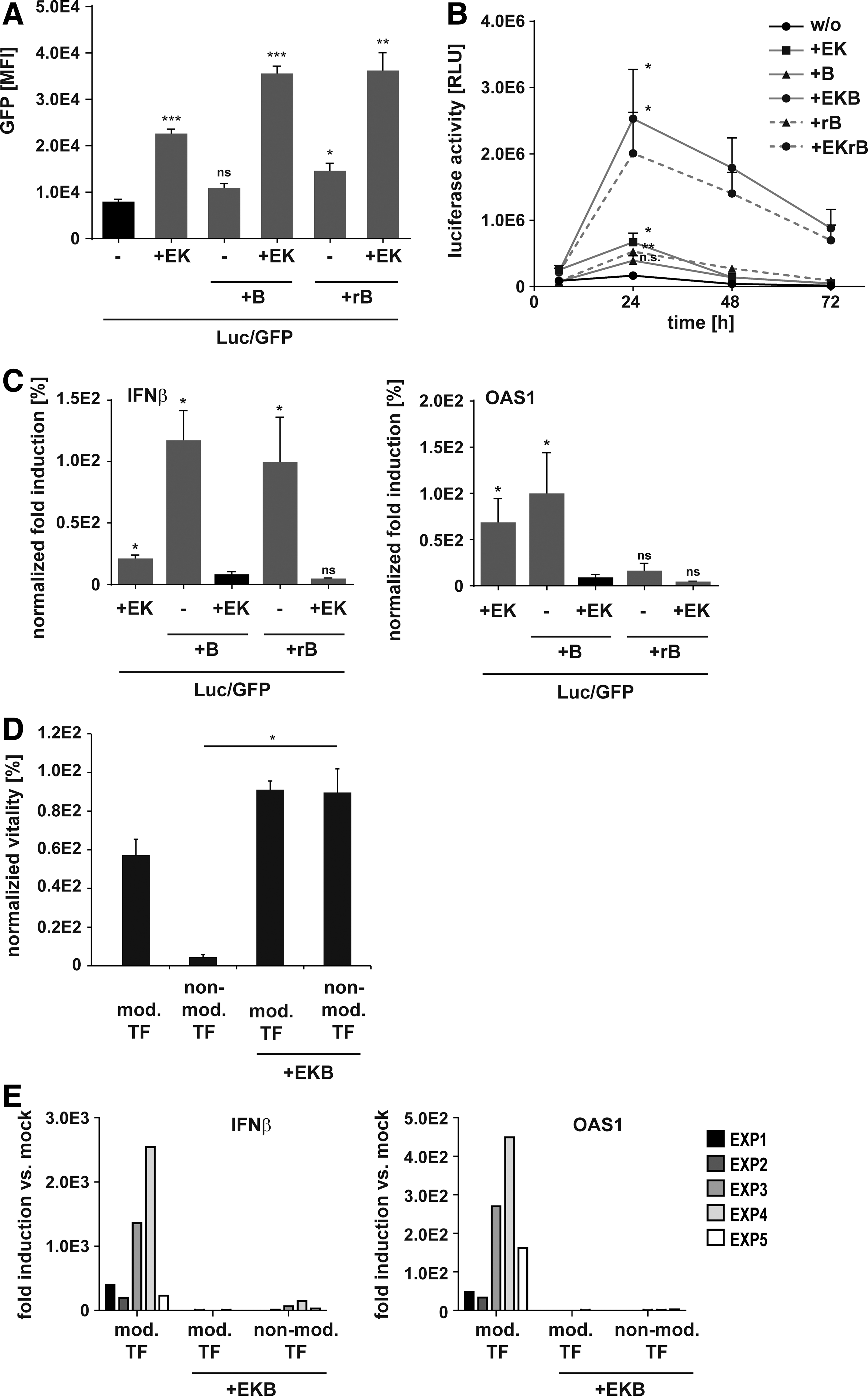
Cotransfection of E3, K3, and B18R mRNAs enhances immunogenic synthetic mRNA translation and inhibits interferon (IFN) response.
