
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease; survival in ALS is typically 3–5 years. No treatment extends patient survival by more than three months. Approximately 20% of familial ALS and 1–3% of sporadic ALS patients carry a mutation in the gene encoding superoxide dismutase 1 (SOD1). In a transgenic ALS mouse model expressing the mutant SOD1G93A protein, silencing the SOD1 gene prolongs survival. One study reports a therapeutic effect of silencing the SOD1 gene in systemically treated adult ALS mice; this was achieved with a short hairpin RNA, a silencing molecule that has raised multiple safety concerns, and recombinant adeno-associated virus (rAAV) 9. We report here a silencing method based on an artificial microRNA termed miR-SOD1 systemically delivered using adeno-associated virus rAAVrh10, a serotype with a demonstrated safety profile in CNS clinical trials. Silencing of SOD1 in adult SOD1 G93A transgenic mice with this construct profoundly delayed both disease onset and death in the SOD1 G93A mice, and significantly preserved muscle strength and motor and respiratory functions. We also document that intrathecal delivery of the same rAAVrh10-miR-SOD1 in nonhuman primates significantly and safely silences SOD1 in lower motor neurons. This study supports the view that rAAVrh10-miR-SOD1 merits further development for the treatment of SOD1-linked ALS in humans.
Introduction
A
Familial ALS, which represents about 10% of all ALS cases, is inherited as a dominant trait, and of these cases approximately 20% arise from mutations in the gene encoding Cu/Zn cytosolic superoxide dismutase 1 (SOD1). 5 An estimated 12–23% of familial ALS and 1–3% of sporadic ALS patients carry a mutation in this gene; 183 mutations in SOD1 have been identified (ALS online genetics database). The precise mechanisms whereby mutant SOD1 proteins are neurotoxic are not fully defined. Evidence suggests that mutant SOD1 acquires toxicity via conformational instability, misfolding, and some degree of aggregation. 6 In turn, this activates multiple adverse events that include the unfolded protein response, 7 endoplasmic reticulum (ER) stress, 8 mitochondrial damage, 9 heightened cellular excitability, 10 impaired axonal transport, 11 and some elements of apoptotic 12 and necrotic 13 cell death. Some data suggest that the misfolded mutant SOD1 protein can spread from cell to cell in a prion-like fashion. 14 Mutant SOD1 can cause toxic misfolding of wild-type SOD1. 12,15
Multiple studies have robustly documented that silencing expression of the mutant SOD1 protein prolongs survival of SOD1-linked ALS mice. Approaches to silence SOD1 have included shRNA, 16 transgene-delivered shRNA, 17 antisense oligonucleotides, 18 and virally delivered silencing elements. Initial viral delivery studies achieved successful attenuation of SOD1 using a lentivirus. 19,20 More recent studies have employed recombinant adeno-associated virus (rAAV), which is now the viral vector of choice for many gene therapy approaches. A 2013 study showed that silencing SOD1 in the high-copy transgenic SOD1 G93A mouse with an rAAV9-shRNA delayed onset and increased survival in mice treated at birth; treatment at later stages also prolonged survival, but without delaying onset. 21 The same group later demonstrated the efficacy of this approach in rats, showing that silencing SOD1 in the upper motor neurons also prolonged survival. 22 More recently, rAAV6 and rAAV9 vectors expressing an artificial microRNA (miRNA) against SOD1 were also shown to prolong survival in P2-treated reduced-copy SOD1 G93A mice, 23 and the survival benefit was 26% with rAAV6 and 14% with rAAV9, a much more modest result than had been obtained in P1-treated animals by Foust et al. 21 In the rAAV6/rAAV9 study, no increase in survival or delay in onset was shown when the reduced-copy SOD1 G93A mice were treated as early adults (P35). 23 Finally, an rAAVrh10 vector expressing an artificial miRNA against SOD1 extended survival of adult SOD1 G93A mice (with a reduced transgene copy number) by about 11% when delivered intrathecally, but did not delay onset or showed any improvement in motor function. 24
The current study was designed to optimize the efficacy of rAAV-mediated RNAi as a treatment for SOD1-linked ALS in adult mice with the expectation that this approach would be clinically applicable. An artificial miRNA molecule was selected to induce silencing. Its expression and efficacy in SOD1 silencing were quantified using two constructs that compared polymerase II (pol II) to polymerase III (pol III) promoters. These constructs are packaged in rAAVrh10, which was selected both because it demonstrated excellent CNS transduction 24 and because it has proven to be safe when delivered to the CNS in humans. 25 In the present study, we have delivered the therapy to adult ALS mice (P56–68 high-copy SOD1 G93A mice) and adult nonhuman primates via intrathecal administration, employing a paradigm that may ultimately be useful as a therapy for mutant SOD1-mediated ALS in humans. This proof-of-concept study documents that expressing miR-SOD1 from a pol II or a pol III promoter significantly prolongs survival after delivery in adult mice, and demonstrates feasibility of intrathecal delivery of the same vector in the nonhuman primate (NHP) Callithrix jacchus.
