
Abstract
Chimeric antigen receptor (CAR) T-cells have shown remarkable results in patients with B-cell leukemia and lymphoma. However, while CAR T-cells have shown complete responses in a majority of patients with acute lymphoblastic leukemia (ALL), lymphomas are more difficult to treat. Different CAR designs and conditioning protocols seem to affect the persistence of patient responses. However, factors that determine if patients receiving the same CARs will respond or not remain obscure. In Sweden, a phase I/IIa trial using third-generation CAR T-cells is ongoing in which we intend to compare tumor biology and immunology, in each patient, to treatment response. CAR T-cell therapy is a powerful tool to add to the treatment options for this patient group but we need to perform the necessary basic research on the multifactorial mechanisms of action to give patients the best possible option of survival. Such studies are also crucial to expand the success of CAR T-cells beyond CD19+ B-cell malignancy. This review will focus on possible barriers of treating lymphoma to define factors that need to be investigated to develop the next generation of CAR T-cell therapy.
Introduction
C
B-cell malignancy is a heterogeneous indication with both solid lesions and circulating cells in blood and bone marrow. Treatment of B-cell malignancy using CAR T-cells presents a unique opportunity to learn mechanisms of action of different CAR designs, to define on and off target toxicity, as well as to understand the limitations of CAR T-cells in terms of sensitivity to immune escape mechanisms and physical barriers of solid tumors.
B-cell Malignancy
B-cell malignancy encompasses a heterogeneous group of cancers derived from B-cells of different differentiation stages. For example, pre-B acute lymphoblastic leukemia (pre-B-ALL) derives from progenitor cells at the pre-B-cell developmental phase in the bone marrow, while diffuse large B-cell lymphoma (DLBCL) derives from B-cells present in the germinal centers of lymphoid tissues. 2 Further, chronic lymphocytic leukemia (CLL) has a mature B-cell phenotype and tumor cells are present in blood, bone marrow, and lymphoid tissues. Nevertheless, they all have in common that they are derived from B-cells and share a few common B-cell linage markers that can be used for targeted therapy. For example, CD20 is expressed on mature B-cells and the CD20-targeting antibody rituximab is currently used together with chemotherapy regimens for CD20+ malignancies. Another linage marker on B-cells is CD19. CD19 is expressed already from the progenitor B-cells to mature B-cells, and to some extent on healthy, but unfortunately not on malignant, plasma cells. Clinical trials using CD19-targeting CAR T-cells have demonstrated remarkable results, mostly in ALL patients but lately also in lymphomas. 3 –5 Another B-cell target is the membrane-bound antibody, and CAR T-cells are being developed that target either the Ig kappa or the lambda chain. 6
B-cell leukemia and lymphoma respond differently to treatment. 7 ALL has rapid progression and can be cured by chemotherapy but patients that relapse or are refractory to chemotherapy have dismal prognosis. For refractory ALL, allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative option, but relapse after HSCT has so far been uncurable. 8 CLL is a slowly progressing chronic disease with varying clinical course and varying response to chemotherapy. For patients with refractory CLL, there are now a new set of signaling inhibitors that target the PI3Kδ and the Bruton's tyrosine kinase (BTK) that inhibits the B-cell receptor-driven proliferation in CLL. 9 DLBCL is an aggressive lymphoma and is initially treated with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). DLBCL commonly responds well, and about 60% of the patients can be cured with R-CHOP. Relapsing patients show increased resistance but may still respond to high-dose chemotherapy and autologous HSCT. 10
Immunotherapy has shown great effect in cancer patients and for B-cell malignancy, and genetically engineered T-cells expressing a CD19-targeting CAR receptor have shown spectacular results during the past few years. 3 –5 T-cells are excellent serial killers that under the right conditions can expand, survive, and kill tumor cells. Furthermore, they can maintain responses if they survive in vivo as effector memory cells in contrast to, for example, antibody-based targeted therapies such as rituximab that do not induce tumor immunity. Further, one single antibody will only have the potential to bind to one tumor cell and induce antibody-mediated cytotoxicity to kill the tumor cell, while CAR T-cells proliferate in vivo and will go from tumor to tumor with sustained cytotoxic activity. Nevertheless, some trials have shown complete responses and other transient partial responses. 11,12 There are many discrepancies that make it difficult to directly compare the published results such as the use of CAR T-cells with different designs, various preconditioning strategies, and the selection of patients (e.g., leukemia versus lymphoma).
