
Abstract
A basic understanding of genome evolution and the life and impact of microorganisms, like viruses and bacteria, has been fundamental in the quest for efficient genetic therapies. The expanding tool box for genetic engineering now contains transposases, recombinases, and nucleases, all created from naturally occurring genome-modifying proteins. Whereas conventional gene therapies have sought to establish sustained expression of therapeutic genes, genomic tools are needed only in a short time window and should be delivered to cells ideally in a balanced “hit-and-run” fashion. Current state-of-the-art delivery strategies are based on intracellular production of protein from transfected plasmid DNA or in vitro-transcribed RNA, or from transduced viral templates. Here, we discuss advantages and challenges of intracellular production strategies and describe emerging approaches based on the direct delivery of protein either by transfer of recombinant protein or by lentiviral protein transduction. With focus on adapting viruses for protein delivery, we describe the concept of “all-in-one” lentiviral particles engineered to codeliver effector proteins and donor sequences for DNA transposition or homologous recombination. With optimized delivery methods—based on transferring DNA, RNA, or protein—it is no longer far-fetched that researchers in the field will indeed deliver the goods for somatic gene therapies.
Introduction
V
It has certainly not escaped the notice of an entire biomedical research community, including the biotech industry, that new powerful genome engineering tools have entered the market. RNA-guided endonucleases, like the Cas9 nuclease derived from the CRISPR/Cas immune system of Streptococcus pyogenes, 2,3 represent the latest addition and perhaps the currently most prominent instrument in a rapidly expanding tool box. Earlier site-directed nucleases include meganucleases, like the I-SceI meganuclease originally identified in mitochondria of Saccharomyces cerevisiae, 4 and human-made designer nucleases like zinc-finger nucleases (ZFNs) 5 and transcription activator-like effector nucleases (TALENs), 6,7 both of which combine a heterologous DNA-binding domain with a bacterial DNA-cleaving protein domain. For decades, tyrosine recombinases, like Cre from the P1 bacteriophage 8,9 and Flp from Saccharomyces cerevisiae, 10 –12 have been instrumental for site-specific recombination in genome engineering. Also, PhiC31 integrase, a serine recombinase encoded by the phage of Streptomyces lividans, has been investigated for translational use of site-directed transgene insertion. 13 With the resurrection of the Sleeping Beauty DNA transposon element from the genomes of salmonids, like Tanichthys albonubes, 14 transposases emerged as effective nonviral tools for gene insertion in human cells. Soon after followed the equally effective PiggyBac transposase, originally derived from the genome of cabbage looper moth (Trichoplusia ni), and a series of transposases derived from other species (reviewed in ref. 15 ). An overview of the major tools and their main activities are listed in Fig. 1. It is beyond the scope of this review, however, to describe molecular details of these technologies, and the reader is referred to recent reviews for further information. 15 –20
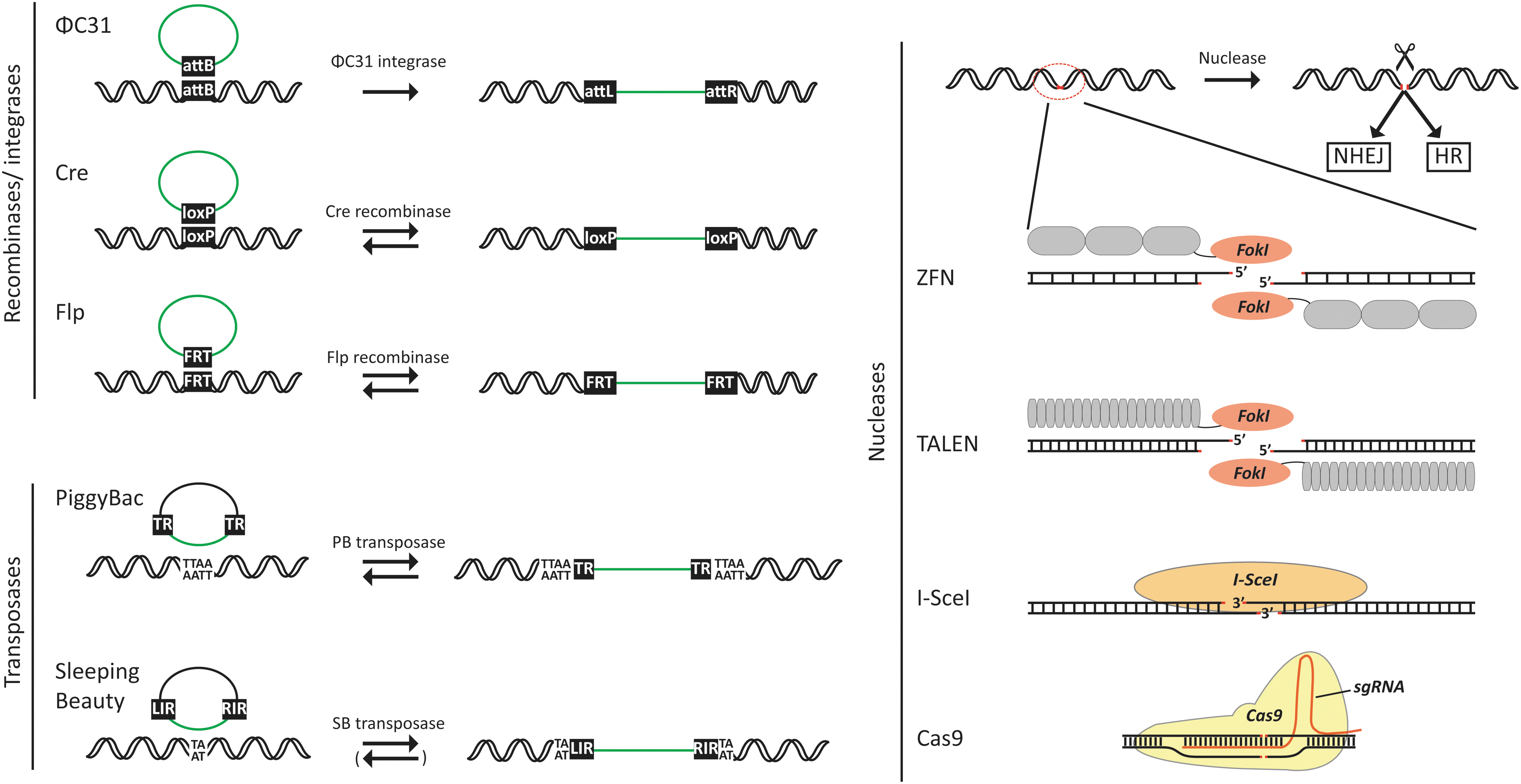
