
Abstract
Current HIV-1 gene therapy approaches aim at stopping the viral life cycle at its earliest steps, such as entry or immediate postentry events. Among the most widely adopted strategies are CCR5 downregulation/knockout and the use of broadly neutralizing antibodies. However, the long-term efficacy and side effects are still unclear. TRIM5α is an interferon-stimulated restriction factor that can intercept incoming retroviruses within one hour of cytosolic entry and potently inhibit the infectivity of restriction-sensitive viruses. The human TRIM5α (TRIM5αhu) generally does not efficiently target HIV-1, but point mutations in its capsid-binding domain can confer anti-HIV-1 activity. Although the mechanisms by which TRIM5αhu mutants inhibit HIV-1 are relatively well understood, their characterization as potential transgenes for gene therapy is lacking. Additionally, previous reports of general immune activation by overexpression of TRIM5α have hindered its broad adoption as a potential transgene. Here we demonstrate the ability of the R332G-R335G TRIM5αhu mutant to efficiently restrict highly divergent HIV-1 strains, including Group O, as well as clinical isolates bearing cytotoxic T lymphocyte escape mutations. R332G-R335G TRIM5αhu efficiently protected human lymphocytes against HIV-1 infection, even when expressed at relatively low levels following lentiviral transduction. Most importantly, under these conditions Rhesus macaque TRIM5α (TRIM5αRh) and TRIM5αhu (wild-type or mutated) had no major effects on the NF-κB pathway. Transgenic TRIM5α did not modulate the kinetics of IκBα, JunB, and TNFAIP3 expression following TNF-α treatment. Finally, we show that human lymphocytes expressing R332G-R335G TRIM5αhu have clear survival advantages over unmodified parental cells in the presence of pathogenic, replication-competent HIV-1. These results support the relevance of R332G-R335G and other mutants of TRIM5αhu as candidate effectors for HIV-1 gene therapy.
Introduction
R
Replacing regions within the CA-targeting domain, called PRYSPRY, of TRIM5αhu with the corresponding sequences from its Rhesus ortholog has resulted in human/rhesus chimeric TRIM5α proteins that can efficiently restrict HIV-1 when transduced into human cells. 27 Modeling studies and genetic screens have also led to the identification of point mutations in the variable region 1 (v1) of the TRIM5αhu PRYSPRY domain that allow it to target HIV-1 for restriction. 23,28 –30 We previously described the R332G-R335G TRIM5αhu mutant as especially efficient at restricting HIV-1. In particular, this double mutant had significantly superior anti-HIV-1 activity compared with the previously described single mutation at position 332. 23,29,31 One major limitation of gene therapy applications is the immune response that often results from introducing foreign proteins into humans. 32 –34 Because the TRIM5αhu mutants differ only slightly from the endogenous form of TRIM5αhu, they are not expected to be immunogenic, thus making them strong candidates for gene therapy applications compared with simian orthologs.
Cytotoxic T lymphocyte (CTL) escape mutations often lead to a fitness cost on viral replication. 35 –38 Some mutations occurring in CA increase the virus susceptibility to TRIM5αhu 39 and may be partially responsible for the control of HIV-1 viremia in HLA B*27 and B*57 individuals. 39,40 It is possible that isolates bearing such CTL-escape mutations might be poorly sensitive to restriction by TRIM5αhu mutants. Along these lines, it is not known whether HIV-1 strains sharply divergent from the few clade B strains used in the vast majority of TRIM5α studies so far would be restricted by TRIM5αhu mutants. Other common caveats in TRIM5α preclinical studies include the fact that very strong transgene promoters are frequently employed and that there is often a high number of integration events per cell following retroviral or lentiviral vector-mediated transduction. Finally, activation of the nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB)/activator protein-1 (AP-1) pathway can result from TRIM5α overexpression, 21,41,42 leading to challenges in developing TRIM5α for anti-HIV gene therapy. Here we demonstrate the antiviral potential of the R332G-R335G mutant of TRIM5αhu against highly divergent viral strains as well as CTL-escape capsid mutants. We show that this mutant efficiently inhibits HIV-1 propagation in human lymphocytes following lentiviral transduction of R332G-R335G TRIM5αhu at a low multiplicity of infection (MOI) and using a relatively weak promoter. Importantly, we show that unlike TRIM5α transfection, stable gammaretroviral vector or lentiviral vector-mediated transduction of R332G-R335G TRIM5αhu does not activate innate immune pathways. Finally, T lymphocytes stably expressing R332G-R335G TRIM5αhu possess a survival advantage in the presence of HIV-1 even when the modified cells are present in relatively low numbers.
Materials and Methods
Plasmid DNAs
pMIP-TRIM5αhu and pMIP-TRIM5αRh are gammaretroviral vectors that express C-terminal FLAG-tagged versions of the wild-type (WT) or mutant proteins along with puromycin as a selection marker. These constructs have been described before. 25,43 Versions of these vectors expressing green fluorescent protein (GFP) in place of puromycin were constructed by transferring the TRIM5α sequence into pMIG 44 using Xho1 and EcoR1. pNL4-3 encodes a fully replication-competent clone of HIV-1. 45 pCMV-Δ8.91-based gag-pol expressing vectors were a kind gift from Greg Towers and Yasuhiro Ikeda. 9 pNL4-3XCS-based proviral vectors containing the Renilla luciferase gene in place of nef, which includes a 580 bp deletion in env and code for NL4-3, NRC1, NRC2, or NRC10 gag-pol, were a kind gift from Allan Hance. 46 pMD-G and pTRIPCMV-GFP have been described previously. 47,48
To produce lentiviral vectors expressing TRIM5α, we first digested pHIV749 with BamHI and DraII and inserted a multiple cloning site generated by PCR using pHIV7 as the template and the following ODNs: HIV_reinsert_fw (5′-CGCGACTCTAGATCATGGATCCTCCGGACTGCACTCTAGATAGGTCACCACCGTCGACTAGCCGTACCT), and HIV_reinsert_rev (5′-ACTATAGGGCGAATTGGGTACC). The resulting plasmid, called pHIV7-empty, was digested with Kpn2I and Eco91I. A PCR fragment containing the CMV-intron (premiR30)-GFP-partial polyA cassette from pSM30-GFP 50 was generated with the primers HIV8miR30_fw (5′-TATTCGCACTGGATACGATCCGGATGATTCTGTGGATAACCGT) and HIV8miR30_rev (5′-AATATCCTCCTTAGTTCCGGTGACCTAGAATGCAGTGAAAAAAATG). This fragment was ligated into pHIV7-empty to generate pHIV8-C-A. A codon-optimized 31-amino-acid-long 2A sequence based on Donnelly et al. 51 (now referred to as 2A31D) was inserted between the intron and EGFP coding sequences; 2A31D contains an AgeI site, start codon, flag tag, and two inverted BsmBI sites, to allow flush attachment of an upstream protein to the 2A31D sequence without codon frame change. This was achieved by ligation of overlapping ODNs and subsequent PCR amplification, yielding a double-stranded fragment of the following sequence: 5′-ATCCACCGGTCGCCACCatgGACTACAAGGACGACGATGAGCAGGAGACGGAAGCCACAGACGTCTCCGTGACCGAGCTGCTGTACCGCATGAAGCGCGCCGAGACCTACTGCCCCCGCCCCCTGCTGGCCATCCACCCCACCGAGGCCCGCCACAAGCAGAAGATCGTGGCCCCCGTGAAGCAGACCCTGAACTTCGACCTGCTGAAGCTGGCCGGCGACGTGGAGAGCAACCCCGGCCCCGTGAGCAAGGGCGAGGAGCTGTTCACCGGG. This fragment was then used in a fusion PCR using the reverse primer ODN 2A-EGFP_rev (5′-AAAGTCCCGGACGTAGCCTTCGGGCATGGCGG). The resulting DNA fragment was then digested with AgeI and PfoI and cloned into pHIV8-C-A to generate pHIV8-C-2A31D-A. Subsequently, the CMV promoter driving transgene expression was replaced with the short human EF1α promoter (EFS promoter) with an added MCS region by fusion PCR. For this, we first generated a long reverse primer by fusion PCR, using pHIV8-C-A as a template and the primer ODNs EFS_brdg_(fw 5′-GCCGCCAGAACACAGGTGCATGCTTAATTAAATTTAAATTTAATACTAGAAGCTTTATTGCGGTAGTTTATCAC) and Intron_rev (5′-TTGCTCACCATGGTGGC). This fragment was used as the reverse primer in a fusion PCR using pLV-tTRKRAB 52 as a template and EFS_fw (5′-ATATATTCCGGAGGCTCCGGTGCCCGT) as the upstream primer. The resulting fragment and pHIV8-C-2A31D-A were digested with Kpn2I and NheI and ligated to generate pHIV8-ES-2A31D-A. Afterward, the woodchuck hepatitis regulatory element (WPRE) was introduced into the 3′ UTR by PCR, using pHIV7-GFP 49 as a template and the primers wpreO_fw (5′-ATATCGGCCGCATCAAGCTGGGCTGCAGG) and wpreO_rev (5′-TATAGGTGACCATCGATGCGGGGAGGC). The PCR product was digested with BsteII and EagI and ligated to pHIV8-ES-2A31D-A, which had been cut with the same enzymes to generate pHIV8-ES-2A31D-W.