Car T-cell Design
The CAR molecule consists of an antigen-recognizing extracellular domain and an intracellular signaling domain. The extracellular portion is typically an antibody single-chain fragment (scFv) directed against a cell surface antigen, while the intracellular domain consists of merged signaling domains from the TcR complex and costimulatory proteins (Fig. 1). T-cell activation is controlled by multiple signaling cascades induced by antigen-presenting cells. For full activation, T-cells need signals from antigen recognition through TcR stimulation in combination with costimulation via a range of proteins such as CD28, CD27, and 4-1BB. The first-generation CAR T-cells mimicked only TcR stimulation by combining a tumor-targeting scFv to the zeta (ζ) chain of the TcR CD3 complex, which allows T-cells to recognize and kill tumor cells in vitro but in vivo persistence was lacking. 1 The second-generation CAR includes a signaling domain from a costimulatory molecule. Costimulatory signaling provides the T-cell with, for example, a better proliferative capacity or increased cytokine production depending on which costimulator that is fused to the CAR. Second-generation CAR T-cells with CD28 as a costimulator were less sensitive to Tregs and their suppressive molecules IL10 and TGFβ. 13 Such CARs have shown effective in ALL but in vivo persistence can be improved for lymphoma. 5,13 The CAR T-cells developed by the University of Pennsylvania included the 4-1BB molecule as a costimulator instead of CD28. This has shown to give an important survival and expansion signal to the CAR T-cells and may explain the persistent responses in their patients. 3 In subsequent trials, initial complete responses were seen in about 90% of the ALL patients and the majority had sustained responses. 