Schematic representation of the most frequently used genomic tools and their action. Site-specific recombinases include ΦC31 integrase and Cre and Flp recombinase. In the shown examples, the recombinases mediate site-directed gene insertion through recombination between recognition sites present in genomic and donor DNA. An arrow pointing toward the right indicates that the process is irreversible, whereas two arrows pointing in opposite directions indicate that the process is reversible. Depending on recognition-site sequences, insertion processes mediated by Cre or Flp can also be irreversible. Illustrated DNA transposases include the most commonly used transposases derived from the piggyBac and Sleeping Beauty transposable elements. DNA transposition is facilitated by recognition of the terminal repeats (designated terminal repeats, TRs, for piggyBac and left and right inverted repeats, LIR and RIR, for Sleeping Beauty) by transposases, leading to transposon excision and subsequent insertion into TTAA tetranucleotide and TA dinucleotide sequences for piggyBac and Sleeping Beauty, respectively. Genomic excision by piggyBac transposase is footprint-free, rendering the process fully reversible, whereas a genetic footprint remains after Sleeping Beauty transposon excision. The right part of the figure illustrates currently available nuclease technologies for introduction of site-directed double-stranded DNA breaks, leading to repair by nonhomologous end joining (NHEJ) or homologous recombination (HR). ZFN and TALEN proteins consist of a site-specific DNA-binding domain (shown in light gray) and an endonuclease motif (FokI) that cuts the DNA as a dimer only. I-SceI is an endonuclease that recognizes and cuts inside an 18 bp sequence, which is not normally present in the human genome. The Cas9 nuclease is guided to a specific DNA sequence by the single guide RNA (sgRNA) that recognizes a predetermined 20 bp sequence, allowing the Cas9 nuclease to cut within the recognition sequence. Color images available online at
Studies of bacterial immunity and the phylogeny of fish transposable elements are examples of basic research creating both the knowledge and inspiration for development of emerging genome engineering technologies. Perhaps with the exception of Cre and Flp, these new tools share a therapeutic potential, which is currently intensively explored and attracting substantial financial investments. However, also common to all of the above protein tools is that they are not naturally transferred between cells and, thus, need to be artificially delivered to cells. For conventional genetic therapies that aim at adding a flawless variant of a mutated disease gene to cells or organs, the goal has been for years to achieve potent gene delivery and at the same time to secure sustained, perhaps even life-long, gene expression. Desirably, a single injection of gene-carrying viral vectors or transfusion of stem cells stably expressing the transgene would be sufficient to provide such long-term treatment. In contrast, for genome engineering purposes the goal would be to deliver the preferred tool, nucleases or transposases, effectively without flooding the cells, and yet in a balanced “hit-and-run” fashion that allows activity of the genome-modifying enzyme only in a short time frame but with sufficient time to carry out the modification.
The delivery remains one of the key challenges that require further attention to unleash the full potential of therapeutic genome engineering. So far, intracellular production from delivered DNA or RNA has been the preferred approach by far. Numerous studies have shown efficacy of tools produced within the treated cells themselves using transfected plasmid DNA or in vitro-transcribed RNA as the source. Also, virally delivered templates may serve as a robust source in hard-to-transfect cell types and for in vivo purposes. In addition, we are starting to see examples of alternative strategies based on the delivery of recombinant protein as well as the first examples of adapting viruses for direct delivery of genome-modifying enzymes. In this mini-review, we discuss some of the advantages and challenges of intracellular production strategies and include considerations related to the delivery of both the engineering tool and the DNA substrate for repair by homologous recombination or DNA transposition. Toward the end of the review, we focus on protein delivery for DNA transposition and editing using novel virus-based approaches.
