
Abstract
Successful therapeutic protein production in vitro and in vivo requires efficient and long-term transgene expression supported by optimized vector and transgene cis-regulatory sequence elements. This study provides a comparative analysis of CpG-rich, highly expressed, versus CpG-depleted, poorly expressed green fluorescent protein (GFP) reporter transgenes, transcribed by various promoters in two different cell systems. Long-term GFP expression from a defined locus in stable Chinese hamster ovary cells was clearly influenced by the combination of transgene CpG content and promoter usage, as shown by differential silencing effects on selection pressure removal among the cytomegalovirus (CMV) promoter and elongation factor (EF)-1α promoter. Whereas a high intragenic CpG content promoted local DNA methylation, CpG depletion rather accelerated transgene loss and increased the local chromatin density. On lentiviral transfer of various expression modules into epigenetically sensitive P19 embryonic pluripotent carcinoma cells, CMV promoter usage led to rapid gene silencing irrespective of the intragenic CpG content. In contrast, EF-1α promoter-controlled constructs showed delayed silencing activity and high-level transgene expression, in particular when the CpG-rich GFP reporter was used. Notably, GFP silencing in P19 cells could be prevented completely by the bidirectional, dual divergently transcribed A2UCOE (ubiquitously acting chromatin-opening element derived from the human HNRPA2B1-CBX3 locus) promoter. Because the level of GFP expression by the A2UCOE promoter was entirely unaffected by the intragenic CpG level, we suggest that A2UCOE can overcome chromatin compaction resulting from intragenic CpG depletion due to its ascribed chromatin-opening abilities. Our analyses provide insights into the interplay of the intragenic CpG content with promoter sequences and regulatory sequence elements, thus contributing toward the design of therapeutic transgene expression cassettes for future gene therapy applications.
Introduction
T
Satisfactory transgene delivery rates are, however, of limited use if transgene expression is not sustainable due to silenced or repressed transcriptional activity. Once the transgene resides within the target cell, it is subjected to complex cellular regulation mechanisms, such as epigenetic processes, which can widely vary amongst different cell systems. 9,10 Acknowledging the fact that various cis-acting sequence elements have a crucial impact on such regulatory mechanisms, 11 the rational design of transgene expression modules aims for the inclusion and optimization of regulatory transgene elements that positively affect gene expression in the target cell of choice. 1
One crucial factor for successful transgene expression is the use of an appropriate promoter. Endogenous housekeeping promoters such as the elongation factor (EF)-1α promoter express at low but constitutive rates. Because of this ability, they have become preferred over viral promoters, such as the most frequently used cytomegalovirus (CMV) promoter, which provides high but often short-lived transgene expression, owing to gene-silencing effects. 1 Ubiquitously acting chromatin-opening elements (UCOEs) have been suggested to overcome transgene silencing. UCOEs are regions containing CpG islands extending over dual divergently transcribed promoters derived from housekeeping gene loci. 12 These elements have been reported to provide stable transgene expression in cell culture systems even when integrated into heterochromatin regions. 13 Thus, they confer considerable usefulness for gene therapy and recombinant therapeutic applications.
Further cis-regulatory sequence elements affecting gene expression performance are intragenic CpG dinucleotides. 14 Genome-wide studies have shown that, in particular, first exons and the 5′ region of exons are rich in CpGs. 15 –17 Further analyses attributed CpG-mediated expression enhancement in mammalian cells to an increased transcription rate, 18 which correlates with alterations in the chromatin structure and RNA polymerase II elongation rates. 19
Because cytosines in particular in promoter regions are the exclusive targets for methylation in vertebrates, 20 CpGs are often avoided in vector elements and also in transgene sequences to prevent silencing 21 and to provide prolonged transgene expression. 14,22 –24 In contrast, it was shown that CpG dinucleotides in the transgene open reading frame can provide improved and long-term transgene expression in mouse tissue. 25
These conflicting observations are suggested to be due to the use of different promoters and transgenes, which vary in codon quality or CpG content, and are flanked by diverse noncoding regions or cis-acting sequence elements. Moreover, different delivery systems were used in these studies, resulting in an episomal state or the random integration of multiple gene copies into the host cell genome.
Therefore, a systematic evaluation and understanding of the interplay of standard promoter sequences, regulatory elements, and coding sequences differing in their CpG content is essential for achieving controlled transgene expression at the desired level and with the preferred kinetic. Such an evaluation requires the targeting of expression modules to a transcriptionally active and epigenetically constant genomic environment under standardized conditions. This standardization can be reached using expression systems that integrate a single transgene copy into a defined locus of the cell line of choice. 26 A commonly used technique to achieve site-specific integration is the Flp-In recombinase system, which is based on the sequence-specific recombinase from Saccharomyces cerevisiae. This system has been applied for ex vivo production of therapeutic proteins 27,28 or the generation of vaccine immunogens. 29
Here, we provide a comparative analysis of highly expressed, CpG-rich, versus poorly expressed, CpG-depleted genes encoding humanized green fluorescent protein (hGFP), which was adopted from previous studies. 18,19 We addressed variable expression capacities of these CpG-modified hgfp genes for different standard promoters, cell lines, and gene delivery strategies. Plasmid DNA-mediated hGFP expression in Chinese hamster ovary (CHO) cells via the Flp-In system served as our model expression system to compare various sequence elements and give insight into the molecular regulation mechanisms on which the expression changes are based. To verify our results under more practicable terms, we tested our CpG-modified transgenes in combination with standard promoter and regulatory elements regarding sustainable transgene expression in pluripotent P19 embryonic carcinoma cells upon lentiviral gene transfer.
Materials and Methods
Cell culture
Flp-In CHO cells (Invitrogen, Carlsbad, CA) stably expressing the lacZ-Zeocin fusion gene were cultured in HAM–F12 (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (FCS), 1% penicillin–streptomycin (Pen/Strep), 2 mM
Generation of stable cell lines
CHO cells were cotransfected with the Flp recombinase-encoding plasmid pOG44 (Invitrogen) and with pFRT-GFP-0 or pFRT-GFP-60 at a ratio of 9:1, using the calcium phosphate coprecipitation technique. 30 Positively transfected cells were selected by gradually increasing the hygromycin B (Invitrogen) concentration in the cell culture medium up to 500 μg/ml.
Lentiviral vector preparation and transduction of cell lines
LVs were produced by transient cotransfection of 1.2 × 107 HEK-293 cells with 30 μg of total DNA of the envelope plasmid pcDNA3.1-VSV-G, the packaging plasmid psPAX2, and the GFP-carrying plasmid at a molar ratio of 1:3:4, employing polyethylenimine (PEI) as previously described. 31 Cells were harvested 48 hr posttransfection and cleared by centrifugation at 3000 × g for 10 min. Supernatants containing the LVs were loaded onto a 30% sucrose cushion in phosphate-buffered saline (PBS) and ultracentrifuged at 130,000 × g for 2 hr. The pellets were resuspended in 300 μl of cold PBS and left on ice for 1 hr. After thorough resuspension of the LV particles, the liquid was transferred into an Eppendorf tube and centrifuged at 14,000 × g at 4°C for 5 min to remove any remaining debris. Lentiviruses carrying hGFP-0 or hGFP-60, respectively, under the control of the CMV, EF-1α, or A2UCOE promoter, were used to transduce P19 cells. To obtain P19 cell populations exhibiting similar levels of hGFP-positive cells, P19 cells were transduced using 2-fold serial dilutions of the respective LV batches. On day 2, hGFP expression was measured by flow cytometry, and those samples that exhibited approximately 30% hGFP-positive cells were chosen for the time-course experiment with measurements every 3 days over 20 days.