11 CAR T-cells have also shown to be effective in different lymphomas but only if the patients are treated with aggressive doses of chemotherapy aiming to reduce regulatory T-cells (Tregs) and myeloid-derived suppressor cells (MDSCs) before infusion. These are immunosuppressive cells that otherwise hamper the function of CAR T-cells. In a study performed by NIH using CD28-containing second-generation CAR T-cells, it was recently shown that the high-dose conditioning required for complete responses resulted in serious toxicity of the lymphoma patients. 5 It is not understood why ALL patients respond much better than lymphoma patients and if other types of CAR T-cell designs may be more favorable for lymphomas.
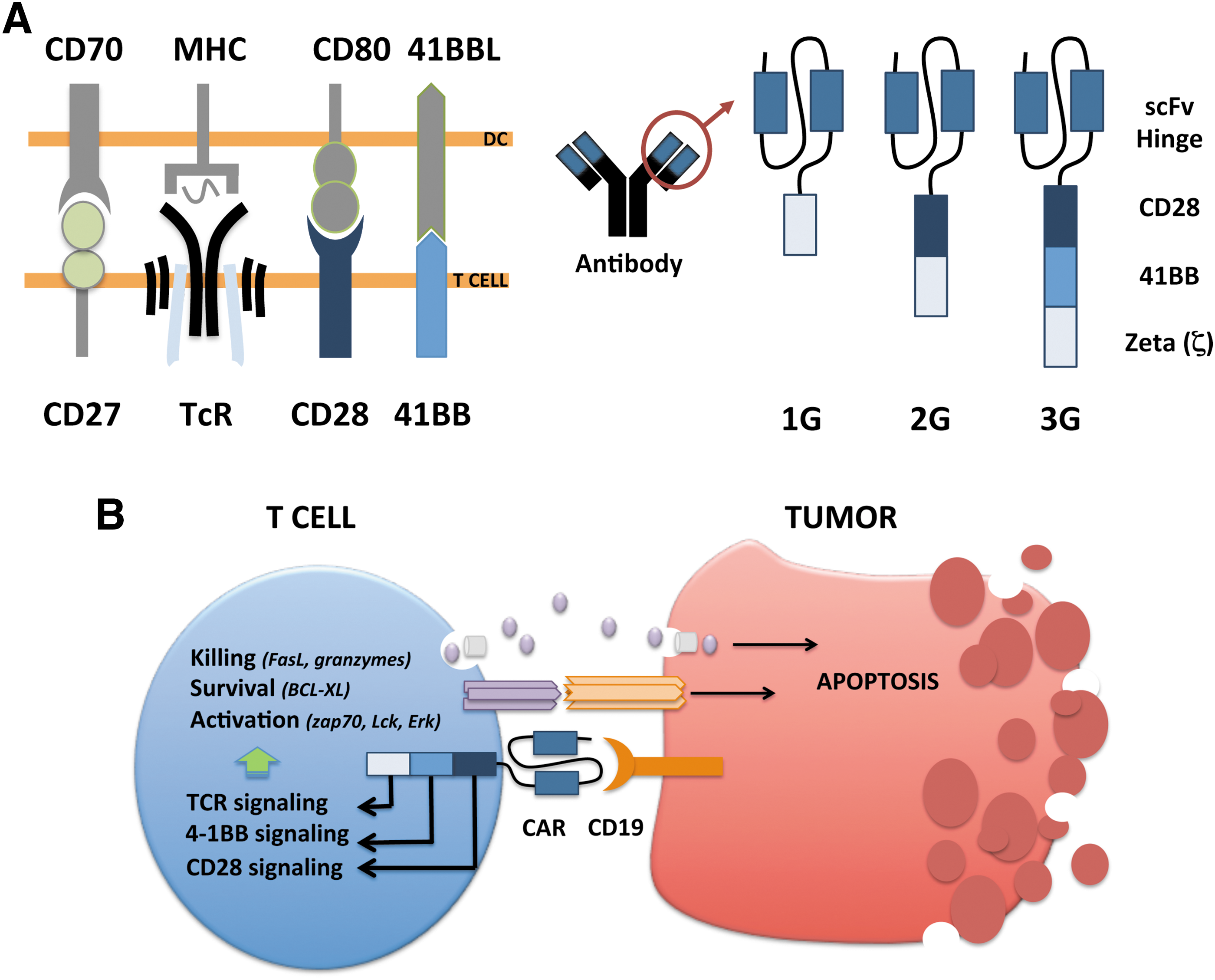
Experimental studies have shown that T-cells stimulated via both CD28 and 4-1BB have greater antitumor activity and longer in vivo persistence than T-cells stimulated by either moiety alone, suggesting that these two molecules have synergistic effects. 14,15 At Uppsala University in Sweden we are currently investigating the clinical capacity of such third-generation CAR T-cells (Fig. 1). We have seen responses in both ALL and lymphoma but the latter is more resistant to therapy, and we are currently investigating biopsies from lymphoma lesions to elucidate mechanisms of resistance that can aid future CAR design or define necessary combination drugs. There are three reasonable explanations to why CAR T-cell therapy is not as effective for lymphoma as for leukemia that need further investigation. First, lymphomas are solid tumors that can provide physical barriers for the CAR T-cell to come in close contact with the tumor cells. Second, regulatory immune cells and inhibitory proteins are concentrated in the vicinity of the tumor. Hence, a solid tumor may have a higher level of immunosuppression at the tumor site to shield the tumor from CAR T-cells. Third, CAR T-cells may lack homing receptors to enter solid tumors. Activated effector T-cells are prone to migrate to the circulation rather than returning to lymphoid tissues, which may further complicate clearance of tumor cells present in lymph nodes.