Plasmid DNA as a Source of Genome-Modifying Enzymes
In its most primitive format, nonviral gene delivery is based on the cellular uptake of plasmid DNA. Plasmid DNA is easily engineered to encode any gene of interest, and transfection of plasmid DNA in cultured cells is a standard technique in the hands of researchers and students worldwide. Because of the negative charge of both plasmid DNA and the surface of cells, the uptake of DNA into cells is restricted, and delivery needs to be supported by complexing DNA with chemicals, like calcium phosphate, that allow effective crossing of the membrane in many cell types 21 or by subjecting the cells to an externally applied force, like an electric field. Hence, nucleofection, a technique based on subjecting cells to a combination of electrical inputs, has become a standard method for transferring DNA to hard-to-transfect cell types. 22 However, penetration of both cell and nuclear membranes using such methods comes at the price of high cellular toxicity and even cell death, and any severe impact on the cells needs to be taken into consideration for downstream applications. Alternatively, plasmid uptake is routinely achieved by wrapping the negatively charged plasmid DNA in a coat of lipids or cationic polymers that may form structures that are often reminiscent of virus particles. 23,24
Transfection of plasmid DNA carrying a gene cassette driven by a strong promoter represented the first obvious choice for establishing intracellular production of genome-modifying enzymes. Hence, initial landmark studies demonstrating transposase-directed mobility of Sleeping Beauty and PiggyBac DNA transposon vectors in cultured human cells were based on cotransfection of plasmids, one encoding the transposase and one carrying the transposon vector. 14,25 Later, in the first clinical trials using Sleeping Beauty–modified cells, T-cells nucleofected with transposase-encoding plasmid DNA as well as a transposon vector carrying the gene for a CD19-specific chimeric antigen receptor were infused into patients with B-lymphoid malignancies undergoing autologous hematopoietic stem cell transplantation. 26 Other studies showed production of induced pluripotent stem cells (iPSCs) using a DNA transfection-based approach for introducing reprogramming factors by DNA transposition in the genome of fibroblasts. 27 –29 In parallel, plasmid transfection provided proof of concept for targeted gene disruption by plasmid-delivered ZFNs, 5,30 TALENs, 31 and CRISPR/Cas9, 32 –34 establishing these tools as the key drivers of the genetic engineering field. Ultimately, nuclease- and DNA transposon-based technologies have been elegantly combined for footprint-free correction of disease-causing mutations in iPSCs. 35 –38 Because of low levels of homologous recombination in some cell types, this editing strategy further adds the genomic incorporation of a selection gene cassette upon homology-directed repair (HDR) of a nuclease-derived double-stranded DNA break. By embedding the selection cassette in a piggyBac transposon located between the appropriate homology arms, the entire element can be removed from correctly targeted clones by subsequent delivery of the piggyBac transposase. Because of the unique features of the piggyBac system, this occurs without leaving any genetic trace, except for the corrected mutation, in the genome of the treated cell. So far, this combined strategy has been based on consecutive transfections of plasmid DNA encoding the nucleases (ZFNs, 38 TALENs, 35,37 or Cas9 nuclease 36 ) and the piggyBac transposase.
Being a standard technique for delivering genes to cultures cells, DNA transfections are easily done and often preferred approaches for ex vivo applications. However, it is well-known that effective plasmid DNA delivery—which is not unlikely to introduce thousands of plasmid copies per cell 39 —is directly associated with a risk of randomly inserting the DNA into the genome of treated cells, providing the cells with the capacity to stably produce transposases or nucleases. Such prolonged expression may impact cell growth, as is the case for Sleeping Beauty transposase 40,41 or perturb genome stability as seen for the PhiC31 integrase. 42,43 It is yet unclear, however, how cells are affected by sustained expression of different types of site-targeted nucleases. The risk of plasmid integration may to some extent depend on the plasmid quality and the ratio of supercoiled, relaxed, and linearized forms in the preparation, but may also be influenced by properties of the treated cells, including the prevalence of double-stranded breaks and the potency of DNA repair pathways, including homologous recombination. The prevalence of genomically integrated helper plasmid encoding nucleases or transposases is often not analyzed or reported, but the concern is supported, for example, by our finding of the transposase-encoding gene cassette in transgenic cloned minipigs generated from skin-derived fibroblasts transfected with components of the Sleeping Beauty system. 44,45
For many applications, relative large amounts of plasmid DNA are used to secure high transfection rates, resulting in the high levels of expression that are often desired for optimal efficacy but which may also increase the risk of random insertion of the plasmid DNA. Importantly, besides a certain gene expression cassette, plasmids contain a bacterial origin of replication and an antibiotic resistance gene required for amplification of the plasmid in bacteria. This raises concerns related to the clinical use of plasmids. Minicircle DNA, derived from plasmid DNA, is devoid of the entire plasmid backbone and may serve as both a protein source and as substrate for inserting DNA. 46 Use of minicircles limits transfer of irrelevant genetic material and should be considered as an alternative to plasmid DNA for DNA-based engineering protocols.