TRIM5α variants were amplified by PCR from the corresponding pMIP constructs, using the primer ODNs 2A-TRIM5a (5′- ATCCACCGGTCGCCACCATGGCTTCTGGAATCCTGG) and 2A-hT5-fus_rev (5′- GCATATTCGGTCACGGAGACGTCTGTGGCTTCCGTCTCTTCACAGAGCTTGGTGAGCACAGAGT) for human variants and 2A-rT5-fus_rev (5′- GCATATTCGGTCACGGAGACGTCTGTGGCTTCCGTCTCTTCACAAAGCTGGGTGAGCACAGAGT) for the rhesus variant. This PCR step also removed the Start codon-FLAG sequence present upstream of TRIM5α in the pMIP constructs. The PCR products were digested with AgeI and BsmBI and cloned into pHIV8-ES-2A31D-W, which had been cut with the same enzymes. The resulting constructs, named pHIV8-ES-T5a(x)-2A31D-W, were used as transgene lentiviral vector plasmids.
Cell lines
Human rhabdomyosarcoma TE671 cells, human embryonic kidney (HEK) 293T cells, and Crandell–Rees feline kidney (CRFK) cells were maintained in Dulbecco's modified Eagle's medium (Gibco) supplemented with 2 mM L-glutamine, 10% fetal bovine serum (Gibco), and 1% penicillin/streptomycin (Thermo Scientific) at 37°C in 5% CO2. The human T-cell lines Sup-T1 and CEM.NKR-CCR553 were maintained at a density between 5×105 and 2×106 cells per ml in RPMI (Corning) that was supplemented with 2 mM L-glutamine, 10% fetal bovine serum, and 1% penicillin/streptomycin at 37°C in 5% CO2. All cell culture reagents were obtained from Hyclone (Thermo Scientific).
Generation of stably transduced cells
TE671 and Sup-T1 cells stably expressing TRIM5α and puromycin N-acetyl-transferase were produced by using pMIP-based retroviral vectors and then maintained in puromycin as described before. 31,54 To generate CEM.NKR-CCR5 cells stably transduced with lentiviral vectors expressing TRIM5α, we produced vector particles as previously described 49 with the following adjustments. HEK293T cells (20–25 million) were seeded in 150 mm plates in 15 ml of medium. Eighteen hours later, the medium was replaced and cells were then transfected with 35 μg of pCMV-G, 25 μg of pCMV-rev, 25 μg of pCHGP, and 35 μg of the pHIV8-ES-T5a(x)-2A31D-W construct using 374.4 μl of CaCl2 in 3 ml total volume. Twenty-five hours postseeding, the medium was replaced and virus particles were collected 24 hr later and purified by ultracentrifugation. CEM.NKR-CCR5 cells were transduced at a density of 105 cells per ml in the presence of 8 μg/ml of polybrene (EMD Millipore), using an MOI of 0.5 as determined by titering the concentrated vector on HEK293T cells. GFP-positive cells were sorted by fluorescence-activated cell sorting (FACS) 25 days later, and gag-pol PCR confirmed that the cells did not carry replication-competent HIV-1.
Production of HIV-1 vectors and viruses
HIV-1-based vectors were produced through transient transfection of HEK293T cells and collected as previously described. 29,31,54 To produce Gag variant HIV-1TRIP-CMV-GFP, the cells were cotransfected with pTRIPCMV-GFP, the respective pCMV-Δ8.91 vector, and pMD-G. To generate the gag variants of HIV-1NL-Luc, the cells were cotransfected with the respective pNL4-3XCS vector and pMD-G. HIV-1NL43 was produced by transfection of pNL4-3. HIV-1 IIIB was amplified by passage in stimulated peripheral blood mononuclear cells and was titered by end-point dilution in MAGI cells, as described before. 55
Single-cycle infection assays
For viral challenges with HIV-1TRIP-CMV-GFP, cells were plated in 24-well plates at 50,000 cells per well and infected the next day with the appropriate HIV-1 vectors at an MOI of 0.1, as normalized by titering the viral stocks in feline CRFK cells. Two days postinfection, the cells were trypsinized and fixed in 1–2% formaldehyde in a PBS solution. The percentage of GFP-positive cells was then determined by analyzing 10,000–20,000 cells with an FC500 MPL cytometer (Beckman Coulter) using the CXP software (Beckman Coulter). For viral challenges with HIV-1NL-Luc, CRFK, and TE671 cells were plated in 24-well plates at 50,000 cells per well and infected the next day with 100 μl of the appropriate HIV-1 vectors, a dose that resulted in measurable luciferase activity in all tested conditions. Two days postinfection, the cells were washed once with PBS and lysed in 100 μl of Glo Lysis Buffer (Promega) following the manufacturer's recommendations. Relative light units were measured using the Renilla-Glo Luciferase Assay System (Promega) according to the manufacturer's instructions and read in a Biotek Synergy HT plate reader.
Replication-competent HIV-1 infection assays and p24 ELISA
In total, 106 CEM.NKR-CCR5 cells were seeded in 1 ml of medium in a 12-well plate and challenged with HIV-1 virus IIIB (NIH AIDS Reagent Program #398; MOI 0.01). After 24 hr, the cells were washed three times with PBS. Subsequently, 0.5–1 ml of the cell suspension was removed every 3 or 4 days and the cell density was readjusted with fresh medium to a volume of 1.5–2 ml. On days 8, 15, and 22, aliquots of the cell suspensions were subjected to low-speed centrifugation (7 min at 250× g) and the supernatants were then used for CAp24 ELISA. CAp24 ELISA was performed using the Alliance kit (Perkin Elmer) according to the manufacturer's instructions. For the experiment with HIV-1 NL4-3, Sup-T1 cells transduced with various pMIG-TRIM5α constructs were infected with 15 μl of virus, a dose previously determined to lead to peak infection in about 12 days. 23 Cells were then maintained in culture at a density between 5×105 and 2×106 cells per ml and aliquots were analyzed by FACS for GFP expression at different time points.