Because cell survival is indispensable for successful reprogramming, we assessed whether EKB mRNAs assure protection of cells during repeated transfection of synthetic mRNA. We used noncytotoxic liposomes to transfect HFFs daily with mRNA cocktails composed of reprogramming TF with and without EKB, all encoded by either nonmodified or modified (Ψ/5mC-modified) RNA. We found that unprotected HFFs died after four daily transfections with nonmodified TF mRNAs, while cotransfection with nonmodified EKB mRNAs rescued HFFs survival. Daily transfection of modified mRNA reduced viability by about 50%, but was also rescued by modified EKB mRNAs (Fig. 1D). Consistent with viability data, fibroblast grew to confluence only in the presence of EKB (Supplementary Fig. S2). Furthermore, analysis of transcript levels in surviving cells after four daily transfections revealed that, as seen for single transfections, cotransfection of EKB mRNAs suppressed IFN response with nonmodified mRNA cocktails (Fig. 1E). Consistent with published data, 17 Ψ/5mC-modified mRNA alone induced IFNβ and OAS1 transcript levels substantially unless EKB mRNAs were cotransfected (Fig. 1E). These data show that cotransfection of EKB mRNAs enables repetitive transfection of immunogenic synthetic mRNA and therefore promised to be suitable for mRNA-based reprogramming.
Reprogramming RNA cocktails containing nonmodified mRNA outperform modified mRNA
To evaluate the effectiveness of nonmodified mRNAs compared with modified mRNAs in reprogramming, we mixed nonmodified or Ψ/5mC-modified mRNAs encoding OCT4, SOX2, KLF4, cMYC, NANOG, and LIN28 (OSKMNL) in equimolar ratios and added EKB mRNAs either nonmodified or Ψ/5mC modified. To enhance the reprogramming capability of our cocktails, we added mature miRNAs 302a–d and 367 to the mixtures. miRNAs can boost the reprogramming process by acting synergistically with reprogramming TF 51 and lentivirally delivered miRNAs 302a–d and 367 were shown to mediate reprogramming. 11 We lipofected HFFs for 5 consecutive days, split the cells onto irradiated feeder cells, and restarted another five-day transfection period two days later. At day 12, cells were split again onto irradiated HFF feeder (refer to Fig. 2A for an overview of the experiment schedule). Splitting at days 5 and 12 was required to prevent overgrowth and detachment of the cells during reprogramming and was necessary to ensure successful reprogramming. To assess conversion to pluripotency, we quantified transcript levels of endogenous pluripotency markers at days 5 and 12. At both time points, marker expression was stronger with the nonmodified mRNA cocktail (Fig. 2B). Colony growth started on day 10 and, after splitting at day 12, colonies re-formed after day 16. On day 19, the nonmodified mRNA cocktail resulted in overgrowth of the plate by alkaline phosphatase-positive (AP+) colonies, while none or only few AP+ colonies had developed with Ψ/5mC-modified mRNA cocktail (Fig. 2C). We found that TF and reporter gene translation in the presence of EKB mRNA was higher in samples transfected with nonmodified RNAs compared with samples transfected with Ψ/5mC-modified mRNA, while both RNA types were equally well transfected into cells (Supplementary Fig. S3). This indicates that a more efficient reprogramming could be related to increased translational efficiency or higher mRNA stability of nonmodified mRNA.

E3, K3, and B18R mRNAs enable reprogramming with nonmodified mRNA.