With most transfection protocols, a proportion of the cells will remain untransfected, whereas other cells will have massive overexpression of the genome-modifying enzyme, resulting in a heterogenous population of cells with variable levels of enzyme expression. This may, for example, result in variable copy numbers of inserted transposon vectors. 47 Heavily transfected cells are likely to undergo the intended genetic modification, but may also sustain the production of the genome tool for days. In our hands, activity of piggyBac transposase encoded by plasmid transfected into human cell lines can be detected up to 14 days after transfection (Kristian A. Skipper and Jacob Giehm Mikkelsen, unpublished observations) because of the persistence and stability of the plasmid. Hence, a DNA-based delivery approach fails at creating a short boost of enzyme activity, which would be attractive in terms of increased safety.
In vivo delivery of plasmid DNA is in general challenging and may require the application of extra physical force to the target cells. Over the last 15 years, gene delivery to mouse liver using hydrodynamic tail vein injection 48,49 has been a key model for studying in vivo genome engineering. DNA transposition in vivo was achieved first by Yant and coworkers by coinjecting Sleeping Beauty transposase and transposon into the tail vein of mice. 50 Hence, treatment with a transposon vector carrying the factor IX gene was therapeutic in a mouse model of hemophilia B. Notably, these first studies also showed that transposition was totally blocked when the transposase was expressed at high levels. 50 It is still not clear whether this phenomenon, referred to as overproduction inhibition (described in ref. 15 ), in the mouse reflects restricted transposon mobility or toxicity in transfected hepatocytes. In accordance, however, we found that the transposase expression level had to be carefully tuned by use of an appropriate promoter when transposon and transposase gene were delivered on a single plasmid, leading to correction of hemophilia B in a preclinical mouse model. 51 Similarly, delivering the DNA components of the Sleeping Beauty system has successfully facilitated phenotypic correction in mouse models of hemophilia A, 52 mucopolysaccharidosis types I and VII, 53 and tyrosinemia type I. 54 Not surprisingly, transfection-based delivery of the piggyBac system works equally well, 55 and activity can be further improved by delivery of plasmids encoding hyperactive transposase variants. 56,57 More recently, hydrodynamic injection of plasmid DNA encoding both the Cas9 nuclease and a guiding RNA (the single guide RNA, sgRNA) was successfully applied to introduce mutations in tumor suppressor genes in the mouse liver 58 and to target the cellular repair machinery to mutations in the fumarylacetoacetate hydrolase (FAH) gene causing tyrosinemia type I. 59
Producing Genetic Tools from Transfected In Vitro-Transcribed RNA
Producing genome-modifying enzymes at the site of action—within the treated cells—has the great advantage that delivery of nucleic acids to cells is well-established, is easy to handle, and only requires standard laboratory facilities. Exploiting cells as a production facility also ensures that the protein is massively produced and correctly processed. Considering the risk of inserting plasmid DNA into the genome of treated cells, transfected in vitro-transcribed mRNA is attracting more and more attention as a source of genome-modifying enzymes. Transfected mRNA has a short half-life in cells and, hence, as desired may establish a short-term boost of enzyme activity without risking sustained production because of plasmid integration or persisting episomal plasmid DNA. Using m7G-capped and UTR-stabilized mRNA encoding the Sleeping Beauty transposase protein, Wilber et al. showed the capacity of this strategy in cultured cells and hydrodynamically injected mice 60 and went on to treat a mouse model of tyrosinemia type I by coinjecting a transposon vector carrying the FAH gene with 100 μg in vitro-generated mRNA. 61 In HeLa cells, in vitro-transcribed mRNA encoding the piggyBac transposase was found to have a half-life of 3 hr resulting in a half-life of the transposase of 12 hr. 62 mRNA transfection has been successfully adapted for engineering primary cells, demonstrated recently by the correction of the IL2RG gene in CD34+ hematopoietic stem cells (from an SCID-X1 patient) transfected with ZFN-encoding mRNA. 63 Notably, to ensure sufficient intracellular ZFN production, stem cells (>106) were nucleofected with 175 μg mRNA. Similarly, electroporation of mRNA encoding the SB100X hyperactive Sleeping Beauty transposase has been established for delivery of a CD19-targeting CAR to primary T-cells 64 and is exploited for production of clinical-grade CD19-specific T-cells. 65
Viral Delivery of Templates for Intracellular Production of Genome-Modifying Enzymes
As natural carriers of genetic information, viruses have become well-established key instruments in conventional genetic therapies 66 and hold a great potential also for delivery of genome-modifying tools. The potential of hybrid systems that combine the best of two worlds—a nonviral tool box and viral gene delivery—was first demonstrated by the capacity of adenoviral vectors to deliver the Sleeping Beauty transposase. 67 In this vector system, we also included an Flp recombinase-encoding expression cassette, allowing optimized DNA transposition from circular substrates generated by Flp recombination of adenoviral vector DNA. This approach was recently successfully adapted for treatment of a hemophilia dog model allowing transposon-based insertion of the canine factor IX gene in the liver of treated dogs. 68 For in vitro use, potent gene delivery by adenoviral vectors has proved useful for delivery of site-targeted nucleases. Delivery of ZFNs targeting the human AAVS1 locus in fibroblasts from Fanconi anemia patients represents a recent example. 69 For viral delivery of TALENs, adenoviral vectors may be particularly appealing, since delivery by one of the obvious alternatives, lentiviral vectors, is restricted by frequent recombination between the repetitive sequences of TALEN DNA binding domain. 70