Materials and Methods
In vitro validation
Human embryonic kidney cells (HEK293, 1E6) were transfected with 2 μg plasmid DNA (Jetprime; PolyPlus) and were harvested at 48 hr posttransfection; total RNA was isolated (Trizol; Life Technologies) and SOD1 transcripts were quantified by RT-qPCR. Experiments were carried in biological triplicates. HEK293 were transfected with 4 μg plasmid DNA (Lipofectamine 2000; Life Technologies) and were harvested at 72 hr posttransfection; Western blot was performed as described previously. 26
Constructs
For the in vivo studies, three constructs were used: a control vector expressing GFP only and two vectors expressing miR-SOD1 driven by either a polymerase II (pol II) or a pol III promoter. The control (CB-GFP) construct is composed of the CMV enhancer, chicken beta-actin promoter (CB) containing the Promega intron, green fluorescent protein (GFP) gene, BGH poly A termination signal. The CB-miR-SOD1 construct contains the same CB-GFP cassette, with the addition of two copies of the artificial miRNA designed to target human SOD1 (miR-SOD1) located within the 3'UTR of the GFP gene. The U6-miR-SOD1 construct contains a first cassette composed of the CMV enhancer, CB promoter containing the SV40 and not the Promega intron, GFP, SV40 poly A, and a second cassette composed of the U6 promoter upstream of one copy of miR-SOD1. The same constructs/vector batches were used for the murine experiments.
Viral vectors and vector batches validation before NHP study
rAAVrh10 vectors were produced, and vector particles and vector genome copies (gc) were quantified by the University of Massachusetts Medical School Vector Core. Titers were quantified simultaneously for all batches.
Before the NHP experiment, vector batches were tested in SOD1
G93A mice for SOD1 silencing. The mice (n = 3 per group) received 2E11 gc of vector intravenously through the lateral tail vein, and liver SOD1 and Sod1 knockdown were assessed by RT-qPCR at 2 weeks postinjection (Supplementary Fig. S1; Supplementary Data are available online at
Animal experiments
All murine experiments were performed at the University of Massachusetts Medical School and were approved by the Institutional Review Board. Male and female adult (P50–68) high-copy SOD1
G93A mice
The C. jacchus experiment was performed at the Harvard Primate Center and was approved by the Institutional Review Board. Marmosets were prescreened for neutralizing antibody (NAb) levels against AAVrh.10 by the University of Massachusetts Medical School Vector Core. In addition, all animals were of similar age (less than 4 years old) and body weight (353–476 g). They received 300 μl of vector intrathecally at lumbar level for a total dose of 6E12 gc/kg body weight. Tail flick reflex was monitored to confirm proper placement of the needle. The animals were euthanized 18–23 days postinjection.
Physiology measurements
Rotarod
Before measurements, mice were trained on an accelerating rod (Med Associates Inc.) until they could remain on the apparatus for 1 min without falling. They were then subjected to rotarod testing on a biweekly basis. The latency to fall from the apparatus was recorded for a duration of 5 min. Three trials were performed for each animal and the longest time taken to fall was recorded.
Grip strength
Two and four limbs' muscle strength was determined by measuring peak force using a digital grip strength meter (Mark-10 force gauge model M4-2) equipped with a hind limb pull bar. Mice were allowed to grip the metal grids, and gently pulled backward by the tail until they could no longer hold the grids. The grip force was observed over 3 trials and the maximum force was recorded. Ventilation was quantified using whole-body plethysmography in unrestrained, unanesthetized mice as previously described. 27 –29 Briefly, mice were placed inside a 3.5″ × 5.75″ Plexiglas chamber, which was calibrated with known airflow and pressure signals before data collection (EMKA Inc.). Data were collected in 10 sec intervals, and the Drorbaugh and Fenn equation 30 was used to calculate respiratory volumes, including tidal volume and minute ventilation. The plethysmography is used with IOX2 software (EMKA Inc.). During both a 30 min acclimation period and subsequent 30–60 min baseline period, mice were exposed to normoxic air (21% O2, 79% N2). At the conclusion of the baseline period, the mice were exposed to a brief respiratory challenge, which consisted of a 10 min exposure to hypercapnia (7% CO2, 21% O2, balance N2). Experiments were conducted on paired control and treated animals (n = 7). For all the physiological parameters, significance was determined with a two-way ANOVA.