Following our experimental setup, transfection of neither miRNAs 302a–d and 367 nor mRNA alone resulted in the formation of colonies (data not shown). To further assess pluripotency of colonies obtained with the nonmodified mRNA cocktail, we analyzed upregulation of endogenous pluripotency transcripts on day 19 by qRT-PCR. All tested transcripts were greatly upregulated in comparison to mock transfected HFFs in three independent experiments (Fig. 2D). Colony morphology was hES cell-like with tightly packed small cells in distinct colonies and well-defined borders (Fig. 2E, F). Colonies stained positive for AP (Fig. 2E) and the hES cell surface marker TRA-1-60 (Fig. 2F) and could readily be passaged. Repeated AP and TRA-1-60 stainings at day 26 after additional passaging and the persistence of hES cell-like morphology proved the stability of pluripotency (Fig. 2E, F). To test the capability of RNA-derived iPS cells to differentiate into tissue of all three germ layers in vivo, we analyzed teratoma formation in immunodeficient (nude) mice. Teratomas grown from 1E6 cultivated RNA-derived iPS cells for six weeks were stained positive for the mesodermal marker Desmin, endodermal marker α-1-fetoprotein (AFP), and ectodermal marker Nestin, which indicates that HFFs were fully reprogrammed to iPS cells (Fig. 2G). We concluded that cotransfection of EKB mRNA enables efficient reprogramming with immunogenic nonmodified RNA.
Four transfections are sufficient for a robust feeder-free reprogramming of human fibroblasts
To further simplify the reprogramming process and to investigate the robustness of the reprogramming protocol using nonmodified mRNA cocktails, we determined the minimal number of daily transfections required for reprogramming (refer to transfection schedule in Fig. 3A). Three transfections resulted already in a few colonies, whereas four to six transfections resulted in robust colony formation (Fig. 3B). Colonies started to form on day 9 and were fully developed and TRA-1-60 positive on day 11 independently of the number of transfections (Fig. 3C). Using this short transfection period, we could even neglect the use of feeder cells. Consistent with the colony formation, hES cell marker inductions were first observed upon three lipofections, while a more robust marker induction required four transfections. Five or six transfections resulted in no additional increase (Fig. 3D). We further analyzed the use of this very short and feeder-free reprogramming protocol for the human newborn foreskin fibroblast cell line NuFF and the adult human fibroblast line HDFa (Supplementary Fig. S4). Interestingly, while being irrelevant for HFFs, a 3-fold excess of OCT4 was needed to reliably reprogram the NuFFs, whereas the HDFa yielded a high efficiency of 1.0–2.0% but only if the seeding density was reduced by 50% compared with the original protocol (Supplementary Fig. S5). Because of this outcome, further improvements were made to the original protocol such as titrating the seeding density and using nonconditioned NutriStem medium. Based on these initial experiments, the combination of excess OCT4 in the reprogramming cocktail, a low seeding density, and using NutriStem medium yielded the highest number of TRA-1-60-positive iPS cell colonies from HDFa (3.0%), which is in part in line with previous studies in fibroblasts using retroviruses. 52 Consequently, only four transfections of our nonmodified mRNA cocktail are required to efficiently reprogram neonatal and adult fibroblasts (for summary see Supplementary Fig. S5).

Development of a minimal robust reprogramming protocol with nonmodified synthetic mRNA and E3, K3, and B18R.
Feeder-free reprogramming of human blood-outgrowth EPCs requires only eight daily transfections
As outlined in the Introduction, blood cells are a highly desirable source for personalized iPS cells. So far it was not possible to efficiently and repeatedly deliver mRNA to cells originating from blood without inducing cytotoxicity. The effectiveness of our reprogramming protocol in different fibroblast lines motivated us to investigate whether also EPCs derived from human blood could be successfully reprogrammed using the same RNA mixture. Circulating EPCs are a rare population in peripheral blood and cord blood, but by using a modified protocol that was recently published, 25 we were able to effectively isolate and establish multiple adherent and expandable EPC primary cultures, some from as little as 1E7 MNCs (Supplementary Fig. S6). Additionally, transfection optimization studies using flow cytometry revealed that nonmodified mRNAs could be efficiently delivered to peripheral blood-derived EPCs (PB-EPCs) without inducing cellular toxicity (Supplementary Fig. S7). PB-EPCs exhibited the typical cobblestone morphology and coexpression of specific markers could be shown by ICC (Supplementary Fig. S8).