NF-κB stimulation assay
TE671 cells stably expressing FLAG-tagged R332G-R335G TRIM5αHu or WT TRIM5αRh or transduced with empty vector were plated in 12-well plates at a cell density leading to 80% confluent cells at the time of transfection. TE671 cells stably expressing R332G-R335G TRIM5αHu or WT TRIM5αRh were transfected with 0.6 μg of the NF-κB luciferase reporter construct (pCEP4-NF-κB-Luc) or the activation-deficient mutant of the reporter construct (pCEP4-ΔNF-κB-Luc). TE671 cells transduced with the empty vector control were cotransfected with R332G-R335G TRIM5αHu or WT TRIM5αRh (2 μg) and the NF-κB luciferase reporter construct (0.6 μg). As a control, TE671 cells transduced with empty vector were cotransfected with R332G-R335G TRIM5αHu (2 μg) and ΔNF-κB-Luc (0.6 μg). The cells were lysed with RIPA buffer 48 hr posttransfection and assessed for luciferase activity using the BrightGlow Luciferase kit (Promega). Luminescence was measured with a Synergy HT multidetection microplate reader (BioTek) and analyzed using the Gen5 software (BioTek). The cellular lysates prepared to quantify luciferase activity were also used to analyze TRIM5αRh expression. Specifically, the three lysates from each triplicate transfection were pooled and analyzed by Western blotting using a rabbit anti-FLAG antibody (Cell Signaling; 1: 2000). Actin was used as a loading control and was detected with mouse anti-actin HRP (Sigma; 1:20,000).
Tumor necrosis factor-α stimulation
For IκBα protein kinetics, CEM.NKR-CCR5 cells were collected unstimulated, after stimulation with TNF-α (10 ng/ml; PeproTech, Rocky Hill, NJ) for 30 min and after a drug-free recovery phase of 2 hr. For the analysis of NF-κB- and AP-1-regulated target genes by RT-PCR, cells were collected unstimulated, after stimulation with TNF-α (10 ng/ml) for 4 and 8 hr, and after a recovery phase of 16 hr following 8 hr of TNF-α stimulation.
RT-qPCR
Total RNA from 5×106 CEM.NKR-CCR5 cells was purified using STAT60 (TEL-TEST) according to the manufacturer's protocols and 7 μg of RNA was digested with 1 μl of Turbo DNase in a 25 μl total volume (AM1907; Ambion). Afterward, 2.4 μg of RNA was reverse transcribed in 46 μl (3.26 ng/μl random hexamers, 0.43 mM dNTP, 10 nM DTT, 1.74 U/μl RNAsin, and 8.7 U/μl MMLV-RT [#28025-013, Invitrogen]) for 10 min at 25°C followed by 15 min at 37°C, and the reaction was stopped by heat inactivation for 15 min at 70°C. qPCR was performed in 20 μl with 25 ng cDNA for detection of GAPDH (200 ng for other target genes) with IQ Sybr Green Supermix (170-8882; Biorad) and 0.5 μM of primer pairs. Primer ODN pairs were as follows: human GAPDH, forward (5′-CCACTCCTCCACCTTTGAC) and reverse (5′-ACCCTGTTGCTGTAGCCA); human and rhesus TRIM5α, forward (5′-AGACATTCTGAAAAGCCTTACGAA) and reverse (5′-ATCAGGAGCTCGAAACACTCTC); EGFP, forward (5′-ATCATGGCCGACAAGCAGAAGAAC) and reverse (5′-GTACAGCTCGTCCATGCCGAGAGT) 56 ; human FasL, forward (5′-TAAAACCGTTTGCTGGGGC) and reverse (5′-CTCAGCTCCTTTTTTTCAGGGG); JunB, forward (5′-GGACGATCTGCACAAGATGA) and reverse (5′-GGGAGTAGCTGCTGAGGTTG); human TNFAIP3, forward (5′-CCCTCATCGACAGAAACA) and reverse (5′-GAACGCCCCACATGTACT); all were provided by IDT. PCRs were performed in 96-well plates (HSP9601; Biorad) using the following settings: 95°C for 9 min, 40× (30 sec at 95°C, 1 min at 59°C, 30 sec at 72°C), and 72°C for 5 min.
Results
Targeting of highly divergent HIV-1 CA constructs by R332G-R335G TRIM5αhu
In order to assess the sensitivity of diverse HIV-1 isolates to mutated human TRIM5α, we first generated a series of HIV-1 vectors carrying gag from 12 different strains in an otherwise isogenic context.
9
The vesicular stomatitis virus G-pseudotyped, GFP-expressing HIV-1 vectors were produced by transient transfection as previously described.
23,54
Strain names, sequence accession numbers, subtypes or clades, and cellular tropism of the HIV-1 isolates used in this study are summarized in Supplementary Table S1 (Supplementary Data are available online at
To analyze the sensitivity of these gag variants to restriction by either TRIM5αRh or R332G-R335G TRIM5αhu, we stably expressed them in human TE671 cells, whereas cells stably transduced with WT TRIM5αhu were used as nonrestrictive controls. As shown in Fig. 1A, all viruses tested were susceptible to restriction by both TRIM5αRh and R332G-R335G TRIM5αhu. Although the efficiency of restriction varied slightly from strain to strain, R332G-R335G TRIM5αhu inhibited all HIV-1 strains by more than 10-fold, with the exception of 93BR029 (3.4-fold). In addition, the pattern of restriction by TRIM5αRh was similar to that of R332G-R335G TRIM5αhu, supporting the idea that they interact with HIV-1 CA in a similar fashion. TRIM5αRh-mediated inhibition was only ∼2-times more efficient than the inhibition mediated by R332G-R335G TRIM5αhu in this assay, demonstrating the potency of the TRIM5αhu mutant approach.

Restriction of highly divergent HIV-1 capsids and of cytotoxic T lymphocyte escape mutants in human cells expressing R332G-R335G TRIM5αhu.
Restriction of HIV-1 bearing CTL escape mutations
Next we tested NRC1, NRC2, and NRC10 isolates, which bear gag mutations that allow for CTL escape but increase sensitivity to endogenous TRIM5αhu, for their sensitivity to restriction by the same TRIM5α variants. For this, we used a different set of vectors based on pNL-Luc, a pNL4-3 derivative that contains a deletion in env and encodes Renilla luciferase in place of nef. The gag and protease sequences of pNL-Luc were replaced with sequences from isolates bearing CTL-escape mutations, yielding the constructs NRC1, NRC2, and NRC10. 39 It was previously established that, compared with NL-Luc, the increase in sensitivity to TRIM5αhu was greatest for NRC10, less so for NRC2, and weak for NRC1. 39 NRC10-VMG is a lab-generated mutant of NRC10 in which three of the mutations that are known to increase sensitivity to TRIM5αhu (V86A, M96I, and G116A) are reverted back to the WT sequence. We measured the infectivity of the various HIV-1 vectors generated in both feline CRFK cells, which do not express any known TRIM5α ortholog, and in human TE671 cells transduced with the various TRIM5α cDNAs (Fig. 1B). The data gathered in CRFK cells allow us to quantify the TRIM5α-independent decrease in viral fitness stemming from the CTL-escape mutations in gag. NRC1 and NRC2 showed slightly decreased infectivity in these cells (<2- and ∼2-fold, respectively), whereas infectivity of NRC10 and NRC10-VMG was significantly lower than that of the control NL-Luc (5.5- and 14-fold, respectively). NRC1 had the same restriction pattern as NL4-3, although the former was slightly less sensitive to R332G-R335G TRIM5αhu.