Flow cytometry
Transfected cells were harvested 48 hr after transfection and resuspended in PBS–1% FCS. Flow cytometry was performed with a FACSCanto II device (with FACSDiva version 6.1.3 software; Becton Dickinson, Franklin Lakes, NJ). Results were evaluated with the FACSDiva version 6.1.3 software. Positive cells were selected using a gate excluding the respective negative cell population, and the mean fluorescence intensity (MFI) refers to cells within this gate. Where indicated, the percentage of positive P19 cells was calculated by histogram subtraction, applying the Overton method in FCS Express V3 (De Novo Software, Glendale, CA) using nontransduced P19 cells as control.
In vitro methylation
Methylation of plasmids was carried out with CpG methyltransferase (M.SssI; New England BioLabs, Ipswich, MA) according to the manufacturer's protocol. Quantitative methylation was verified by digestion of 1 μg of methylated DNA with the CG methylation-insensitive restriction enzyme SacI and the CG methylation-sensitive restriction enzyme ApaI (both from New England BioLabs) for 1 hr at 37°C. Plasmids were purified with a PCR purification kit (Qiagen, Venlo, The Netherlands).
Bisulfite conversion and sequence analysis
A QIAamp DNA mini kit (Qiagen) was used to isolate genomic DNA from cells according to the manufacturer's instructions. Sodium bisulfite treatment of genomic DNA was performed to convert unmethylated cytosine to thymine residues, using an EpiTect bisulfite kit (Qiagen) according to the manufacturer's instructions. Primers used for bisulfite-treated DNA amplification were designed on the basis of converted sequences: Bis-CMV5′ fwd, TTGTATGAAGAATTTGTTTAGGG; Bis-CMV5′ rev, TAATACCAAAACAAACTCCCAT; Bis-CMV3′ fwd, GGATTTTTTTATTTGGTAGTATATTTA; Bis-CMV3′ rev, CTCTAATTAACCAAAAAACTCTACTTATAT; Bis-hGFP60-5′ (915) fwd, TTGTTATTATGGTGAGTAAGGG; Bis-hGFP60-5′ (1359) rev, TAATTATACTCCAACTTATACCCCA; Bis-hGFP60-3′ (1206)-fwd, AGGAGTGTATTATTTTTTTTAAGGA; Bis-hGFP60-3′ (1685)-rev, TAAATATCTACAAAATTCCACCACA.
Primer-binding sites are devoid of CpGs to allow equal amplification of methylated and unmethylated DNA. PCR products of bisulfite-converted DNA were separated by gel electrophoresis, and bands of the appropriate size were cut out and purified with a QIAquick gel extraction kit (Qiagen). Sequence alignment was conducted with the software SeqMan II (version 5.0.3; DNASTAR, Madison, WI) and chromatograms were analyzed with the software Chromas (version 2.32; Technelysium, South Brisbane, QLD, Australia). The methylation levels of CpG dinucleotides were determined by measuring the ratio of each of the cytosine peak heights to the sum of respective cytosine and thymine peak heights in automated DNA sequencing traces, according to a technique published by Jiang and colleagues. 32
Quantitative PCR
Quantitative PCR (qPCR) was performed to evaluate gene copy numbers or to quantify nucleosome-depleted (FAIRE) and immunoprecipitated (ChIP) DNA. A DyNAmo Flash SYBR green qPCR kit (Finnzymes, Vantaa, Finland) and TaqMan genotyping master mix (Life Technologies, Carlsbad, CA) were used for qPCR applications according to the manufacturers' protocols. Quantitative amplification was carried out in a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA). Product specificity was assessed on the basis of melting curves. Fluorescence was measured and expressed as crossing point (Cp) when exceeding the background fluorescence of the PCR master mix, using StepOne software version 2.2.2 (Applied Biosystems).
For relative quantification analyses of hgfp transgene copy number in CHO Flp-In cells, the DyNAmo Flash SYBR green qPCR kit (Finnzymes) was used. The following primers were used: hgfp-TSS-fwd (AGAGAACCCACTGCTTACTGGCTTA), hgfp-TSS-rev (GCTAGCCAGCTTGGGTCTCCCTA), hgfp-ORF-fwd (GGGTGGTGCCCATCCTGGT), hgfp-ORF-rev (GTGGTGCAGATGAACTTCAGGGT), β-Actin-fwd (ACCACCATGTACCCAGGCATTG), β-Actin-rev (GAGCCACCGATCCACACAGAGT), rDNA-fwd (GGCGGACTGTCCCCAGTG), and rDNA-rev (GTGGCCCCGAGAGAACCTC). For hgfp copy number determination in P19 cells, a predesigned custom TaqMan copy number assay (4400294; Life Technologies) targeting the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) was applied. For relative quantification, this assay was combined with the TaqMan copy number reference assay (4403316; Life Technologies) specifically binding to the endogenous mouse telomerase reverse transcriptase-encoding gene (mtert).
PCR efficiencies (E) were determined by evaluation of serial dilutions of the respective templates. E can be calculated from the slope of the standard curve: E = 10–1/slope. Primers were designed such that E was approximately 2. Data were analyzed by the 2−ΔΔC T method.
FAIRE
Formaldehyde-assisted isolation of regulatory elements (FAIRE) analysis was done essentially according to a published protocol. 33 Approximately 3 × 107 exponentially growing Flp-In CHO cells stably expressing the hGFP variants were cross-linked for 7 min at room temperature with 1% formaldehyde added directly to the culture medium. The reaction was quenched by the addition of glycine to a final concentration of 125 mM. Cells were scraped off, washed twice with ice-cold PBS, and collected by centrifugation (700 × g, 5 min, 4°C). The cell pellet was snap-frozen at –80°C for storage or directly resuspended in buffer IA (10 mM HEPES–KOH [pH 7.9], 85 mM KCl, 1 mM EDTA, 1 × protease inhibitor cocktail [Roche, Indianapolis, IN]) and lysed on ice for 10 min in buffer IB (10 mM HEPES–KOH [pH 7.9], 85 mM KCl, 1 mM EDTA, 10% Nonidet P-40, 1 × protease inhibitor cocktail [Roche]). The cell lysate was centrifuged at 700 × g for 5 min and cell nuclei were lysed in buffer II (50 mM Tris–HCl [pH 7.4], 1% sodium dodecyl sulfate [SDS], 0.5% EMPIGEN BB, 10 mM EDTA [pH 8.0], 1 × protease inhibitor cocktail [Roche]). Samples were sonicated with a Bioruptor sonicator (Diagenode, Liège, Belgium) to yield approximately 200- to 500-bp DNA fragments. Cell debris was spun at 16,100 × g for 5 min and the clarified supernatant was treated with RNase A at a final concentration of 0.33 μg/μl for 1–2 hr at 37°C. Twenty-five percent of the sheared chromatin was isolated, treated with proteinase K (0.5 μg/μl) at 56°C for 1 hr, and reverse cross-linked overnight at 65°C. Released DNA was isolated by adding an equal volume of phenol–chloroform–isoamyl alcohol (25:24:1) in Phase Lock Gel light tubes (5 Prime, Hilden, Germany). The remaining 75% of sheared chromatin was directly extracted with phenol–chloroform in the same way without prior proteinase K treatment and reverse cross-linking.