To reprogram PB-EPCs, eight daily transfections were necessary using the reprogramming cocktail containing nonmodified mRNAs coding for OSKMNL and EKB as well as miRNAs 302a–d and 367 while cultured in EPC medium (Fig. 4A). On day 5, morphology of PB-EPCs already started to change and rapidly progressed further on day 7. The medium was transitioned to NutriStem medium on day 10, while colonies exhibited morphologies similar to human hES cells and were positively stained for TRA-1-81 (Fig. 4B). By day 15 all nonreprogrammed PB-EPCs died off because of the selective environment of the medium, and therefore only fully reprogrammed PB-EPCs remained and could be picked and expanded under feeder-free conditions (Fig. 4B). Efficiency of reprogramming was calculated to as high as 0.25% of input cells. Again, as seen for NuFFs and HDFa, a threefold excess of OCT4 in the reprogramming cocktail seems to enhance efficiency and robustness of reprogramming (Supplementary Fig. S9). Further, we wanted to demonstrate that our nonmodified RNA reprogramming technology can be extended to endothelial cells isolated from different origins.
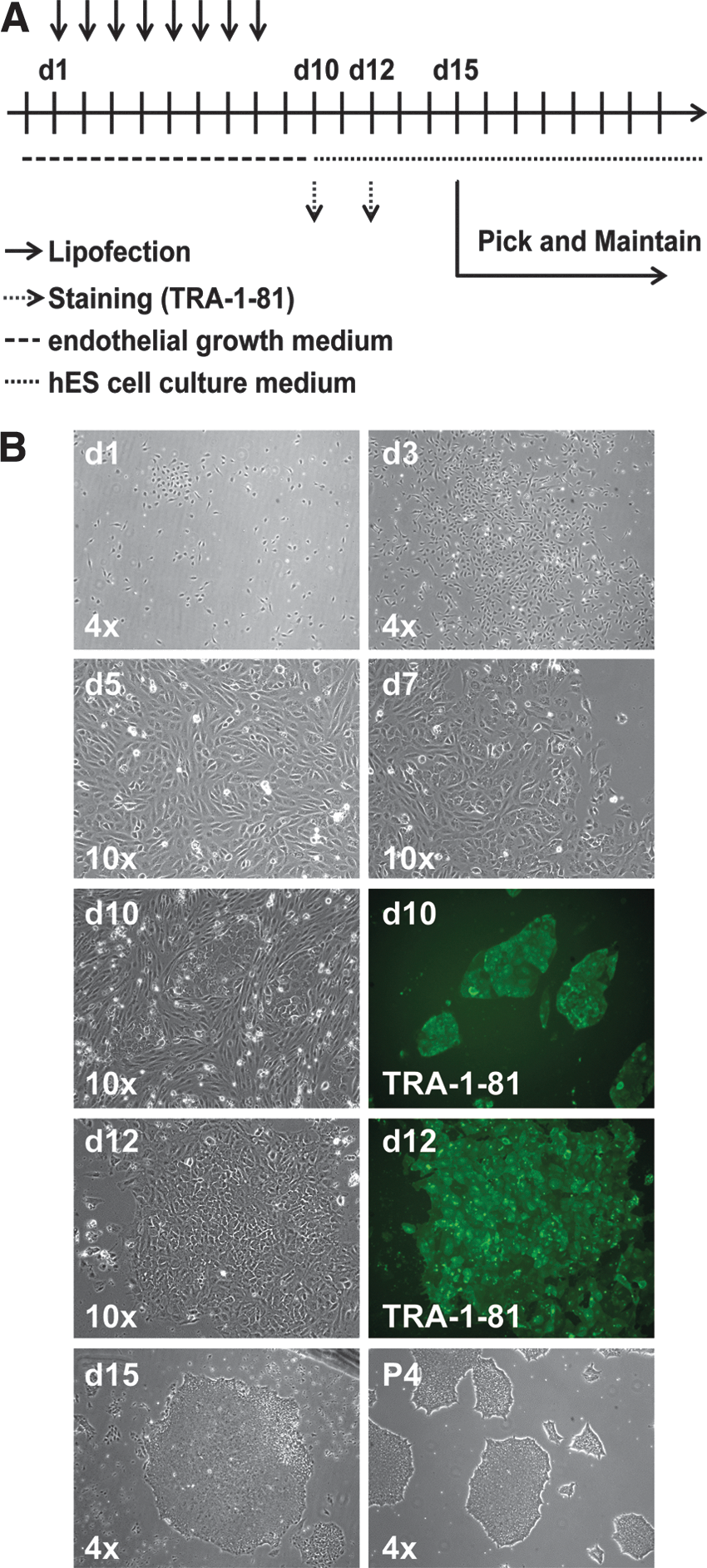
Human EPC reprogramming timeline and morphology progression using nonmodified synthetic mRNAs.