In a similar fashion, adeno-associated vectors (AAVs) have been adapted for nuclease delivery. Both in neonatal and in adult mice, AAV-delivered ZFNs targeting a defective human F9 gene (engineered into the mouse genome) were found to induce stable levels of human factor IX expression in the presence of a donor sequence codelivered by an AAV vector. 71,72 In addition, potent AAV-directed gene delivery to the liver of adult mice was recently adapted for delivery of the Cas9 nuclease and an accompanying guide RNA. Identifying and exploiting a shorter Cas9 variant derived from Streptococcus aureus, referred to as saCas9, Ran and coworkers demonstrated 40% disruption of the Pcsk9 gene in the AAV-treated mouse liver. 73 This expands on previous findings by Ding et al., demonstrating rates of Pcsk9 gene mutagenesis that are higher than 50% by adenovirus-delivered spCas9 (derived from Streptococcus pyogenes). 74 As the PCSK9 protein is involved in degradation of the LDL receptor, knockout of the Pcsk9 gene was found in both cases to lower cholesterol levels in the treated mice. In addition, AAV-based transfer of the SB100X gene led to the finding that AAV-delivered DNA may serve as a substrate for DNA transposition. 75 A related strategy was recently adopted for delivery of the piggyBac system, 76 and a recent report suggests clinical applications of such AAV/piggyBac hybrid systems based on the AAV-directed transfer of piggyBac transposase to the growing liver of newborn mice. 77 A third viral delivery system is based on herpes simplex virus-derived vectors, which have been adapted for transfer of the Sleeping Beauty system and may be particularly attractive for insertion of transposons in CNS. 78
Integrase-defective lentiviral vectors (IDLVs) 79 represent another powerful platform for delivery of transposition and editing systems. We initially explored the IDLV platform for delivery of Flp recombinase and found that DNA generated by reverse transcription of IDLV-delivered vector RNA can serve as a substrate for Flp recombination. 80 Interestingly, we found that both 1- and 2-LTR circles, generated from linear lentiviral DNA intermediates, served as donors for recombination. Using a similar approach, we demonstrated effective Sleeping Beauty DNA transposition by codelivering IDLVs expressing the transposase and carrying the transposon element, respectively, to human cells. 81 These findings were published back to back with a study by Vink et al. reporting similar observations. 82 Notably, in this way the natural lentiviral integration machinery was replaced by a transposase-directed insertion pathway, resulting in an integration profile that was significantly different from that of conventional lentiviral vectors. 81 We now know that the piggyBac transposase may equally well utilize IDLV-delivered substrates for DNA transposition. 47 Along the same lines, episomal DNA intermediates established by reverse transcription of IDLV-delivered vector RNA serve as effective donors for HDR by homologous recombination. 63,69
It has been suggested, though, that the use of free-ended nonviral or IDLV DNA as donors for homologous recombination may be associated with additional illegitimate recombination events. 83 For use in knockout libraries and for screening purposes, lentiviral delivery of the Cas9 and sgRNA genes is well-established and used by many laboratories. 84,85 In its current format, this vector integrates into the genome of transduced cells and establishes sustained expression of both nuclease and sgRNA. This is a powerful tool that may be further adapted for potential in vivo use. Proof of principle for the in vivo use of virus-based systems for delivery of genome-engineering enzymes is now emerging, and future studies should address how the delivery of effector proteins and recombination substrates is perfectly timed and balanced. Currently, viral systems suffer from their inherent capacity to establish prolonged expression of transposases and nucleases, and tight, inducible expression systems would have to be implemented for improved safety. Likewise, delivery carefully targeted to the tissue or organ of interest appears equally important for safe translational use.
Direct Delivery of Proteins for Genetic Engineering
Intracellular production of genome-modifying proteins has been proven both effective and clinically relevant. So if it ain't broke, why fix it? Well, we are not yet familiar with the cellular effects or potential toxicity of sustained production of nucleases, and researchers in the field will keep scrutinizing alternative and perhaps more gentle ways, of delivering genomic tools. Perhaps we can further optimize the window of action in cells, better control the localization of the foreign protein within cells, and even establish sufficient genome-modifying activity by directly delivering the protein to cells without reaching the high protein levels achieved by transfection-based methods.