As previously observed by Battivelli et al., 39 NRC2 and NRC10 showed marked increases in sensitivity to TRIM5αhu. Specifically, infectivity of NRC2 and NRC10 in TE671 cells expressing TRIM5αhu was reduced 13-fold and 58-fold, respectively, compared with their respective CRFK controls. Restriction of these two variants by R332G-R335G TRIM5αhu and TRIM5αRh was also efficient. In particular, NRC2 and NRC10 were restricted 30-fold and 125-fold by R332G-R335G TRIM5αhu, respectively, whereas the NL-Luc control was restricted slightly more than 10-fold. As expected, the NRC10-VMG revertant showed decreased sensitivity to all three TRIM5α variants when compared with NRC10. In summary, some CTL-escape HIV-1 gag mutants are markedly more sensitive to restriction by R332G-R335G TRIM5αhu than TRIM5αhu.
Survival advantage of lymphocytes expressing HIV-1-restrictive TRIM5α variants
The previous experiments used replication-incompetent vectors that can only complete the early stages of infection. Typical HIV-1 spreading assays performed in our earlier studies 23,29,31 also do not faithfully represent the situation of in vivo gene therapy protocols, in which only a minority of the cells are protected by the presence of the antiviral transgenes. We designed experimental conditions that allowed a minority of human lymphocytic TRIM5α-expressing Sup-T1 cells to expand because of their resistance to HIV-1-mediated killing. For this experiment, we mixed cells stably expressing various restrictive or nonrestrictive TRIM5α variants with parental cells at an approximate ratio of 1:99 (Table 1). The transduced cells also expressed GFP, allowing for convenient quantification by FACS. We then infected the various cocultures with a single dose of NL4-345 that results in a replication peak at about 2 weeks postinfection as measured by reverse transcriptase assay. 23 For the first two time points at which we performed FACS, the proportion of GFP-positive cells was close to 1% in all cultures, as expected. However, HIV-1 spread very efficiently in all cultures, and there were too few surviving cells for us to perform cytometry between days 11 and 18 postinfection. We nonetheless maintained these cells, and more than a month after the beginning of the experiment, recovery was sufficient for us to analyze the proportion of GFP-expressing cells in all cultures.
Survival advantage of Sup-T1 cells expressing TRIM5α variants following HIV-1 infection
Mixed cultures containing ∼1% of cells transduced with MIG-TRIM5α and 99% of unmodified parental Sup-T1 cells were infected with the pathogenic NL4-3 strain of HIV-1. Cells were maintained in culture for 39 days postinfection, and a fraction of each culture was collected at various times for flow cytometry analysis. Data show the percentage of cells expressing GFP, which is translated from the same mRNA as TRIM5α.
Vector refers to the “empty” MIG expressing only GFP.
Around the peak of HIV-1 propagation, the mortality rate in the various cultures was too high to collect enough cells for flow cytometry analysis.
The results were strikingly different depending on whether the initial mixed culture contained cells expressing HIV-1-restrictive TRIM5α or not. In the nonrestrictive controls, that is, the cells transduced with WT TRIM5αhu or with the “empty” MIG vector, we could barely detect any cells expressing GFP, showing a total lack of survival advantage of the transduced cells. Although it is not clear how the GFP-negative cells survived the infection in our experiment, such phenomena have been linked to the absence of positive cellular factors such as the transcription factor NF-κB. 59 In all other cases, the proportion of GFP-expressing cells had markedly increased, ranging from ∼20% (cells expressing R332G TRIM5αhu) to close to 90% (cells expressing TRIM5αRh). In addition, the percentage of GFP-positive cells was proportional to the level of protection conferred by the respective TRIM5α variants: TRIM5αRh>R332G-R335G TRIM5αhu>R332G TRIM5αhu. 23,29,31 Although the exact proportion of TRIM5α-expressing cells that survived HIV-1 infection in the first 4 weeks of infection is not known, this experiment suggests that R332G-R335G TRIM5αhu can confer protection not only against HIV-1 infection but also against cytopathic effects stemming from the active spread of the virus in the unmodified cells.
Lack of detectable NF-κB activation in TE671 cells stably expressing TRIM5α
TRIM5α overexpression by transient transfection can activate the transcription factors NF-κB and AP-1, 21,22,41,42,60 and in some cases results in type I interferon production. 21,60 However, it was not known whether stable overexpression of TRIM5α following retroviral transduction constitutively activates these pathways, which could result in unregulated inflammation. To investigate this possibility, we used cells that were stably transduced with R332G-R335G TRIM5αhu or TRIM5αRh. In parallel, we used control cells, which we transiently transfected with the vector plasmids encoding R332G-R335G TRIM5αhu or TRIM5αRh. To assess NF-κB activation, we transfected the cells with a construct expressing luciferase under the control of an NF-κB-inducible promoter, as described previously. 41,42 Two days posttransfection, luciferase activity and TRIM5α expression levels were assessed (Fig. 2). Basal NF-κB activity was detectable in control cells transfected with the NF-κB-luc construct, as compared with cells transfected with the control plasmid bearing a deletion in the NF-κB binding domain (an ∼3-fold increase). NF-κB activation levels were statistically indistinguishable in cells stably transduced with R332G-R335G TRIM5αhu or with TRIM5αRh compared with the control cells (Fig. 2, top panel). However, when the control cells were transiently transfected with the TRIM5α-expressing retroviral vector plasmids, we observed a large increase (∼10-fold) in NF-κB activation levels. This result suggests that expression of TRIM5α by transient transfection, but not following stable transduction, stimulates NF-κB (see Discussion). Interestingly, levels of TRIM5α expression in the transfected cells were similar to those in the stably transduced cells, as judged in a Western blotting analysis (Fig. 2, bottom panel). It is possible, however, that a minor fraction of the transiently transfected cells express very high levels of TRIM5α, and that NF-κB activation occurs mostly in this subset of cells.