Indeed, by using the same reprogramming protocol as used for PB-EPCs, HUVECs and cord blood-derived EPCs (CB-EPCs) could be successfully reprogrammed. HUVECs required six transfections resulting in reprogramming efficiencies of TRA-1-60-positive iPS cells up to 3%, while CB-EPC required six transfections to achieve a frequency of 0.002% and extending it to eight transfections achieved an increased reprogramming efficiency of 0.016% (Supplementary Fig. S10). EPC-derived iPS cell cultures stayed stable and clean of differentiated cells when continued to culture in xeno-free hES cell medium (Fig. 4B and data not shown). Characterization of PB-EPC-derived iPS cells showed that they express key pluripotency markers such as OCT4, NANOG, SSEA-4, REX-1, TRA-1-81, and TRA-1-60 (Fig. 5A). To further assay their pluripotent potential, we injected two different PB-EPC-derived iPS cell lines into immune compromised mice and after 10 weeks teratoma were formed. Differentiation to tissue of all three germ layers was visualized by H&E staining (Fig. 5B). Karyotype analysis as well as cell identity determination of PB-EPC-derived iPS cell lines (>13 passages) were normal as demonstrated for the parental primary PB-EPC line (Fig. 5C and data not shown). Taken together, our data show that nonmodified reprogramming mRNA leads to a fast-paced and high-quality reprogramming of blood-derived EPCs.

Characterization of PB-EPC-derived iPS cells.
Discussion
For the clinical application of iPS cells, it is crucial to have reproducible, robust, yet fast derivation methods, which do not employ viral or DNA-based delivery vectors. The RNA reprogramming technology presented in this article is able to convert both human adult fibroblasts (four transfections) and blood-derived EPCs (six to eight transfections) into iPS cells within two weeks. Given the undoubtedly superior availability of patient blood cells to any other patient cell type, our technology represents a significant advancement toward iPS cell generation for cell-based therapies.
Rapid reprogramming of these cell types was achieved by cotransfection of a potent synthetic mRNA/miRNA cocktail that promotes reprogramming through increased translation of nonmodified mRNAs while thoroughly inhibiting cytotoxicity. The approach overcomes immunogenic hurdles inherent to mRNA-mediated reprogramming as previously shown in human fibroblasts 36,38 by the use of three VACV immune evasion proteins (EKB). The idea to suppress the innate immune response in order to allow repetitive transfer and expression of RNA encoding reprogramming TF was first described by Angel and Yanik. 36 Others have addressed RNA immunogenicity and cytotoxicity as well. siRNA knockdown of key cellular defense factors enabled repeated nonmodified RNA transfections but reprogramming remained ineffective. 36 The pace and quality of reprogramming with our methods indicate that blocking innate immunity with viral proteins is more effective than siRNA-mediated knockdown, which could relate to more pleiotropic effects of the VACV proteins compared with target-specific siRNAs. Besides siRNA, the use of modified mRNA to reduce immunogenicity of mRNA has become a well-established reprogramming technology for human fibroblasts 16 –18,34,53,54 and also enabled the generation of iPS cells from adipose tissue. 55 This modified RNA reprogramming additionally requires media supplementation with rB18R to protect cells from residual secreted IFN and at best a minimum of 8–12 daily transfections. 16,18
To circumvent the addition of recombinant proteins in our reprogramming approach, we replaced rB18R with B18R mRNA. Although rB18R alone compared with B18R mRNA showed a more efficient downregulation of OAS1 transcripts, rB18R and B18R mRNA were similarly effective in combination with E3 and K3 mRNAs. The better performance of rB18R to B18R mRNA as a single agent is probably because of the immediate protective effect of the recombinant protein supplement. Modified RNA reprogramming was described faster than all other nonintegrating reprogramming protocols ranging from 15 days for miRNA 12 to 30 days for protein transduction 14 till the formation of iPS cell colonies. However, modified mRNA failed to reprogram fibroblast cells in our protocol, unless they were combined with EKB-coding mRNA and stem cell-specific miRNAs. This indicates that the reduced number of transfections and short timeline of our protocol result from the incorporation of the very potent although immunogenic nonmodified RNAs.