DNA from the aqueous phase of both chromatin fractions (with/without reverse cross-linking) was subsequently precipitated by the addition of ammonium acetate (pH 7.5) to a final concentration of 2.5 M and an equal volume of isopropanol followed by overnight incubation at −20°C. The precipitate was collected the next day by centrifugation (16,000 × g, 30 min, 4°C), washed with 70% ethanol, air dried, and resuspended in 200 μl of water. Quantification of purified DNA was carried out by qPCR on the StepOnePlus instrument (Applied Biosystems), using the DyNAmo Flash SYBR green qPCR kit from Finnzymes according to the manufacturer's instructions. Primers were designed to cover the transcription start site (TSS) (TSS fwd, 5′-AGAGAACCCACTGCTTACTGGCTTA-3′; TSS rev, 5′-GCTAGCCAGCTTGGGTCTCCCTA-3′), a region of the open reading frame (GFP ORF 954 fwd, GGGTGGTGCCCATCCTGGT; GFP ORF 1074 rev, GTGGTGCAGATGAACTTCAGGGT), and rDNA as internal control (rDNA fwd, 5′-GGCGGACTGTCCCCAGTG-3′; rDNA rev, 5′-GTGGCCCCGAGAGAACCTC-3′). Product specificity was assessed on the basis of melting curves. Data were analyzed by the 2−ΔΔC T method. All results were normalized to rDNA and referred to GFP-0 cultivated under selection pressure. They are presented as the ratio of DNA recovered from cross-linked cells divided by the amounts of the same DNA in the corresponding non-cross-linked samples. FAIRE analysis was performed with four independent chromatin preparations each.
Cloning of lentiviral transgene vectors
The lentiviral (LV) vectors pHR′SINcPPT-EF1α-eGFP-WPRE and pHR′SINcPPT-UCOE-EGFP-WPRE (kindly provided by F. Zhang, Institute of Child Health, University College London, London, UK) served as basis for LV construction. The UCOE element was created as previously described. 34 The element EF-1α-eGFP was released from pHR′SINcPPT-EF1α-eGFP-WPRE via EcoRI and SbfI. The elements CMV-hGFP-0, CMV-hGFP-60, EF-1α-hGFP-0 and EF-1α-hGFP-60 were obtained by amplification from the plasmids pcDNA5-CMV/EF-1α-hGFP0/60, with primers introducing the restriction sites EcoRI and SbfI. CMV-hGFP-0, CMV-hGFP-60, EF1α-hGFP-0, and EF1α-hGFP-60 were subcloned into pHR′SINcPPT-WPRE via EcoRI and SbfI to obtain pHR′SINcPPT-CMV-hGFP-0-WPRE, pHR′SINcPPT-CMV-hGFP-60-WPRE, pHR′SINcPPT-EF1α-hGFP-0-WPRE, and pHR′SINcPPT-EF1α-hGFP-60-WPRE. Using pHR′SINcPPT-CMV-hGFP-0-WPRE and pHR′SINcPPT-CMV-hGFP-60-WPRE as a template, hGFP-0 and hGFP-60 were amplified and cloned into pHR′SINcPPT-UCOE-EGFP-WPRE via SalI and NdeI, thereby replacing eGFP and creating pHR′SINcPPT-UCOE-hGFP-0-WPRE and pHR′SINcPPT-UCOE-hGFP-60-WPRE.
Results
Differential transgene expression of CpG-modified hGFP in mammalian Flp-In cells
Because of opposing observations from various studies regarding the impact of CpG dinucleotides on transgene expression, 18,25,35 –37 the critical contribution of the intragenic CpG frequency is still unclear. In this study, gfp genes optimized for human codon usage (hgfp) were used as reporter genes 38 for transgene expression analyses controlled by different promoters, in various cell lines, and under standardized conditions. On the basis of the hgfp sequence comprising 60 CpG dinucleotides (hgfp-60) in its open reading frame (ORF), a synthetic hgfp-0 gene lacking CpG dinucleotides was used as described previously. 18 The reporter genes code for the same amino acid sequence and do not contain any introns. The modifications had only a negligible impact on GC content (55% for hgfp-0, and 61% for hgfp-60) and the codon adaptation index (0.93 for hgfp-0 and 0.96 for hgfp-60). hgfp gene variants were stably transfected into CHO cell lines, using the Flp recombinase-mediated recombination system (Flp-In). 39 The Flp-In system allows site-specific integration of a single-copy transgene, 26 which makes the established cell lines suitable for comparisons between transgene variants and enables their analysis within the same genomic environment. In accordance with results obtained from previous studies, 18 flow cytometric analysis revealed lower hGFP expression for the CpG-depleted as compared with the respective CpG-rich gene variants in stably transfected CHO Flp-In cells. CMV promoter-driven expression was decreased 6- to 7-fold, and EF-1α promoter-driven expression was decreased 4- to 5-fold, for hGFP-0 (Fig. 1, bottom, first time point: compare solid and open symbols).
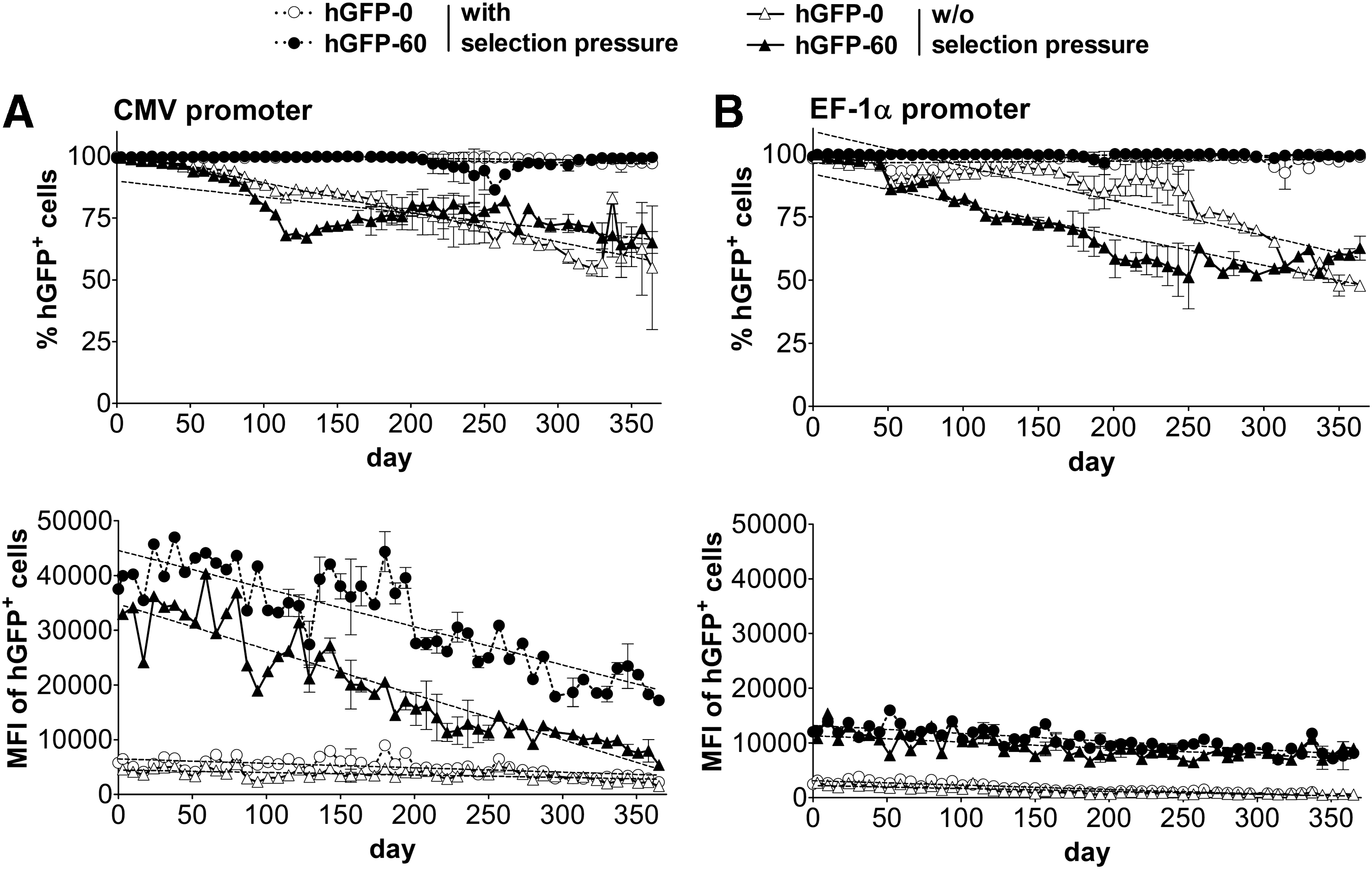
Long-term expression of humanized green fluorescent protein (hGFP) in stably transfected Chinese hamster ovary (CHO) Flp-In cells cultured with or without selection pressure as analyzed by flow cytometry. The expression level of polyclonal CHO Flp-In cells stably transfected with hgfp variants driven by
Impact of selection pressure on long-term expression of hGFP CpG variants