Physical Barriers of Car T-cell Infiltration
It is known that T-cells are present in most tumors and that the number of tumor-infiltrating T-cells is correlated to a positive overall survival. 16 However, when investigating the localization of the T-cells within a tumor lesion, it is evident that most T-cells remain in the tumor stroma and only few cells, or none, have infiltrated into the parenchyma. Several factors may be restricting infiltration into the parenchyma. For example, blood vessels are dysfunctional in tumor lesions and may not express the necessary receptors for T-cell attachment, rolling, and diapedesis such as ICAM-1, VCAM-1, and P/E-selectins. It may also be the T-cells that lack their counterpart receptors such as the integrins α4β1 (VLA-4) and LFA-1, but also PSGL-1 and CD43 to migrate into the parenchyma. 17 Endothelial cells can be activated via CD40 to upregulate receptors for attachment, which may aid T-cell transmigration. 18 A combination of CD40-targeted therapies may therefore be of interest for CAR T-cell therapy of lymphoma. Moreover, CD40 stimulation on dendritic cells (DCs) provides Th1-mediated immunity, which would further complement CAR T-cell therapy. 19 In fact, Curran et al. recently published CAR T-cells that express recombinant CD40 ligand (CD40L) aiming to support the T-cell survival in the tumor milieu. 20
Expression of CD40L may also enhance T-cell infiltration into the tumor if it activates the endothelial cells. However, in a mouse model, constitutive CD40L expression in lymphoid cells led to a lymphoproliferative disorder and the in vivo use of CAR T-cells expressing CD40L needs to be evaluated with care. 21 CAR T-cells may also be engineered to express chemokine receptors to facilitate homing to the tumor. For example, the tumor often releases CCL2 and CXCL5, which aid recruitment of macrophages, MDSCs, and neutrophils to the tumor. 22 By overexpressing the CCR2 or CCR4 on CAR T-cells, they may better home to tumor lesions. Nevertheless, lymphoma situated in lymph nodes may be targeted by other means. Naïve T-cells in blood have access to lymph nodes via specialized high endothelial venules (HEVs) but for effector cells this process is far less efficient. Instead, effector and memory cells enter lymph nodes via the lymphatics. 23 T-cells enter the lymphatics via HEVs and binding to selectins is crucial. The chemokine CCR7 is important to home T-cells to the lymph nodes and CCR7 is expressed on naïve and memory T-cell populations, while effector cells usually lack CCR7. 24 Hence, protocols that expand CAR T-cells with an effector/memory phenotype with CCR7 expression will preserve their capacity to migrate into lymph nodes. 25 Nevertheless, the T-cells can lose CCR7 expression in vivo and CCR7 expressed in trans with CAR may be needed to maintain access to lymph nodes.
Tumor cells and tumor stroma, such as M2 macrophages, often express VEGF to stimulate angiogenesis 26 and this holds true also for lymphoma. 27 It has been demonstrated that VEGF prevents T-cell infiltration into tumors and VEGF blockade by the tyrosine kinase inhibitor (TKI) sunitinib upregulated chemokines, which was followed by an increased T-cell infiltration. 28 Sunitinib is also known to inhibit MDSCs in renal cell carcinoma, 29 which would further potentiate CAR T-cell therapy if used in combination settings. However, the effect of sunitinib on T-cells needs further analysis since many signaling pathways downstream of the TcR complex and costimulatory molecules may depend on tyrosine kinases.
Physical barriers could also entail the extracellular matrix in solid tumors that may impede T-cell penetration, especially in tumors with a dense nature, for example, because of collagen-producing fibroblasts. 30 Interestingly, the expression levels of genes associated with remodeling of extracellular matrix and inflammatory responses were higher in DLBCL patients who were later cured by chemotherapy compared with the expression levels in nonresponders. For example, the matrix metalloproteinase-12 produced by macrophages was increased in the patients who were later cured and these patients also had a higher number of infiltrating T-cells. 31 In an elegant study by Caruana et al., CAR T-cells engineered to constitutively express heparanase showed better capacity to infiltrate tumors and an improved overall survival in a mouse model. Collectively, the data demonstrated that physical barriers may be circumvented by appropriate CAR T-cell design. 32
The Hostile Tumor Microenvironment
Immunosuppression as a means to escape antitumor immune responses is the most difficult obstacle for effective immunotherapy. The tumor can release inhibitory substances like TGFβ and IL10 that directly hamper T-cell proliferation and cytotoxic function. These substances can also inhibit antigen-presenting cells, leading to hampered activation of tumor-reactive T-cells. Further, TGFβ plays a crucial role to drive differentiation of naïve T-cells into Tregs and IL10 promotes differentiation of M2 macrophages. Tregs will then contribute with more TGFβ, IL10, and also other suppressive agents like IL35 and adenosine. 33 M2 macrophages are anti-inflammatory, proangiogenic, and protumorigenic.