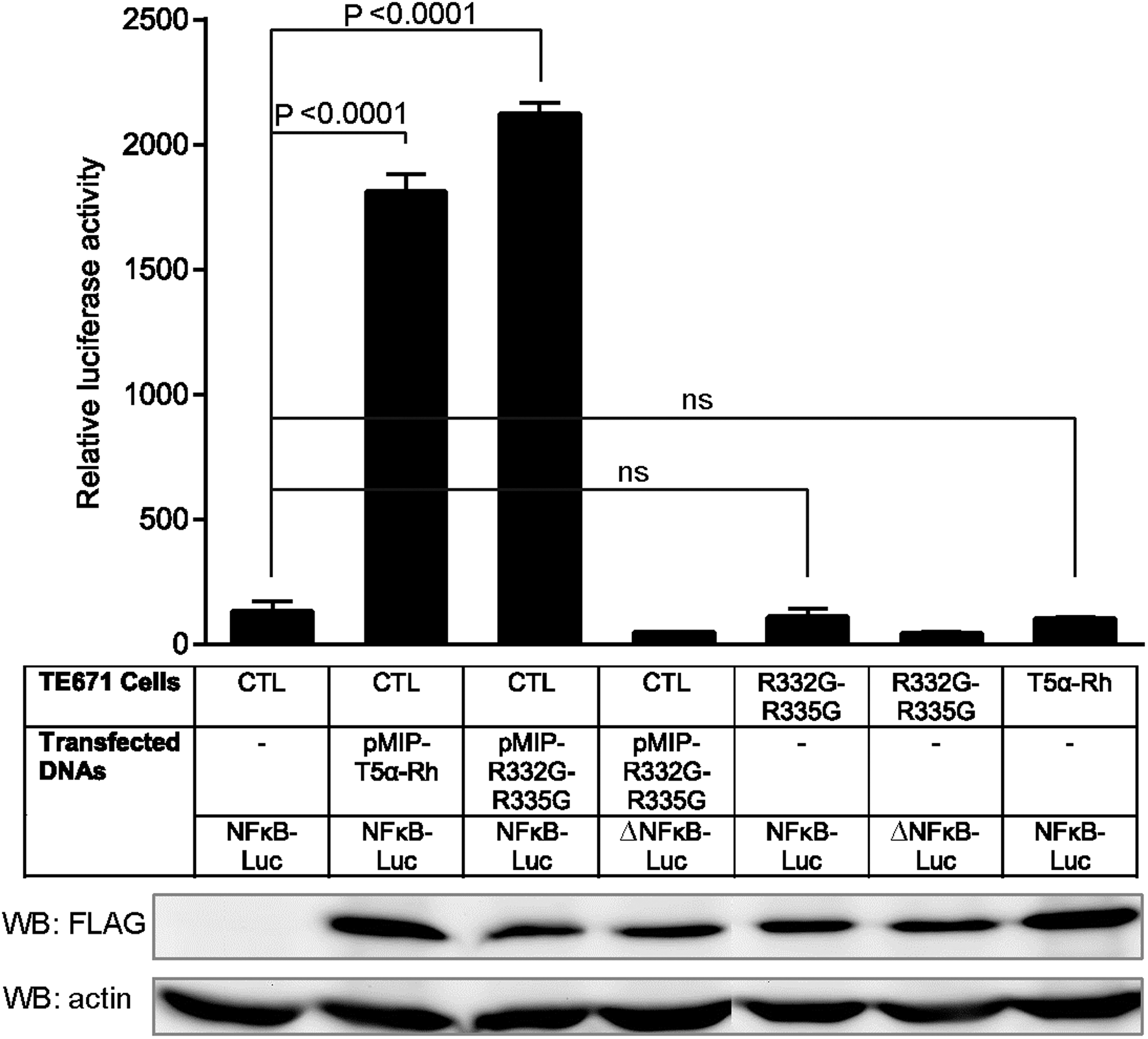
Activation of the NF-κB pathway by transient transfection of Rhesus or human TRIM5α but not by their stable transduction. TE671 cells stably expressing FLAG-tagged R332G-R335G TRIM5αhu, WT TRIM5αRh, or control cells (CTL) were transfected in triplicate with a plasmid expressing luciferase from an NF-κB-dependent promoter, or with the control plasmid (“ΔNFκB-Luc”) bearing a mutation in the NF-κB binding site. Empty vector-transduced cells were also cotransfected with pMIP plasmids expressing FLAG-tagged TRIM5αRh or R332G-R335G TRIM5αhu. Two days later, cells were lysed and luciferase activity was quantified (top panel). p-Values were determined by the one-way ANOVA test followed by Turkey's multiple comparison test. ns, nonstatistically significant. Lysates were also analyzed by Western blotting using an antibody against FLAG to detect exogenous TRIM5α expression and a loading control antibody against actin (bottom panels).
Stable expression of TRIM5α from a lentiviral vector with predominantly single integration
We next tested the anti-HIV-1 and innate immune activation properties of TRIM5αhu mutants, using a vector system in which TRIM5α expression is relatively low and cell-to-cell variation is minimal. For this experiment, we designed a lentiviral vector plasmid, pHIV8-ES-2A31D, to express TRIM5α and GFP under control of the short human EF1α promoter. GFP translation was possible because of the presence of a ribosome-skipping 2A domain C-terminal of TRIM5α. We transduced T lymphocytic CEM-based CEM.NKR-CCR5 cells 53 with the “empty” lentiviral vector, expressing only GFP, or with a vector that also expresses TRIM5α (Supplementary Fig. S1). In addition to TRIM5αRh, WT TRIM5αhu, and R332G-R335G TRIM5αhu, we also tested the single mutant R332G TRIM5αhu in this novel experimental model. To demonstrate comparable transduction and expression rates, GFP fluorescence intensity was analyzed (Supplementary Fig. S1A, B). Results show that transduction levels and GFP expression levels were similar across the various TRIM5α-expressing constructs at both time points tested. Cells transduced with the “empty” vehicle showed higher levels of GFP expression, which is probably because of the smaller size of the GFP-only transgene cassette. In addition, analysis of the GFP expression profiles of the GFP-positive cells after FACS sorting at day 29 postinfection showed the presence of a single peak when cells were transduced with one of the TRIM5α-expressing vectors, suggesting that most cells had only one copy of the integrated vector (Supplementary Fig. S1A).
In contrast, two or more peaks were apparent for cells transduced with the empty vector. Although the basis for this observation is unclear, it is unlikely to result from multiple integration events because less than 5% of the transduced cells were GFP positive (not shown). In order to determine the levels of transgenic TRIM5α expression relative to endogenous TRIM5αhu, we quantified total TRIM5α mRNA using SybrGreen RT-PCR (Supplementary Fig. S1C, D). The primers used in this experiment can amplify WT or mutant TRIM5αhu sequences, as well as TRIM5αRh (see Materials and Methods). Using this approach, we determined that transduction with the empty vector had no impact on endogenous TRIM5α mRNA expression levels, whereas transduction with the various TRIM5α-expressing vectors resulted in an ∼6-fold to ∼12-fold increase in mRNA levels (Supplementary Fig. S1C, D). It is unclear why about two times less TRIM5α mRNA was detected in cells transduced with R332G-R335G TRIM5αhu compared with cells transduced with TRIM5αhu or TRIM5αRh (Supplementary Fig. S1D), even though GFP expression levels were similar (Supplementary Fig. S1B). A likely possibility is that the long half-life of GFP 61 masked differences in protein translation levels. The TRIM5α protein, on the other hand, has a notoriously short half-life of about 40 min. 62 It cannot be excluded, however, that the mutations affected reverse transcription or PCR to some degree, by decreasing access to the random hexamers used in the reverse transcription step or the specific primers used in the PCR step. In addition, mutations in this PRYSPRY region have been shown to modulate protein stability. 63 In conclusion, transgenic TRIM5α was moderately overexpressed, consistent with predominantly single integration events in our experimental system, with the R332G-R335G variant possibly being expressed at slightly lower steady-state levels.
TAK1 is not activated in TRIM5α-transduced cells
Loss-of-function experiments have demonstrated that transforming growth factor-activated kinase 1 (TAK1) is important for the activation of NF-κB following overexpression of TRIM5α 21,60 or Bst2/Tetherin, 64,65 or treatment with the proinflammatory cytokine TNF-α. 66 The TAK1 kinase complex, which comprises TAK1 as well as TAK1-binding proteins 1 and 2 (TAB1 and TAB2), is activated by K63-linked polyubiquitin chains that are catalyzed by the E3 proteins TRAF667 and TRIM5α. 21,41 TAK1 phosphorylates IκB kinase (IKK), which in turn phosphorylates IκBα, leading to NF-κB activation. 67,68 TAK1 is central to many innate immune pathways, including those downstream of innate sensors of the Toll-like receptor (TLR) and retinoic acid-inducible gene 1 (RIG-I) families. 68 TAK1 stimulatory functions require phosphorylation at Ser412. 69,70 We used Western blotting to analyze the total and Ser412-phosphorylated forms of TAK1 in stably transduced CEM.NKR-CCR5 cells (Fig. 3A). Quantification from three independent experiments showed that stable overexpression of the various TRIM5α variants used did not significantly modulate total levels of TAK1, compared with the untransduced cells (Fig. 3B). Levels of Ser412-phosphorylated TAK1 were slightly higher in cells transduced with WT and R332G TRIM5αhu, compared with the other cell lines, but this effect did not reach statistical significance (Fig. 3B). In conclusion, the results from this experiment support a model where the levels of TAK1 are unaltered in cells stably overexpressing TRIM5α.