Sustainability of transgene expression in CHO Flp-In cell lines was initially maintained by the application of selective antibiotic pressure, using hygromycin B. The chromatin structure at the promoter and ORF of CpG variants is thereby anticipated to remain permissively open and the DNA unmethylated because of the constant transcription of the hygromycin resistance gene hygromycin-phosphotransferase (hpt), the stop codon of which is located 2.7 kb upstream of the hgfp-controlling CMV promoter. It has been shown that the physical neighborhood of housekeeping genes leads to similar transcriptional activity, 40,41 so that enforcing active hpt is expected to support active hgfp transcription. To address whether intragenic CpG dinucleotides might negatively affect expression levels on selection pressure removal, due to intragenic transgene methylation and chromatin compaction, CHO Flp-In cells stably transfected with hgfp variants were maintained both with (+hygromycin) and without (–hygromycin) antibiotic selection pressure over the period of 1 year (Fig. 1). To determine a possible impact of promoter origin (cellular vs. viral), expression capacities of CMV promoter- and EF-1α promoter-driven transcription were examined in parallel and assessed by quantification of GFP fluorescence by flow cytometry.
The fraction of hGFP-expressing CHO Flp-In cells containing any of the promoters was almost constantly 100% when cultivated under selection pressure and could be maintained in a reasonably high percentage of CHO Flp-In cells without selection pressure after 1 year (CMV promoter: 57 and 74% remaining GFP+ cells for hGFP-0 and hGFP-60, respectively; EF-1α promoter: 54 and 52%, respectively) (Fig. 1). Surprisingly, intragenic CpG dinucleotides did not lead to accelerated loss of hGFP-positive cells compared with CpG-lacking gene variants, either with the CMV promoter or the EF-1α promoter. The expression efficiency, quantified as the mean fluorescence intensity (MFI), decreased over time—although to a different extent—in all cell lines examined. The MFI decreased faster in cell lines expressing hGFP-60 (decrease of 25 × 103 MFI units/year with, and 30 × 103 without, selection pressure, resembling a decrease from the initial MFI down to 43 and 14% after 1 year, respectively) compared with hGFP-0 (decrease of 2.9 × 103 MFI units/year with, and 1.7 × 103 without, selection pressure, resembling 55 and 63% of the initial MFI after 1 year, respectively) when controlled by the CMV promoter. The MFI decrease rates were lower for the EF-1α promoter-controlled variants, regardless of selection pressure (hGFP-60: 5.3 × 103 and 4.2 × 103 MFI units/year, i.e., down to 60 and 63%; hGFP-0: 2.9 × 103 and 1.9 × 103 MFI units/year, i.e., 6 and 13% with and without selection pressure, respectively). Interestingly, despite maintaining hGFP expression in all cells under selection pressure (% positives), the expression levels nevertheless decreased over time, thus indicating ongoing gene silencing. However, increasing the CpG content of the hgfp gene under the control of the EF-1α promoter led—within certain limits—to good resistance against such silencing.
To estimate the relative contributions of CpG content and promoter type to the variance in hGFP expression levels, we performed two-way analysis of variance (Supplementary Table S1; supplementary data are available online at
Several mechanisms underlying the observed reduction in expression are conceivable, especially transgene loss, methylation of the promoter and/or the ORF, and chromatinization. Likely, all of these processes—and possibly others—together may contribute to various degrees to the observed phenotypes. To shed light on the contributions of these gene control mechanisms, long-term hGFP-expressing CHO Flp-In cells were subjected to transgene regulation studies. Only transgenes controlled by the CMV promoter were included in these studies as they showed the most pronounced loss of function in absolute terms.
Relative copy number and methylation status of hgfp in correlation to expression levels with and without selection pressure
CHO Flp-In cells expressing hGFP-0 and hGFP-60, respectively, cultivated without selection pressure were subjected to fluorescence-activated cell sorting (FACS) 1 year after withdrawal of selection pressure. Both cell lines were sorted according to their respective MFIs into the subpopulations “no,” moderate (“mod”), and maximum (“max”) hGFP expression (Supplementary Fig. S1). Genomic DNA of each of the cell populations was isolated to determine hgfp copy numbers and DNA methylation levels of the expression cassette. The removal of selection pressure did not lead to changed transgene copy numbers in moderately and maximally hGFP-0/hGFP-60-expressing cell populations, compared with CHO Flp-In cells maintained under selection pressure, as determined by quantitative PCR (Fig. 2). Notably, the cell populations characterized with a complete deficiency of hGFP protein (“no” gene expression) still contained the hgfp transgene in a substantial fraction of cells (30% in hGFP-0 and 60% in hGFP-60; Supplementary Table S2), implying that the loss of function is only in part due to gene loss and must additionally be due to epigenetic repression. Therefore, the DNA methylation level of CpG dinucleotides was determined after bisulfite treatment by measuring the ratio of each methylated cytosine peak height to the sum of the respective unmethylated plus methylated cytosine peak heights in automated DNA sequencing traces, according to a technique published previously. 32 Whereas cells expressing hGFP-0 and hGFP-60 maintained in the presence of selection pressure exhibited virtually no methylation either in the promoter (Fig. 3) or in the ORF (Supplementary Fig. S2), cells cultivated in the absence of selection pressure showed gradually increasing levels of DNA methylation both in the promoter and in the ORF, and in inverse correlation to their expression performance (Supplementary Table S2).