One possible explanation for the greater overall performance using nonmodified mRNA compared with less immunogenic modified mRNAs in our experimental setup could be the higher translational efficiency or stability of mRNA composed of conventional nucleosides. This is in line with the observation that reporter gene and TF expression in presence of EKB was higher when encoded on nonmodified than Ψ/5mC-modified mRNA. However, more efficient reprogramming by nonmodified RNA was probably not only related to increased TF expression, but—despite EKB—also to stronger Toll-like receptor (TLR)-mediated signaling in cells transfected with nonmodified mRNA. 56 TLR signaling was previously described as being beneficial for reprogramming 35 and is thought to promote epigenetic changes necessary during reprogramming. Cells detect synthetic RNA with constitutively expressed sensor molecules; therefore, it is conceivable that the first transfection of nonmodified mRNA activates innate immune signaling until effective levels of EKB proteins are reached. This initial innate immune response may prime the target cells for reprogramming without being cytotoxic.
Additionally, our reprogramming cocktail contained miRNA cluster 302/367, which proved essential for successful reprogramming with the nonmodified mRNA cocktail. Both RNA species therefore seem to cooperate synergistically, which is in line with publications emphasizing the role of miRNAs in the generation of iPS cells and their concerted action together with reprogramming TFs. 51,57 –59
Fibroblast isolation from adults requires at least small surgical intervention like skin punch biopsies, which limits their availability as a personalized source for patient- and disease-specific iPS cells for research and therapy. The establishment of a robust, integration-free reprogramming protocol for the generation of blood-derived iPS cells shown here for the first time is a pivotal step toward availability of clinically applicable iPS cells. Viral- and DNA-based reprogramming methods have been applied to blood cell types but were inefficient, and reprogramming using integration-free RNA technologies was not reported successful so far. In this study we successfully achieved reprogramming of blood-derived EPCs using the same nonmodified synthetic mRNA/miRNA cocktail that proved superior in fibroblast reprogramming compared with the modified RNA cocktail. The properties of our reprogramming cocktail with respect to innate immune inhibition were therefore pivotal for successful reprogramming of PB-EPCs and only eight transfections resulted in the efficient generation of TRA-1-81-positive PB-EPC-derived iPS cell colonies in as few as 10 days. By using either fresh or frozen endothelial cells isolated from different sources such as CB-EPCs and HUVECs, we were able to reduce the number of needed transfection possibly because of their more immature state compared with PB-EPCs.