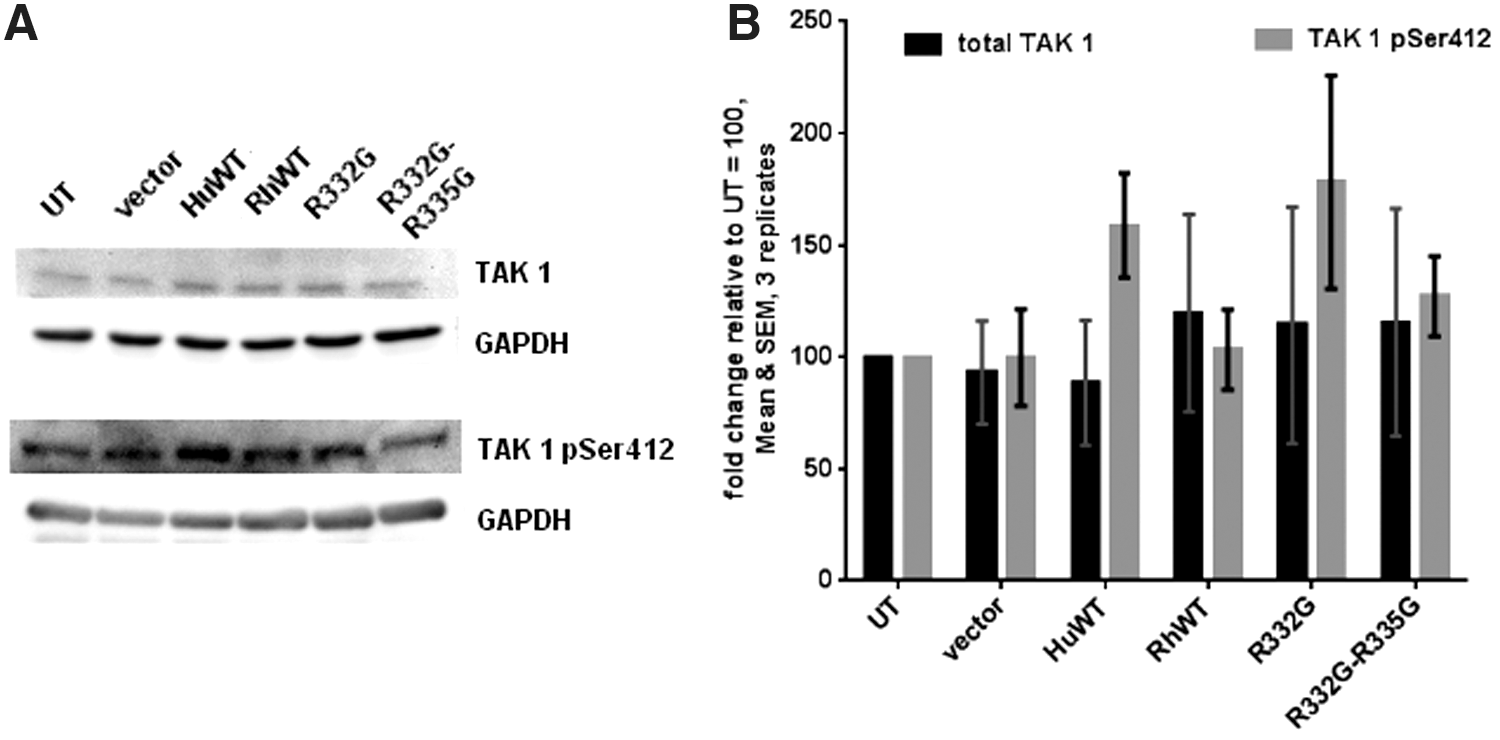
Stable TRIM5α transduction does not affect TAK1 expression levels or its Ser412 phosphorylation.
No effect of transgenic TRIM5α on IκBα kinetics following TNF-α treatment
Although TRIM5α transduction did not appear to significantly modulate TAK1 activation levels, there was still a possibility that it could modulate the kinetics of the NF-κB/AP-1 pathway. To test this, we treated the various CEM.NKR-CCR5 cell lines with TNF-α and then analyzed kinetics of IκBα and of NF-κB-dependent gene expression. IκBα binds to NF-κB in the cytoplasm, preventing its nuclear translocation and transcriptional activity. 71 TNF-α causes the phosphorylation and degradation of IκBα, 72 allowing NF-κB to reach the nucleus, where it can potentially activate hundreds of genes. 73,74 Among these are IκBα, leading to a negative feedback loop that contributes to ending the effects of TNF-α. 75,76 By controlling the duration of NF-κB activation, the negative feedback loop indirectly controls NF-κB transcriptional activity profiles as well. 73,77 Therefore, we used an experimental setting that would allow us to study both the activation and recovery phases. We observed that, as expected, TNF-α stimulation resulted in a significant decrease in IκBα levels after 30 min of treatment (Fig. 4A). Upon repeated experiments, we found that stable transduction with WT or mutant TRIM5αhu or with TRIM5αRh expressing transgenes did not alter the capacity of TNF-α to downregulate IκBα expression (Fig. 4B). Gel densitometry analyses showed that, on average, IκBα expression was reduced ∼2-fold as a result of the treatment. After 2 hr of posttreatment recovery, IκBα expression returned to normal or slightly elevated levels. One exception was the untransduced cells, in which IκBα expression was about three times higher compared with pretreatment levels. This result could derive from the fact that IκBα pretreatment levels are higher in cells transduced with the lentiviral vectors used (encoding TRIM5α or not) than in untransduced cells (see Fig. 4A). It is possible that low levels of innate activation are induced by the lentiviral transduction itself, independent of TRIM5α expression. 78 In conclusion, the downregulation/recovery profiles seen in cells transduced with the various TRIM5α variants were similar to the controls, further supporting the assertion that the TRIM5α variants do not affect NF-κB activation.