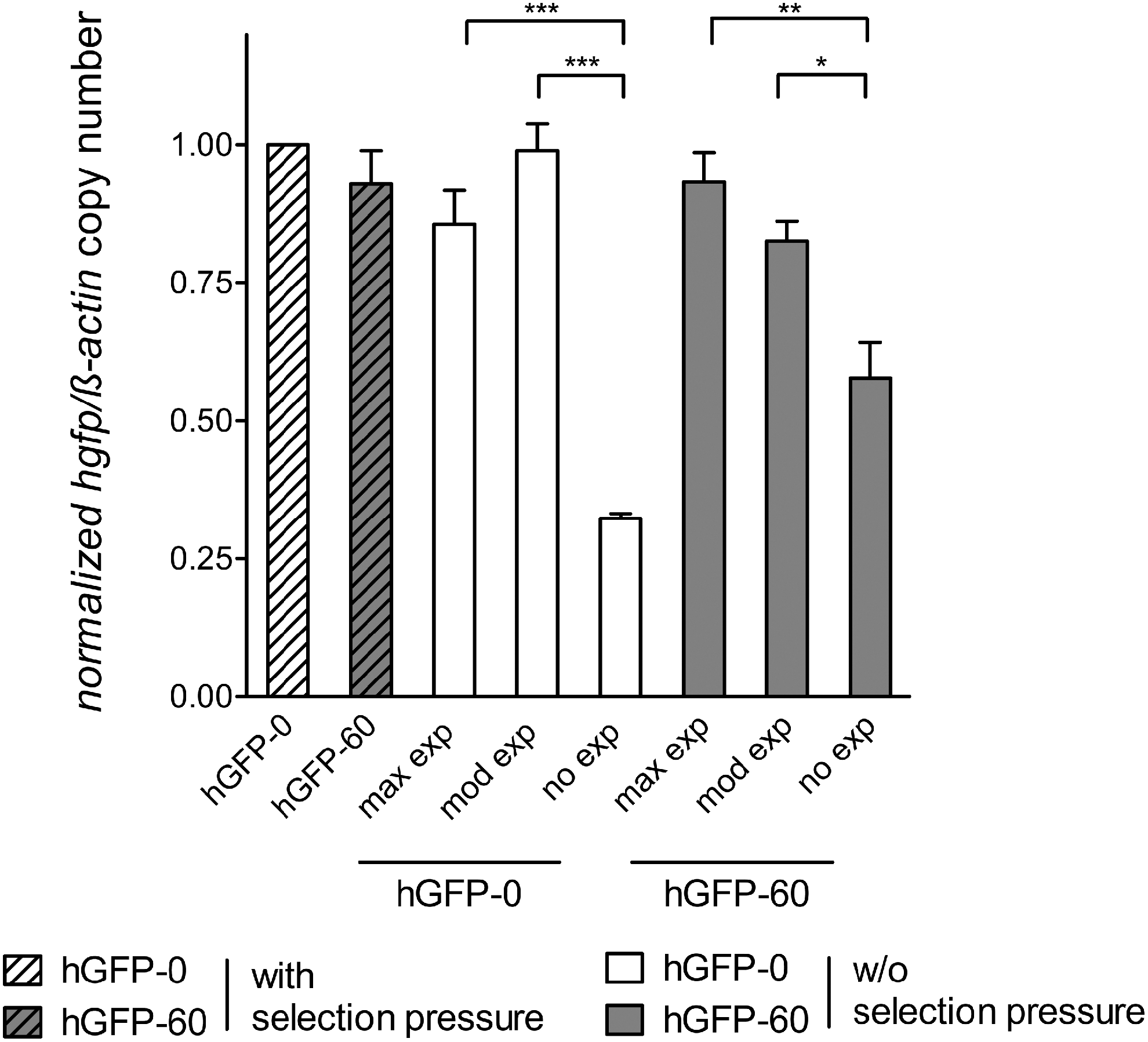
hgfp copy numbers relative to β-actin in CHO Flp-In cells stably expressing hgfp variants under the control of the CMV promoter with or without selection pressure. Genomic DNA of cells sorted into subpopulations (no, moderate [mod], and maximum [max] gene expression) was isolated and subjected to quantitative PCR. Primers encompassing the transcription start site (TSS) of hgfp were used to determine the copy numbers of hgfp transgenes. All C T values were normalized to the corresponding C T values of β-actin and fold changes were evaluated by the 2−ΔΔC T method. hGFP-0 expressed under selection pressure was set to the value 1; the remaining gene variants were scaled accordingly. The mean and standard deviations of two DNA preparations of triplicates each are shown. Significance was calculated by ANOVA/Tukey's multiple comparison test (*p < 0.05; **p < 0.01; ***p < 0.001).
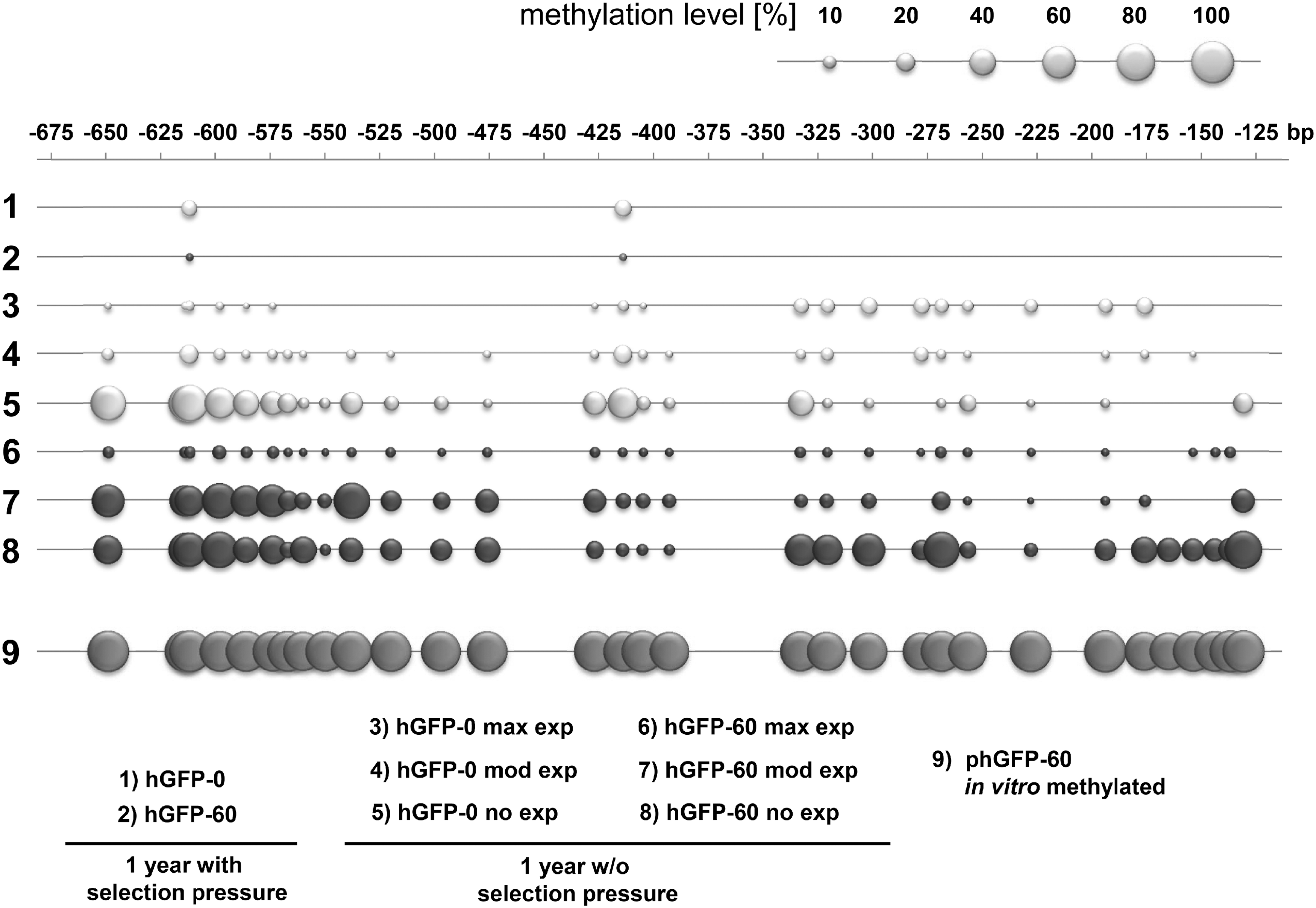
Methylation levels of the CMV promoter. Genomic DNA of CHO Flp-In cells expressing hGFP variants with selection pressure and of CHO-hGFP cells without selection pressure sorted into fractions (no, moderate [mod], and maximum [max] gene expression), was isolated, and subjected to bisulfite sequencing. In vitro methylated phGFP-60 served as a positive control. The methylation level is reflected by the size of the bubbles, as shown in the scale above the diagram. Numbers above the charts represent the distance from the hgfp start codon. Examined cell lines are characterized below the diagrams. The methylation level of CpGs was determined by measuring the ratio of the cytosine peak height to the sum of cytosine and thymine peak heights in automated DNA sequencing traces. 37
Correlation of expression efficiency and chromatin structure
To assess the extent to which differences in DNA methylation translate into changes in chromatin structure 42 and ultimately expression yields, we determined the chromatin density of stably hGFP-expressing CHO Flp-In cells maintained under variable selective conditions. For this purpose, cells were subjected to FAIRE. 33 The FAIRE procedure results in preferential enrichment of nucleosome-depleted genomic regions that can be quantified by real-time PCR. The amount of extracted nucleosome-free DNA detected at the TSS and ORF of hgfp clearly correlated with the presence of selection pressure, indicating that the abolishment of selection pressure induced a significantly increased chromatin density at the transgene expression cassette (Fig. 4). In the absence of selective conditions, no impact of intragenic CpG content on the amount of extracted nucleosome-free DNA was observed. Thus, without selection pressure, we were unable to detect any differences in chromatin density, either at the TSS or in the hGFP ORF. Quantification of isolated nucleosome-free DNA furthermore revealed that intragenic CpG depletion led to a higher chromatin density at the hgfp ORF in CHO Flp-In cells growing under selective conditions, thereby supposedly impeding transcription efficiency, whereas a high intragenic CpG content maintained an open chromatin structure at the ORF in CHO Flp-In cells under selective conditions.

Inverse chromatin densities of CHO Flp-In cells stably expressing hGFP variants in vivo as analyzed by formaldehyde-assisted isolation of regulatory elements (FAIRE). Enrichment for nucleosome-depleted chromatin by FAIRE extraction was performed, and DNA from the aqueous phase was quantified by real-time PCR using primer pairs specific for
CpG-dependent differential transgene expression in murine embryonic carcinoma cells (P19)
Given the progress based on zinc finger nucleases 43,44 or, more recently, transcription activator-like effectors 45 and the CRISPR/Cas9-system, 46 site specific and controlled single copy integration of expression modules may not be an unachievable goal anymore. In this respect, the insights provided by the DNA-plasmid-delivery Flp-In system might support the design of optimized expression modules to achieve sustainable transgene expression in vivo. Currently, retroviral and lentiviral vectors (RVs and LVs) are frequently applied in gene therapy trials, 47 –49 and stem cells are the major source for regenerative medicine. 50 –52 Because of the high gene-silencing potential of stem cells, 52,53 expression sustainability of transgene elements delivered by RV/LV vectors into stem cells must be elaborately tested before their application.