An emerging applicability of recombinant protein
Because of degradation of protein and the lack of a source encoding the protein, an approach based on direct protein delivery guarantees time-restricted activity within treated cells limiting potential toxicity. Cellular uptake can be facilitated by cell-penetrating peptides (CPPs), also known as protein transduction domains, which can be fused to proteins of interest. 86 However, initial attempts to deliver recombinant Sleeping Beauty and piggyBac transposases to cells in vitro did not lead to effective DNA transposition. 87,88 This could reflect that the purified protein was not active because of suboptimal folding or that recombinant transposase protein was trapped in endosomes. 89 In contrast, however, an intrinsic cell-penetrating capacity of ZFNs facilitated robust uptake of CCR5-targeted ZFNs in cultured mammalian cells, resulting in targeted gene disruption frequencies of 24% in human cells subjected to consecutive treatments with recombinant protein. 90 Along the same lines, a recent study showed gene disruption at the CCR5 locus in a series of human cell types treated consecutively with a mix of CPP-conjugated recombinant spCas9 and sgRNA complexed with CPPs in condensed nanoparticles. 91 This DNA-free method led to mutation rates of up to 16% depending on the cell type. In related work, Kim and coworkers obtained high gene disruption frequencies of up to 79% in human cells electroporated with ribonucleoprotein (RNP) complexes consisting of the recombinant spCas9 protein and in vitro-transcribed sgRNA. 92 Notably, off-target effects were reduced, and cleavage activity was evident immediately after RNP transfection. In addition, the spCas9 protein was only vaguely detectable 24 hr after RNP transfection, whereas plasmid-encoded spCas9 was detectable for at least 3 days after transfection. Hence, protein transfection created a short window of Cas9 enzyme activity.
Adapting lentiviruses for direct protein delivery
In recent years, we have studied the use of engineered lentiviral particles as an alternative strategy for delivering genome-modifying proteins. Viruses are evolutionarily set up to bring their own tool kit and may lend inspiration to new ways of delivering proteins. Hence, the rationale is to load HIV-derived lentivirus particles with foreign proteins of interest and exploit the cell-penetrating capacity of viruses to deliver genome-modifying enzymes to cells and potentially directly inside the nucleus of transduced cells. Different strategies for tricking lentiviruses to incorporate proteins that are not normally part of the virus particle have been explored previously. Early studies aiming at tracking the path of viruses from the cell membrane to the nucleus took advantage of the incorporation of Vpr, an HIV-1 accessory protein, into assembling virus particles. By fusing reporter proteins, like GFP, to Vpr, it was possible to follow the intracellular migration of the virus core. 93,94 This approach, based on the incorporation of approximately 700 copies of Vpr per virus particle, 95 has been adapted also for delivery of I-SceI meganuclease, 96 Cre recombinase, 97 and a series of other proteins (reviewed in ref. 98 ). However, therapeutic translation of such strategy may also suffer from the reported toxicity of Vpr, 99 and—as Vpr is not required for effective lentiviral transduction—Vpr is not normally included in lentiviral vector preparations.
Retroviral particles consist of structural and enzymatic proteins that are incorporated in the assembling virus as Gag and GagPol polypeptides. After budding of the virus, the individual proteins are released from the polypeptides by the viral protease, allowing the components of the virus to “fall into place” in the mature particle. HIV-1 particles have been estimated to contain 5000 copies of each of the structural proteins in Gag, whereas markedly fewer GagPol molecules (∼250) are incorporated in each virus particle. 95 It is an attractive option, hence, to fuse foreign protein with Gag. Studies dating back to the early 1990s provided evidence that protein fused to the Gag polypeptide of gamma-retroviruses can become incorporated in the viral structure. 100,101 Voelkel et al. utilized this strategy to deliver Flp recombinase in particles derived from murine leukemia virus. 102 In the context of lentiviral particles, work by the group of Jun Komano has been instrumental in showing the robust delivery of proteins, including β-lactamase and caspase 3, fused to the matrix protein at the N-terminus of Gag and GagPol. 103,104 Furthermore, foreign proteins fused to the p6 protein at the C-terminal end of Gag 105 –108 and inserted between the matrix and capsid proteins 109 have been successfully incorporated in lentiviral particles.