The high reprogramming efficiency of HUVECs may be because of their higher proliferative rate compared with the much slower growing CB-EPC line and is in line with previous publications showing that the proliferation potential of a cell directly influences its reprogramming capabilities. 60,61 Nevertheless, six transfections seem to be the limiting number required for CB-EPCs to reprogram, whereas the limiting number for multiple PB-EPC lines is eight possibly because of their more mature state and slower growth rate compared with CB-EPCs. However, in terms of efficiency, we only reprogrammed one CB-EPC line in this study and therefore donor variability or cell intrinsic properties could also be responsible for these differences and therefore require further investigation. Remarkably, blood EPCs present thereby a unique source of cells for derivation of clinical-grade iPS cells using RNA reprogramming technologies. Subsequent differentiation to hematopoietic stem cells (HSCs) would be a therapeutic opportunity to treat myeloproliferative hematopoietic malignancies in which the disease-causing somatic mutations are restricted to hematopoietic lineages. 62,63 HSCs derived from RNA-reprogrammed iPS cells could replace allogeneic bone marrow transplantations, thereby alleviating the risk of graft-versus-host disease while providing genomically uncompromised and healthy HSCs. In regard to clinical applications, it should be mentioned that all active substances used in our protocol are RNAs, which can ease clinical translation because only one class of biological—RNA—has to be produced in clinical grade and used throughout. Nevertheless, the genomic integrity and stability of RNA-reprogrammed EPC-derived iPS cell lines should further be investigated.
With our approach we were able to significantly shorten the transfection period to generate iPS cells with RNA. However, further shortening to a single RNA transfection is desirable. This was achieved with alphaviral self-replicating RNA, 15 but although persistent replication of alphaviral RNA is unlikely because of cellular antiviral defense, clinical-grade iPS cells have to be proven free of residual contamination with self-replicating vectors. In this regard, mRNA is undoubtedly safe and a single transfection reprogramming using mRNA seems also attainable because we achieved low-efficient reprogramming with only three transfections. Future efforts should therefore focus on further stabilization of mRNA vectors to increase half-life and translational efficiency or the development of transfection reagents providing more sustained delivery. Optimization of TF mRNA stoichiometry, addition of novel reprogramming factors, 64 –67 or removal of recently identified inhibitors of reprogramming with cotransfected siRNA 68 can be easily tested with our protocol and may lead to a single transfection protocol.
As stated in the result section, the original protocol could be further improved across other fibroblast cell lines by adjusting mRNA molar stoichiometries to elevate OCT4 transcript levels in the RNA mixture and titrating cell seeding densities. There are multiple conditions that can be used to reprogram different fibroblast and endothelial cell sources although the range of the resulting iPS cell colonies reaches from a few to more than thousands. To present one common reprogramming protocol that works well across different fibroblasts lines and endothelial cells, we would recommend titrating the starting cell density for each cell line using a 3-fold excess of OCT4 (3:1:1:1:1:1) and only NutriStem medium throughout the experiment (there is an initial EPC medium step before transitioning to NutriStem in case of EPC reprogramming) (for a summary see Supplementary Fig. S9).
Overall, our study shows that the use of nonmodified RNA in combination with the inhibition of IFN response by mRNA-encoded viral factor EKB is a powerful RNA technology to enhance in general RNA-based transfer of genetic information. Together with stem cell-specific miRNA, it leads to RNA-based reprogramming that allows for astonishingly simple and fast generation of integration-free iPS cells from fibroblasts and from even harder to reprogram cells such as blood-derived EPCs.
Footnotes
Acknowledgments
This work was grant supported by the German Federal Ministry of Education and Research (BMBF; Support Code 01 GN 2004). Responsibility for the content of this publication rests with the authors. We would like to thank Jennifer Groß and Drs. Anne Kölsch, Astrid Spruß, and Stefan Jacobs for their excellent technical assistance and advice. Further we like to thank the RNA synthesis unit of TRON for providing synthetic mRNAs. We also like to thank Dr. Amer Rana and Chris Huang at the University of Cambridge for the assistance in establishing EPC derivation and culture techniques in Stemgent labs.
Author Disclosure
M.A.P., A.B., and U.S. are employees of BioNTech RNA Pharmaceuticals GmbH, a company that develops mRNA-based therapeutics. In addition, M.A.P., T.B., S.H., Ö.T., and U.S. are inventors on patent applications that cover optimization of RNA-based therapeutics and RNA-based reprogramming techniques. B.H., S.E., J.M., and K.Y. are in the research and development program at Stemgent Inc. in Cambridge, MA. The remaining authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.