Pluripotent embryonic carcinoma stem cells (P19), exhibiting a high potential of epigenetic regulation, 54 were transduced with SIN-LVs harboring hgfp variants to investigate the susceptibility of the transgenes to become silenced depending on intragenic CpG frequency. Three different promoter–transgene combinations were compared for their ability to confer expression of hGFP CpG-rich and CpG-depleted variants within this system: the CMV promoter, the EF-1α promoter, and the ubiquitously acting chromatin-opening element from the human HNRPA2B1-CBX3 locus (A2UCOE). A2UCOE was reported to sustain stable transgene expression in cell culture systems because of its chromatin-opening feature, even in the absence of selection pressure, or when integrated into heterochromatin regions. 12 SIN-LVs were generated by transient transfection of HEK-293 cells with the envelope plasmid pcDNA3.1-VSV-G, the packaging plasmid psPAX2, and an LV plasmid carrying either hgfp-0 or hgfp-60 under the control of the CMV, EF-1α, or A2UCOE promoter (Fig. 5A).

Level and kinetics of hGFP expression in transduced P19 cells depend on promoter choice. P19 cells were transduced with dilutions of SIN-LV vectors [schematics in
P19 cells were transduced with the various SIN-LVs, employing serial dilutions. Two days later, hGFP fluorescence was quantified by flow cytometry. Cell populations transduced with the lentiviral vectors in the respective dilution that led to approximately 30% of hGFP-positive cells were selected for further propagation. By this procedure, a sufficient GFP starting signal is available for assessment of potential silencing activity, while at the same time an expected low multiplicity of infection (MOI of about 0.3) is used in order to avoid multiple integration events in the majority of cells. To verify the number of integrations, we isolated whole DNA from a fraction of the cells on day 5 and determined the integrated vector copy number (VCN) per cell by quantitative PCR targeting the WPRE, normalized for cell number as determined by mtert qPCR. Whereas the measured values for cells transduced with constructs controlled by the EF-1α or A2UCOE promoter matched expectations quite well (see Supplementary Table S3), cells transduced with the CMV promoter-driven hGFP variants exhibited a high average VCN/cell of 34 for hGFP-0 and 3.2 for hGFP-60. Thus, only by employing an inadvertently high MOI, which leads to a high copy number of the hgfp gene per cell and consequently to a higher overall expression level, it was possible to reach the observed value of 30% hGFP-positive cells on day 2. This implies a rapid and efficient silencing of CMV promoter-controlled transgene expression in P19 stem cells, although less pronounced for the CpG-enriched sequence.
Changes in expression levels of hGFP variants in P19 cells using different promoters
hGFP expression levels were regularly monitored over a period of 20 days by flow cytometry (Fig. 5B and Supplementary Fig. S3).
Despite comparable proportions of hGFP-expressing cells at the start of the experiment (between 28 and 37%), the stability of hGFP expression exhibited high variations between different promoters and CpG variants. The fraction of hGFP-expressing cells rapidly declined within only 5 days when using the CMV promoter (hGFP-0, 13-fold; hGFP-60, 5-fold reduction from day 2 to day 5) and remained at a rather low level thereafter. Accordingly, expression levels also decreased. EF-1α promoter-driven hGFP expression declined at a slower but constant rate to reach a similar low proportion of hGFP-positive cells after 20 days (hGFP-0, 14-fold; hGFP-60, 5-fold from day 2 to day 20), while the expression levels in positive cells were more or less stable. In marked contrast, the number of hGFP-positive cells with the A2UCOE element remained nearly unchanged and exhibited notably stable expression levels.