Given the relatively few full-length GagPol polypeptides included in each lentiviral particle, it is perhaps surprising that proteins fused to the integrase protein at the C-terminus of GagPol are quite efficiently delivered to transduced cells. A trilogy of studies from Seppo Ylä-Herttuala's group in Kuopio, Finland, documented hitch-hiking of integrase-fused proteins, including the mCherry reporter and p53, 110 as well as the I-PpoI homing endonuclease (derived from the slime mold Physarum polycephalum), 111 which recognizes a 15 bp sequence present in the about 600 genes encoding 28S ribosomal RNA in the diploid human genome. Because of the wealth of recognition sites and robust lentiviral transfer of the protein cargo, delivery of I-PpoI was found to be cytotoxic in a manner dependent on the enzyme activity. 111,112 However, by fusing the integrase to a mutated catalytically inactive I-PpoI variant, it was possible to promote targeted lentiviral insertions into ribosome RNA genes. 111 It is important to note also that vectors carrying an integrase fusion protein maintained the capacity to transfer vector RNA. 110
Our own endeavors to develop lentiviral protein transduction for genome engineering were directed first at incorporating the hyperactive piggyBac transposase hyPBase 113 in lentiviral particles. 47 A schematic representation of this approach is provided in Fig. 2. Using the packaging construct design developed by Aoki et al., 103 we fused hyPBase to the N-terminal end of Gag and separated the two domains with a HIV-1 protease cleavage motif. Also, Gag was modified by adding an N-terminal myristoylation signal derived from the Lyn kinase. Upon maturation of particles, hyPBase was released from the polypeptide and induced high levels of DNA transposition (higher than a standard plasmid cotransfection protocol) in cells treated with the virus. Notably, this activity was dependent on viral uptake mediated by the vesicular stomatitis virus glycoprotein present on the surface of the particles and intracellular release of engulfed viruses by the endosomes. Also, the hyPBase protein and vector RNA containing the transposon element could be copackaged in IDLVs, allowing effective codelivery and robust transposition from reverse-transcribed DNA in a panel of cell lines and in primary cells. 47 We learned from this work that robust DNA transposition was indeed achieved despite the fact that the hyPBase could only be detected in distinct foci within cells. Interestingly, we reproducibly found that clones generated by transduction with hyPBase-loaded viruses contained only a single copy of the transgene as opposed to the variable copy numbers in clones generated by a DNA transfection approach.
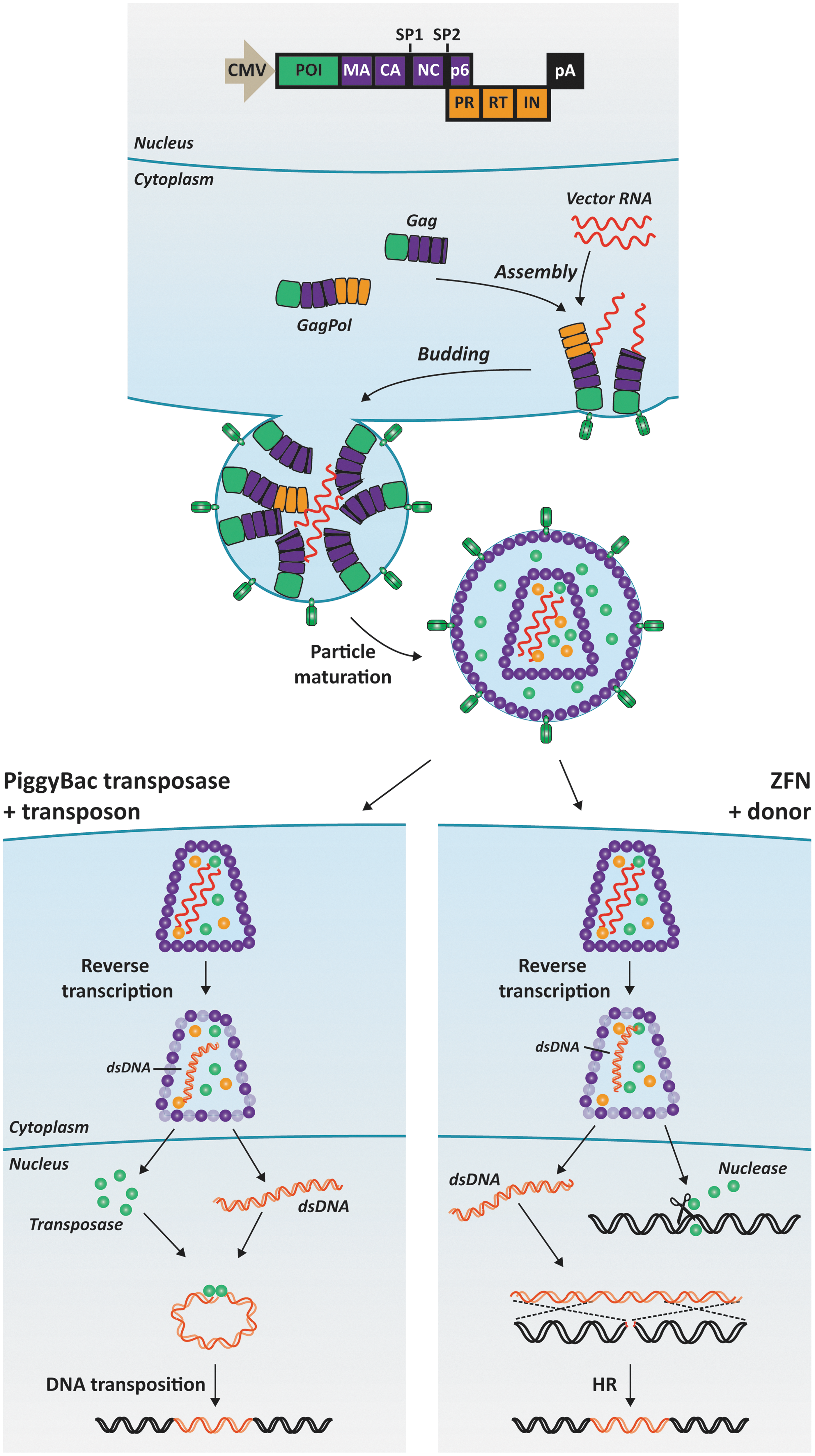
The principles of lentiviral protein transduction. The schematic representation illustrates the production of lentiviral particles loaded with proteins of interest (POIs) as well as the release and action of transduced piggyBac transposases (left) and ZFNs (right) within lentivirus-treated cells. The POI is fused to the matrix protein (MA) in the N-terminus of the Gag and Gag-Pol polypeptides. Approximately 5000 Gag polypeptides and much fewer Gag-Pol polypeptides are assembled during production of a single virus particle. After budding from the cells, the polypeptides are processed by the viral protease (PR), allowing maturation of the particle. The POI is separated from the remaining virus proteins within the virus particle and is capable of exerting its enzymatic function upon internalization in cells exposed to the viral particles. Vector RNA (shown in red wavy lines) can be packaged in virus particles in parallel with foreign proteins and can be engineered to contain the DNA transposon (left) or the donor sequence for homologous recombination, HR (right). Color images available online at