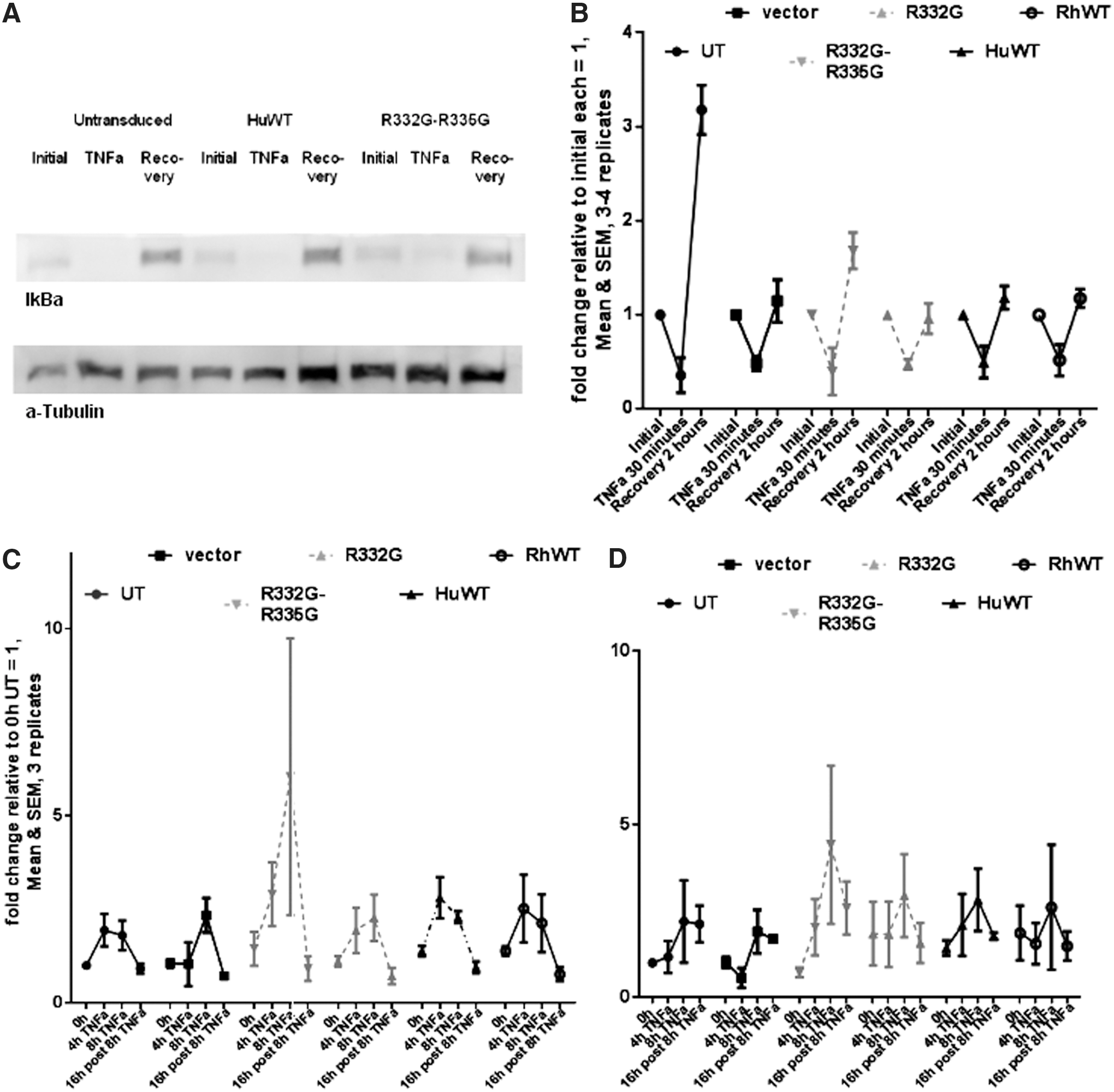
TRIM5α transduction has no significant impact on the kinetics of IκBα protein levels or on the regulation of the AP-1/NF-kB pathway. Sorted, stably transduced CEM.NKR-CCR5 cells expressing the indicated TRIM5α variants or transduced with the vector expressing GFP only (“vector”) were subjected to TNF-α treatment for 30 min. An aliquot of each culture was then lysed for protein analysis, while the remainder was kept in culture for 2 additional hours in the absence of TNF-α.
No effect of TRIM5α on the kinetics of expression of genes relevant to the NF-κB and AP-1 pathways
Treatment with TNF-α activates the AP-1 transcriptional complex through mechanisms that are both independent of and dependent on NF-κB. 79 Because TRIM5α overexpression can activate both NF-κB and AP-1, 21,41 we investigated whether stably transduced TRIM5α would modulate TNF-α-induced expression of factors relevant to both pathways. We treated the various CEM.NKR-CCR5 cell lines with TNF-α for 4 or 8 hr and then maintained an aliquot of each culture for 16 additional hours in the absence of the drug. qRT-PCR was performed to analyze mRNA expression levels for tumor necrosis factor, alpha-induced protein 3 (TNFAIP3, also known as A20) (Fig. 4C), and JunB (Fig. 4D). Transcription of TNFAIP3 is stimulated by NF-κB, and TNFAIP3 negatively regulates the NF-κB pathway, by using an IκBα-independent mechanism. 80 Transcription of JunB, a component found in some AP-1 complexes, is stimulated by both NF-κB and AP-1, and JunB regulates AP-1 activity. 81,82 We observed an ∼2- to ∼2.5-fold increase in TNFAIP3 mRNA levels after 4 or 8 hr of treatment with TNF-α in most cell lines (Fig. 4C). In cells stably expressing R332G-R335G TRIM5αhu, activation seemed at first glance more efficient than in the cells transduced with the other TRIM5α variants or with the empty vector (Fig. 4C). However, the effects seen with this TRIM5α variant were not significantly different from the results obtained with the other variants. Expression levels returned to normal by 16 hr after the end of TNF-α treatment (Fig. 4C). JunB showed delayed activation kinetics compared with TNFAIP3. In most cell lines analyzed, higher JunB mRNA levels were found after 8 hr in presence of TNF-α but not after 4 hr (Fig. 4D). In some cell lines analyzed, JunB levels were still higher than normal 16 hr after drug withdrawal (Fig. 4D). We observed some variation among the various cell lines tested, but these effects were not statistically significant. Likewise, the JunB activation profiles were similar in cells stably expressing TRIM5α and in untransduced or empty vector-transduced cells (Fig. 4D). In summary, stable transduction with TRIM5αhu (WT or mutant) or TRIM5αRh had no major impact on the capacity of TNF-α to downregulate IκBα or upregulate NF-κB/AP-1-dependent genes.
R332G-R335G TRIM5αhu strongly restricts HIV-1 IIIB spreading in CEM.NKR-CCR5 cells
Finally, we analyzed how efficiently R332G-R335G TRIM5αhu inhibits HIV-1 over multiple rounds of replication. For this, we infected the various CEM.NKR-CCR5 lines generated with IIIB, a highly pathogenic strain of HIV-1. 83 p24 CA in supernatants was quantified by ELISA at days 8, 15, and 22 postinfection (Fig. 5). As expected, the HIV-1 levels in control, empty vector-transduced cells increased markedly between days 8 and 15, and then between days 15 and 22. Signs of cell death were visible at day 15 and most cells were dead at day 22 (not shown). Compared with these control cells, transduction of WT TRIM5αhu had no effect on HIV-1 propagation, as expected. At all three time points, HIV-1 amounts in the supernatants of cells overexpressing WT TRIM5αhu were similar to the control cells, or even higher (Fig. 5). Transduction of R332G TRIM5αhu decreased HIV-1 replication levels by approximately 10-fold, as seen on days 8 and 15. However, HIV-1 spread efficiently in these cells; by day 22, most cells had died and the amount of virus detected in the supernatants was similar to that of the control. In contrast, cells stably expressing R332G-R335G TRIM5αhu or TRIM5αRh were highly resistant to the spreading of HIV-1 infection. At all time points analyzed, CAp24 levels were lower by several orders of magnitude in these two cell lines, compared with the empty vector-transduced cells or compared with the cells stably transduced with WT TRIM5αhu (Fig. 5). Interestingly, CAp24 levels were highest at day 15 for R332G-R335G TRIM5αhu, and then decreased by day 22. This opens the possibility that virus replication was slowed by R332G-R335G TRIM5αhu, allowing uninfected cells to outgrow infected cells. In summary, R332G-R335G TRIM5αhu and TRIM5αRh but not R332G TRIM5αhu very efficiently restricted the spreading of HIV-1 infection in CEM cells.