Besides the production of IL10 and TGFβ, M2 macrophages produce CCL22, which attracts CCR4+ Tregs. Further, they express PDL1 and can inhibit activated PD1+ T-cells. 34 The tumor also produces prostaglandin E2, which drives expansion of MDSCs. MDSCs are immature myeloid cells at different differentiation stages and these cells also have suppressive capacity. 35 They have many inhibitory mechanisms; for example, they release arginase-1 and upregulate nitric oxide synthase 2, both involved in the metabolism of L-arginine. T-cells deprived of L-arginine reduce CD3ζ and lose their proliferative capacity. 36 Tregs, MDSCs, and M2 macrophages can together create a very hostile milieu for T-cells, which is concentrated to the tumor lesions but affect the whole patient. 33 In CLL, immunosuppression occurs early in disease with malfunction of T-cells, natural killer (NK) cells, and monocytes. 37 Lymphoma lesions are infiltrated with CD163+ M2 macrophages and their presence is associated with worse prognosis. 38,39 The presence of MDSCs is correlated to a poor overall survival. 40 However, the role of Tregs in B-cell malignancy is contradictive and has even been associated to a better prognosis. 41
Tregs may have a dual role in tumors derived from immune cells since their main function is to suppress such cells. Hence, while suppressing antitumor immune responses, Tregs may as well suppress the tumor. In previous work, we demonstrated that Tregs in patients with B-cell leukemia or lymphoma had increased levels of Tregs and that their Tregs expressed cytolytic markers. In vitro, these Tregs could kill B-cell tumors. 42 Nevertheless, the net outcome of all suppressive cells and cytokines in B-cell lymphoma is an immunosuppressive environment that will hamper T-cell efficacy. In a pilot study we have demonstrated that Tregs are elevated in CLL and DLBCL, while the pediatric ALL patients had similar Treg levels as age-matched controls. 42 However, a high level of TGFβ has been associated to high-risk ALL. 43 Nevertheless, these data support a mechanism that explains why this patient group seems to be the best responder to CAR T-cell therapy.
Conditioning of Patients Receiving Car T-cell Therapy
CAR T-cell therapy has limited effect if the patients do not receive preconditioning therapy. Preconditioning chemotherapy is often given to patients receiving immunotherapy to decrease Tregs and MDSCs that may otherwise hinder the intended immune activation. Further, chemotherapy-induced lymphocyte or myeloid cell depletion may induce bone marrow cytokine production that restores the immune cell populations and favors the activation of antitumor responses. The most commonly used protocol was developed at NIH when using fludarabine and cyclophosphamide before infusion of in vitro–expanded melanoma-specific T-cells. 44 This protocol is also used before infusion of CAR T-cells 5 but because of the toxicity, other regimens have been used as well depending on the indication. 4,5 Metronomic cyclophosphamide has been given to patients undergoing immunotherapy in an attempt to control suppressive immune cells over time. 45 Such supportive chemotherapy protocols may be of great value if they do not hamper the desired antitumor responses. One such supportive chemotherapy of interest may be gemcitabine. Gemcitabine is a nucleoside analog that replaces cytidine during DNA replication, which leads to growth arrest and apoptosis. Gemcitabine also targets ribonucleotide reductase, thereby blocking the function of this enzyme. Several studies have shown that patients treated with gemcitabine had significantly lower levels of the immunosuppressive molecule TGFβ, Tregs, and MDSCs but an increased number of DCs, monocytes, and activated T-cells. 46,47
CLL is commonly treated with fludarabine, bendamustine, and lenalidomide. 48 While both fludarabine and bendamustine are immunosuppressive per se, lenalidomide enhances the degradation of Ikaros 1 and 3. Since Ikaros 1 represses production of IL2, its inhibition will release IL2 production and enhance the function of both T and NK cells. 49 Interestingly, lenalidomide inhibits Tregs. 50 Hence, lenalidomide may also be of interest as combination treatment with CAR T-cells. There are also other interesting possibilities with the new generation of cancer therapeutics such as the BTK inhibitor ibrutinib. Ibrutinib inhibits ITK that drives the development of Th2-type CD4+ T-cells. By inhibiting ITK, Th1 T-cells that are essential in the antitumor responses were promoted. 51 Signaling pathway inhibitors such as BTK and PI3K inhibitors as well as lenalidomide are evaluated also in DLBCL in phase II–III trials. 10 Thus, combining standard-of-care treatments with CAR T-cell therapy may be an interesting and accessible option to enhance efficacy in both CLL and non-Hodgkin's lymphoma.