For transgene expression controlled by the EF-1α promoter, a 2-fold increased MFI was observed for hGFP-60 compared with hGFP-0, whereas expression by the CMV promoter and A2UCOE promoter, respectively, was not affected by intragenic CpG dinucleotides. Notably, high-level hGFP expression was still observed after 20 days in the remaining fraction of hGFP-positive cells (5.7%) transduced with the EF-1α promoter/hGFP-60 expression module. EF-1α promoter/hGFP-60 expression levels exceeded the MFIs observed in hGFP-positive cells transduced with A2UCOE/hGFP-60 and A2UCOE/hGFP-0 by more than 3-fold. To verify the reproducibility of the obtained results, analogous assays were conducted with P19 cells transduced at higher MOIs, resulting in an overall higher percentage of hGFP-positive cells at the start of the experiment but a comparable hGFP expression profile according to MFIs for all vectors used in this study (Supplementary Fig. S4).
Partial prevention of hgfp silencing in P19 cells by DNA methyltransferase inhibition
To test whether the loss of function was associated with DNA methylation, P19 cells were supplemented with DNA methyltransferase (DNMT) inhibitor 5-aza-2′-deoxycytidine (5′aza) on day 20 after transduction, followed by 2 days of incubation and subsequent flow cytometric analysis (Fig. 6). An increased proportion of hGFP-positive cells was detected when they were maintained in the presence of 5′aza. The effect was most pronounced for the CMV promoter-driven construct, with a 16- and 6.8-fold increased expression for hGFP-0 and hGFP-60, respectively, comparing the day 22 values without and with 5′aza treatment. The effect was weaker for EF-1α, with nearly no change for the hGFP-0 variant (1.5-fold) and a 3.8-fold increase for the hGFP-60 variant. Consistent with the enduring expression under the control of the A2UCOE element, 5′aza treatment had little influence on the number of GFP-positive cells (1.3- and 1.6-fold change for hGFP-0 and hGFP-60, respectively). The cell populations cultured without 5′aza supplementation showed little change in the fraction of hGFP-positive cells.
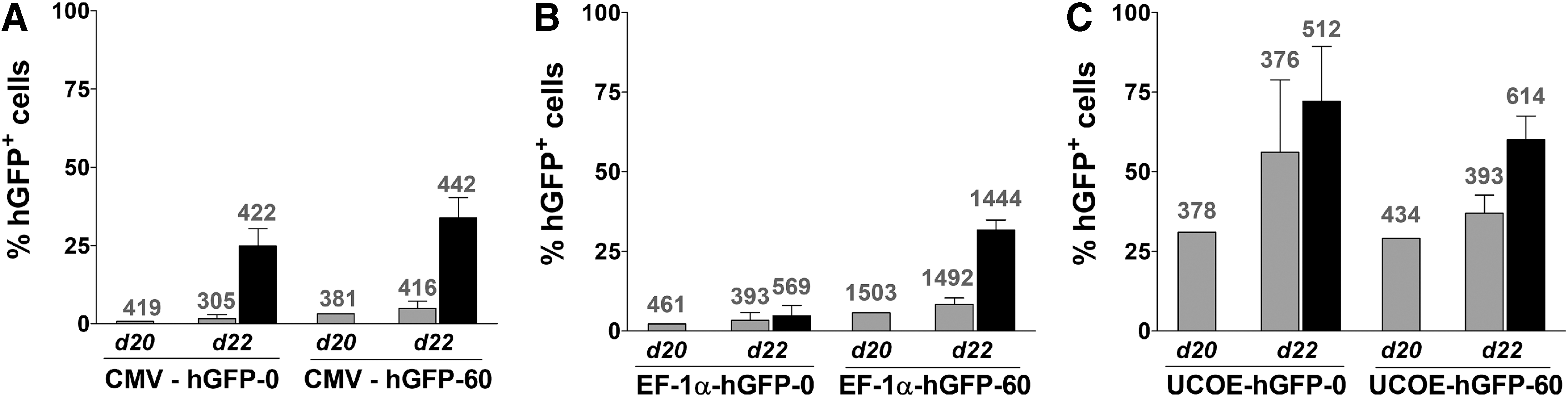
Effect of the DNA methyltransferase (DNMT) inhibitor 5-aza-2′-deoxycytidine (5′aza) on hGFP expression in P19 cells. 5′aza (5 μM) was added via the culture medium to a subset of each cell population on day 20 after infection. The hGFP expression of cells supplemented with 5′aza (solid columns) and of cells without 5′aza (shaded columns) was assayed 2 days later (day 22 after transduction). Columns represent the percentage of hGFP+ cells (calculated by the Overton method), and numbers above the columns indicate the respective MFI of hGFP-positive cells. The CMV promoter
Discussion
The generation of improved expression vectors for recombinant protein production and gene therapy applications requires a detailed understanding of transgene regulation mechanisms. An important regulatory function of transgene expression has been ascribed to CpG dinucleotides, which are actually significantly underrepresented throughout the mammalian genome as a consequence of their high susceptibility to mutation. 55 Despite this negative selection, evolutionary processes have maintained a high CpG frequency within exons, particularly first exons and the 5′ region of exons, compared with introns in human genes. 15 –17,56 It seems obvious that the evolution of this intragenic CpG overrepresentation must confer to gene expression and its regulation any selective advantage over other nucleotide combinations. Depending on the origin, their genomic surrounding, developmental stage, and type of cell, CpG dinucleotides can have either gene-silencing effects 35 –37,57 or can in contrast be beneficial for expression levels. The latter can be due to various effects, such as increasing mRNA stability 58 or increasing de novo mRNA synthesis of transgenes. 19,25 For hgfp genes with modified CpG content in the open reading frame, we have previously shown, using T7 polymerase-driven transcription in the cytoplasm of mammalian cells, that modulation of the CpG content does not affect mRNA stability or translational efficiency. 18 To avoid the spreading of repressive epigenetic marks surrounding the integration locus into the proximal transgene and to prevent loss of the transgene through the outgrowth of transgene-lacking cells, transgene expression is optimally conducted under selection pressure. Of course, this selective force primarily preserves the resistance gene, which can be considered a housekeeping gene because of its essential function for survival under selective conditions. However, it is likely that the regulatory mechanisms acting to keep the resistance gene's chromosomal region in an open state extend to the transgene that is in close proximity (in this case 2.7 kb downstream), as it has been shown that housekeeping genes often occur in clusters where the genes have similar transcriptional profiles. 40,41
Regarding recombinant protein production, there are, however, applications that need cells to grow under antibiotic-free conditions, such as to minimize cellular stress 59 or to avoid the contamination of cells with antibiotics in industrial fermentation processes. 60 More importantly, possible future gene therapy applications using retro- or lentiviral gene transfer, site-specific zinc finger nucleases, 44 transcription activator-like effectors, 45 or the CRISPR/Cas system 46 will depend on expression systems that are highly efficient without selective conditions. Here, we could show that selection for hygromycin resistance also preserved the maintenance of hGFP in CHO Flp-In cells. Almost 100% of the cells could clearly be classified as hGFP-expressing, although the expression level (quantified by flow cytometry and given as relative MFI units) declined over time. Several mechanisms could conceivably account for the observed inactivation of the expression module. By analysis of gene copy numbers (Fig. 2), we could rule out gene loss, which is in line with 100% of the cells being hGFP positive. The methylation analysis (Fig. 3) showed that the promoter and ORF are nearly free of methylated cytosines, thus also ruling out promoter methylation. Therefore, the progressing decrease in hGFP expression levels is most likely due to increased chromatin density and/or repressive histone modifications. Withdrawing the selection pressure resulted in gradually decreasing amounts of hGFP-positive cells and again a concomitant decrease in MFI. Whereas intragenic CpG enrichment accelerated inactivation of the CMV promoter-controlled transgene, hgfp transcription driven by the EF-1α promoter could surprisingly resist gene silencing more effectively with an increased intragenic CpG content. Defective hGFP expression by the CMV promoter coincided with a clear transgene loss, which was almost 2-fold higher for hgfp-0 compared with hgfp-60. Two mechanisms may be responsible for the gradual increase in hgfp-negative cells. First, negative cells might expand over time because of a growth advantage, which might be minimal but manifests over the very long time scale of the experiment. Second, and less likely, cells might lose again the transgene cassette by genomic deletions. In addition, the decline in expression levels is due to increased transgene methylation levels of the CMV promoter of both transgene variants and the ORF of hgfp-60. Because of the tendency of de novo DNMTs to spread bidirectionally in the genome, 61 it is assumed that the excessive amount of intragenic CpG dinucleotides in hGFP-60 attracts many de novo DNMTs, leading to ORF methylation, which then spreads into the promoter, thus causing a higher methylation rate in the hgfp-60-controlling CMV promoter compared with hgfp-0. Considering the inverse rates of DNA methylation and transgene loss between hgfp variants, we suggest that the reduced methylation targets of hgfp-0 compared with hgfp-60 were compensated by an increased frequency of complete transgene loss.