Based on these findings, we went on to establish a similar technology for genome editing. The rationale was to codeliver designer nucleases and the donor sequence for homologous recombination in “all-in-one” viral particles (Fig. 2) and thereby achieve editing without overloading treated cells with site-targeted nucleases. Incorporating pairs of ZFNs targeting the human CCR5 and AAVS1 loci, we achieved levels of gene disruption up to 17% in primary fibroblasts and up to 24% in primary keratinocytes. 114 Our initial findings suggest that such targeting can be achieved with reduced off-target activity of the ZFNs, but further genome-wide analyses still remain to consolidate these observations. In a reporter cell line harboring a mutated eGFP gene, treatment with viral particles loaded with eGFP-targeted ZFNs and the donor sequence for accurate HDR repair induced correction of the mutation in more than 8% of the cells. These findings documented the potential of exploiting lentiviral particles for both protein and donor sequence delivery in genome editing.
Concluding Remarks
As the gene therapy community is gearing up to meet in Helsinki, Finland, to discuss somatic therapies for disease at the annual ESGCT meeting, the controversial and unprecedented use of gene editing in human embryos has surfaced. 115 The actual performance of such experiments and the mindset behind therapeutic germ line alteration is out of line with ethical practice and prevalent moral norms. We are here to understand evolution, not to change it. For a scientific field that has moved quickly and may soon develop treatments for patients, public acceptance will certainly not benefit from the work of researchers failing to resist the temptation to alter human embryos. Generating a slippery slope toward genetic enhancement protocols is certainly not a way for our field to deliver the goods.
With the speed and hype of the developments within the genome editing field, it is no longer far-fetched, however, that gene correction technologies will indeed deliver and make it to the clinic. For genomic editing or gene insertion by DNA transposition, getting the enzyme into cells is a job half done, and delivery of the donor for repair, or the substrate for DNA transposition, needs to be not only equally effective, but also perfectly timed to support correction by homologous recombination rather than repair by error-prone nonhomologous end-joining. And, yes, limitations certainly apply to the current technologies; DNA transposition is not yet efficiently targeted and copy numbers are difficult to control, and CRISPR/Cas9 activity may still be associated with the risk of introducing off-target alterations in the genome. While the details are sorted out, ways to deliver the goods—proteins, guide RNAs, and donors—will remain at the center of the discussion. Indeed, we will find that not only the performance but also the safety and potential cytotoxicity of genome-modifying technologies is closely linked to the delivery strategy.
Quoting Charles Darwin on their recent 2015 release, famous Finnish symphonic rock band, Nightwish, celebrates the grandeur of evolution and the “endless forms most beautiful.” 116 Learning from evolution and embracing tools adapted from endless living forms, somatic genetic therapies have become reality. Individualized, gene-correcting treatments may be waiting just around the corner. See you in Helsinki.
Footnotes
Acknowledgments
Recent work utilizing and exploring genome engineering technologies in the laboratory of J.G.M. is made possible through support of the Danish Council for Independent Research/Medical Sciences (Grant DFF-4004-00220), The Lundbeck Foundation (Grant R126-2012-12456), the Hørslev Foundation, Aase og Ejnar Danielsens Fond, Grosserer L.F. Foghts Fond, Agnes og Poul Friis Fond, Oda og Hans Svenningsens Fond, Snedkermester Sophus Jacobsen & Hustru Astrid Jacobsens Fond, and Familien Hede Nielsens Fond. J.G.M. is head of Gene Therapy Initiative Aarhus (GTI-Aarhus) funded by the Lundbeck Foundation and a member of theAarhusResearchCenter for Innate Immunology (ARCII) established through funding by the AUIdeas program at Aarhus University.
Author Disclosure
No competing financial interests exist.