The capacity of CAR T-cells may be enhanced by other immunotherapies. For example, the checkpoint blockade antibodies targeting the CTLA-4 and PDL1/PD1 pathways 52 may prevent CAR T-cell exhaustion in lymphoma lesions. Another proposed combination is the use of oncolytic viruses and CAR T-cell therapy. 53 This is of high interest since most viruses are by nature immunostimulatory and attract T-cells to the site of infection. Oncolytic viruses conferring expression of immunostimulatory proteins to the tumor area may even further promote CAR T-cell efficacy. For example, we have shown that adenoviruses expressing CD40L in the tumor enhance Th1 immunity with infiltration of T-cells at the same time reducing Tregs and MDSCs and promoting M2 to M1 switch. 54 –56 In human bladder cancer patients, an adenovirus expressing CD40L induced large T-cell infiltrates in the bladder wall. 57
Future Considerations
There is an immediate need to solve CAR T-cell accessibility and survival in lymphoma and more research is clearly needed concerning the homing of CAR T-cells to lymphoid tissues and enhancing their capacity to migrate in the tumor extracellular matrix. Even if high-dose chemotherapy before CAR T-cell infusion can lead to complete responses, some patients will die from such harsh preconditioning and it also limits the number of patients who can receive CAR T-cell therapy. Nevertheless, there are other agents available to target tumor-induced immune inhibition. For example, the currently used lenalidomide or signaling pathway inhibitors may be used alongside with CAR T-cells. Further, combining CAR T-cells with other immunotherapeutics such as checkpoint blockade antibodies or oncolytic viruses may increase their survival in the tumor lesions and support efficacy. The combination with other immune therapeutics is very interesting to broaden the immune activity against the tumor since both checkpoint blockade antibodies and oncolytic viruses will activate not only the CAR T-cells but also the naturally occurring tumor-recognizing T-cells. This could prevent escape mutant tumor cells that are not positive for the CAR target. In the trials using CD19-targeting T-cells, CD19-negative clones have expanded and caused progressive disease. 58 There are also novel agents being developed blocking IL35 that may support CAR T-cell therapy by reducing the inhibitory effect of Tregs that may be of value in the future. 59 Such treatments may as well release the ongoing immune responses and not only support the CAR T-cells.
The lessons learned from the clinical use of CD19-targeting CAR T-cells may be valuable for other indications in terms of CAR design and suitable preconditioning or supportive combination treatments as discussed in this review. Suitable targets are easier to determine for cells of hematopoietic origin, especially B-cells, but trials are ongoing to target also solid malignancies with CAR T-cells targeting Her2, GD2, IL13Ra2, and mesothelin. ROR1 is also an interesting target present both on B-cells and on cancers of epithelial origin. 60 A study was performed in nonhuman primates that demonstrated a good safety profile 61 and a clinical trial to evaluate efficacy in CLL patients is listed at the ClinicalTrials website but not yet recruiting patients. In light of the results of CAR T-cell treatment of lymphomas, there will likely be a greater focus on the tumor micromilieu (including endothelium, stroma, as well as immune cells) in future trials to find means to increase infiltration of T-cells into the parenchyma and to change the environment to allow T-cell survival and activation. Instead of only providing preconditioning, solid malignancies will likely benefit from supportive combination treatments that affect the tumor microenvironment for weeks or months after CAR T-cell infusion.
Footnotes
Acknowledgments
The authors want to acknowledge Prof. Malcolm Brenner and Prof. Gianpietro Dotti at Baylor College of Medicine, Houston, for the collaborative project using CAR T-cells in Sweden. The CAR T-cell research in Uppsala is supported by AFA Insurance AB, the Swedish Cancer Society, the Swedish Childhood Cancer Society, Lions Cancer Fund at Uppsala University Hospital, and the Swedish State Support for Clinical Research.
Author Disclosure
Prof. Loskog is the CEO and board member of Lokon Pharma AB, chairman of Vivolux AB and RePos Pharma AB, and scientific advisor at NEXTTOBE AB, and has a royalty agreement with Lokon Pharma AB and Alligator Bioscience AB. Prof. Enbland and Dr. Karlsson have no conflicts of interest.