Analogous to DNA methylation, chromatin density at the TSS and ORF of hgfp inversely correlated with the application of selection pressure. Interestingly, hgfp-0 showed significantly higher chromatin density at the ORF compared with hgfp-60. It is assumed that the increased chromatin density resulting from intragenic CpG depletion is a major contributor to impeded transcription efficiency observed in CHO Flp-In cells under selective conditions. Overall, the results imply that chromatin structure plays a crucial role in CpG-mediated transcription regulation in CHO Flp-In cells, which complements results from our previous studies. 18
To test the expression performance and sustainability of CpG-modified hgfp variants in a gene therapy-relevant system, pluripotent embryonic carcinoma cells (P19), exhibiting high potential for epigenetic regulation, 54 were transduced with SIN-LVs incorporating hgfp variants. The stability of hGFP expression over time revealed substantial differences depending on the driving promoter. Whereas the CMV promoter is widely used because of its strong gene expression potential in several tissues, 62,63 it is also known to confer highly variable expression depending on the cell type, 64 which seems most critical in embryonic stem cells. 65,66 Indeed, the disposition of the CMV promoter to undergo extensive epigenetic repression in embryonic stem cells is emphasized by our finding that considerably more SIN-LVs, as determined by measuring the integrated vector copy number 5 days posttransduction, were necessary to obtain a similar fraction of hGFP+ cells on day 2 as compared with the other promoters studied. This indicates that the majority of expression cassettes had been silenced rapidly. Therefore, inadvertently we employed an excessive MOI for transduction, but most probably no hGFP signal at all would have been measurable for comparable low MOIs of about 0.3 as were applied in the case of the EF-1α and A2UCOE settings. The characteristics of such a high silencing activity of CMV promoter-controlled transgenes in P19 cells were more pronounced in hgfp-0. Thus, reducing the intragenic CpG content, which is a widely used strategy in transgene expression applications, 24,35,36 did not prevent gene silencing in this cell type. In fact, the extremely low transcriptional activity of CpG-lacking transgene expression even seemed to promote the rapid silencing of CMV-controlled hGFP expression.
The human EF-1α promoter, claiming robust and constitutive retroviral transgene expression, appeared to be more suitable to mediate hGFP expression in P19 cells compared with the viral CMV promoter. Nevertheless, EF-1α-mediated transgene expression also declined gradually over a period of 20 days. Interestingly, the EF-1α-controlled expression level of hGFP in P19 cells was increased by more than 2-fold in hgfp-60 compared with hgfp-0. Consistent with CMV promoter vectors, EF-1α promoter-mediated hGFP expression therefore seems to benefit from intragenic CpG content, leading to delayed gene repression. Several cis-regulatory sequence elements have been proposed to avoid transgene silencing, such as locus control regions (LCRs), chromatin insulators, or scaffold/matrix attachment regions (S/MARs). 67 –69 In addition to these elements, ubiquitous chromatin-opening elements (UCOEs) consisting of divergently transcribed promoters of housekeeping genes, surrounded by a methylation-free CpG island with chromatin-opening abilities, were demonstrated to induce stable transgene expression. A UCOE derived from the human HNRPA2B1-CBX3 locus (A2UCOE) was shown to confer stable levels of transgene expression in various cell lines, including the murine pluripotent iPSC-7 line 70 and the embryonal carcinoma P19 cell line. 34,71
We did indeed detect a constant fraction of hGFP-expressing P19 cells, and efficient expression of hGFP-0 and hGFP-60 when controlled by the A2UCOE element over a period of 20 days. Moreover, a high ratio of hGFP-positive cells related to vector copy number was observed, indicating a low silencing tendency. Remarkably, gene expression was equally efficient between hGFP-0 and hGFP-60. A2UCOE can provide a transcriptionally active environment through its chromatin-opening features. It is assumed that the methylation-free CpG islands of A2UCOE interact with active histone modifications and that the bidirectional transcription by the A2UCOE promoter is associated with an inherent chromatin-opening function. 71 This antisilencing activity can also be mediated by the CBX3 promoter part alone and is associated not only with less CpG methylation of the promoter, but also with a higher level of activating and lower levels of repressing histone modifications. 72 On the basis of these observations, and in concordance with the previously described findings, the chromatin-opening features of A2UCOE seem to overcome the establishment of a more repressive chromatin state induced by the lack of CpG dinucleotides in hgfp-0. Indeed, it was shown that A2UCOE-driven transgene expression in human fetal liver hematopoietic stem cells was stable over a period of at least 10 months and clearly outperformed other promoters (EF-1α and the phosphoglycerate kinase-1 promoter [PGK]). 73
Previous studies demonstrated a clear correlation of DNA methylation and declined transgene expression in P19 embryonic carcinoma cells when controlled by a viral promoter. 71 The supplementation of DNMT inhibitor 5-aza-2′-deoxycytidine (5′aza) to P19 cells carrying stably integrated hgfp variants indeed revealed a clear contribution of DNA methylation events to transgene repression. In contrast to CMV and EF-1α promoter-mediated gene expression, A2UCOE-driven transgenes were almost not affected by the demethylating agent. The inability of 5′aza treatment to reestablish initial high levels of hGFP-expressing cells is assumed to be due to further epigenetic silencing effects, such as histone modifications, which are not affected by 5′aza. This hypothesis is supported by similar observations in previous transgene expression analyses conducted in P19 cells. 71
In conclusion, the data clearly demonstrate that the stability of transgene expression in SIN LV-transduced P19 carcinoma stem cells depends on the choice of promoter and transgene sequence. Most notably, transgene expression in embryonic stem cells can, if controlled by the appropriate promoter, benefit from an augmented intragenic CpG frequency, as shown by the human EF-1α promoter. The results gained in this and previous work imply that this effect results from destabilization of the chromatin structure. These chromatin changes are assumed to result from a complex epigenetic regulation network triggered by intragenic CpG changes. The exact mechanism of this phenomenon remains to be elucidated in future experiments.
Thus, our data suggest that the impact of intragenic CpG dinucleotides on transgene expression varies depending on promotor choice and targeted cell type. It will therefore be interesting to expand the current study to a larger set of cell line and promoter combinations in order to identify general patterns, which inform the choice of promotor and gene design for ex vivo production and in vivo gene therapy purposes.
Footnotes
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.