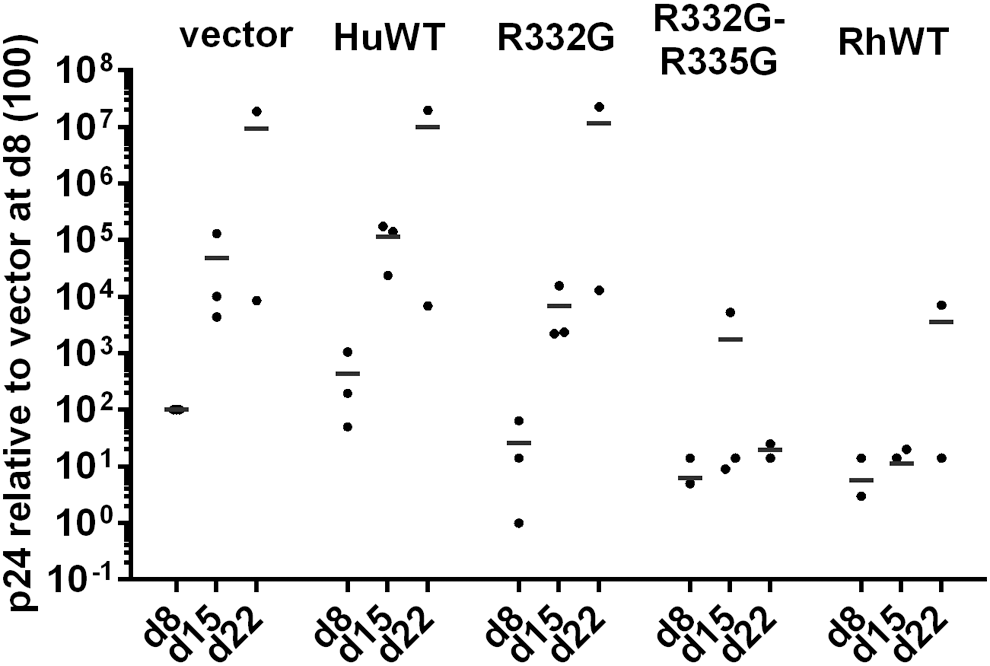
Different TRIM5α variants show markedly different potency of HIV-1 restriction in propagation assays. CEM.NKR-CCR5 cells stably transduced with the indicated TRIM5α variants, or transduced with the empty parental vector as a control (vector), and sorted for GFP expression, were infected with HIV-1 IIIB (MOI 0.01). CAp24 ELISA was performed on supernatants at days 8, 15, and 22. Shown are relative p24 amounts in 2–3 independent experiments, normalized to the value obtained for the vector control at day 8 (×100).
Discussion
Currently, there is only one proven long-term effective treatment against HIV infection. This functional cure is achieved through gene therapy, more precisely through replacement of the WT HIV-1 coreceptor CCR5 with a naturally occurring deletion. 84 Although CCR5 loss-of-function approaches are promising because of the stable endogenous sequence, the virus usually exhibits a switch from CCR5 affinity early in the infection to CXCR4 in the late or mature phase of infection. 4 The possible use of CXCR4 deletion to overcome the therapeutic obstacle has been studied. 85 However, unlike CCR5, CXCR4 is crucial for several immune functions that have no redundant mechanisms. 86 Other common attempts to inhibit HIV-1 via gene therapy have included combinatorial vectors that aim at the virus through RNAi, ribozymes, or other approaches. 87,88 Although RNAi is highly potent, it usually acts at the late stages of the infection, cannot completely prevent viral release, and is often rendered inefficient by mutational escape of the virus. 89,90 Approaches that also use initial cellular entry restriction have therefore become very interesting to the HIV-1 gene therapy community. Although overexpression of peptides that block virus fusion via interaction with the gp41 pocket has been proposed, 91,92 mutational escape can occur as well 93,94 and the immunogenicity of gp41-derived peptides remains to be tested. Restriction factors such as TRIM5α are thought to play important roles in protecting against nonhost retroviruses and limiting cross-species transmission. 95 –98 We and others have previously shown that point mutations in the PRYSPRY domain of the human TRIM5α protein can increase restriction of lab-adapted HIV-1 viral strains. 23,28,30,99,100 Because those constructs differ only slightly from their endogenous counterpart, they would probably not be immunogenic, thus making them interesting candidates for therapeutic treatment of HIV-1. However, it was not yet known whether these TRIM5α mutants would target CA from clinical isolates of HIV-1. Our results show that all strains from a diverse panel of HIV-1 variants are potently restricted by R332G-R335G TRIM5αhu and by TRIM5αrh.
CTL escape mutations were previously shown to increase sensitivity to TRIM5αhu. 39,46 Specifically, mutations in the NRC10 strain affect the CypA-binding loop of CA (V86A and M96I), which is known to play an important role in TRIM5α-mediated restriction. 20,101 –103 In addition, the residue at position 86 was already shown to play a crucial role in the sensitivity to both TRIM5αRh 57,58 and TRIM5αhu mutants. 31 Our results show that the effects of CTL escape mutations on TRIM5α sensitivity can also be extended to a more restrictive human TRIM5α variant like R332G-R335G. This suggests that the highly restrictive TRIM5α variants used in gene therapy applications could help control the rate at which CTL escape mutations appear, and make it more difficult for the virus to escape both host immune responses and transgene-mediated inhibition at the same time.
Unregulated immune activation by transgenic, overexpressed TRIM5α was, potentially, a major impediment to the development of TRIM5αhu mutants as anti-HIV-1 transgenes. This concern was first raised several years ago, when it was demonstrated that transfecting TRIM5α could activate NF-κB and AP-1 through the activation of TAK1 and the generation of K63-linked ubiquitin chains. 21,41,42,60 Most of these previous experiments were done by transfecting HEK293 cells, however. Here we confirm that NF-κB can be activated by transfecting TRIM5α into TE671 cells as well. Importantly however, we could not detect any NF-κB activation in these cells when TRIM5α was expressed through stable gammaretroviral vector-mediated transduction rather than through transient transfection. NF-κB activation in transient transfection conditions might result from very high levels of TRIM5α expression in a fraction of the cells. In addition, DNA transfection itself causes an innate immune response mediated by IFI16 and NF-κB. 104,105 Furthermore, stable lentiviral vector-mediated transduction of TRIM5α in lymphocytes did not lead to any major change in TAK1 activation or in the dynamics of the NF-κB pathway following TNF-α treatment. Altogether, our results strongly suggest that stable transduction of TRIM5α does not activate or modulate this immunity pathway. Pertel et al. also reported that NF-κB could be activated upon interaction between TRIM5α and an incoming restriction-sensitive virus. 21 Although we have not attempted to reproduce this observation in this study, it should be pointed out that this effect, if present, would contribute to the specific (virus-triggered) TRIM5α antiviral activity and thus would be desirable.
R332G-R335G TRIM5αhu was significantly more efficient than the single mutant R332G at inhibiting HIV-1 spreading infection and at increasing the survival of gene-modified cells. These results confirm our previously published data obtained in single-cycle infection experiments using HIV-1 vectors 23,29,31 or in spreading infection of a different HIV-1 strain (NL4-3) in a different T-cell line (Sup-T1). 23,31 Collectively, our data also show that, in lymphocytes, R332G-R335G TRIM5αhu restricts HIV-1 about as efficiently as TRIM5αRh does. We also note that, the two mutations being located close to one another, it should be feasible to introduce them both simultaneously into the human genome, through the use of recently developed genome-editing methods such as those based on clustered regularly interspersed, short palindromic repeats (CRISPR)/CRISPR-associated nuclease 9 (CRISPR-Cas9). 106,107
Footnotes
Acknowledgments
We thank Greg Towers (University College London, UK), Yasuhiro Ikeda (Mayo Clinic, MN), and Allan Hance (INSERM, Paris, France) for sharing plasmids. We are also grateful to Haitang Li and Kathleen Riopel for technical help, and to Mélodie B. Plourde and Natacha Mérindol for helping with redaction of the article. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CEM.NKR-CCR5 (cat # 4376, contributed by Alexandra Trkola) and HIV-1 IIIB (cat # 398, contributed by Robert Gallo). This work was supported by the Canadian Institutes of Health Research Operating grant MOP-102712 (L.B.), National Institutes of Health grant 5R1A1042552 (J.J.R.), and California Institute of Regenerative Medicine grant TB1-01176 (J.J.R.). M.V. was supported by an FRQS Master's Training Award.
Author Disclosure
Lionel Berthoux and Quang Toan Pham are co-owners of a U.S. patent (8,623,815 B2) covering antiviral applications of TRIM5αhu Arg335 mutations. No competing financial interests exist for the other authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.