Immunostaining of NHP tissue
Agarose-embedded fixed spinal cord tissue was sectioned at 40 μm and floating sections were immunostained for GFP. Briefly, the endogenous peroxidases were saturated for 15 min in 0.5% H2O2, 10% methanol. The tissue was then permeabilized and blocked for 1 hr in 1% Triton X-100 and 10% normal goat serum (ab7481; Abcam). Sections were incubated in a 1:2000 dilution of primary antibody (anti-GFP antibody ab13970; Abcam) for 5 days at 4°C, then in a 1:400 dilution of secondary antibody (Biotinylated Goat Anti-Chicken IgG Antibody, BA-9010; Vector Labs) for 2 hr at room temperature, then in the avidin/biotinylated enzyme complex (Vectastain Elite ABC kit; Vector Labs PK-6105) for 1 hr at room temperature, and finally in the peroxidase substrate (NovaREDd kit Peroxidase Substrate Kit, SK-4800; Vector Labs). Tissue sections were then dehydrated and mounted before imaging on a Leica DM5500B using Leica Application Suite (Leica Microsystems).
Laser-capture microdissection of motor neurons and RNA isolation
Laser-capture microdissection of motor neurons was performed by an experienced technician at the Harvard NeuroDiscovery Center (∼2000 cells per section per animal). RNA was subsequently isolated using Ambion RNAqueous-Micro kit (Life Technologies).
RT-qPCR
First-strand cDNA was synthesized with the High Capacity RNA-to-cDNA kit (Life Technologies). Commercial hsa SOD1 and hsa HPRT were used for the in vitro validation, and hsa SOD1 and mmu Hprt TaqMan assays were used for the murine experiment. Custom primers and FAM-labeled probes were designed for detection of C. jacchus SOD1 and HPRT mRNA, but not gDNA. A custom small RNA TaqMan assay was designed for detection of mature miR-SOD1, and a commercially available assay was used for detection of snoRNA-135 (primer and probe sequences or commercial references available in Supplementary Table S2). All TaqMan assays and master mixes were ordered from Life Technologies and used according to the manufacturer's recommendations. RT-qPCR data were analyzed with the ΔΔCt method. 31
RNA multiplexed branched fluorescent in situ hybridization
Frozen spinal cord tissue was cryosectioned at a thickness of 20 μm. Multiplexed branched fluorescent in situ hybridization was performed according to the manufacturer's recommendations (ACDbio). Briefly, the sections were fixed for 15 min at 4°C in 10% neutral-buffered formalin and pretreated with a protease-based solution for 30 min at room temperature to increase availability of the target mRNA. DNA probes were hybridized with the target mRNAs (GFP in C1, Alexa488; SOD1 in C2, Atto 550; ChAT in C3, Atto 647) for 1 hr at 40°C and bound probe signal was subsequently amplified through branching before imaging on a Leica DM5500B using Leica Application Suite.
Biodistribution in NHP
Genomic DNA (gDNA) was isolated (DNeasy Blood and Tissue kit; Qiagen) from frozen tissue and ddPCR (Biorad) was performed according to the manufacturer's recommendations using 50 ng of DNA as input and TaqMan assays detecting GFP and Cja albumin (Supplementary Table S2).
Neutralizing antibody response to AAVrh.10
Serum sampled before injection and at weeks 1 and 3, and CSF sampled at week 3 were heat-inactivated for 30 min at 56°C. AAVrh.10-CMV-LacZ (3E8 gc/well) was incubated with 2-fold serial dilutions of the samples for 1 hr at 37°C, 5% CO2. The mixture was then added to 1E5 Huh7 cells previously infected with an adenovirus (100 vp/well) and incubated for 18–22 hr at 37°C, 5% CO2. Cells were washed and developed with Galactor-Star kit (Life Technologies). Luminescence was measured with a luminometer (Synergy HT, Biotek). The NAb titers are expressed as the highest dilution that inhibited β-galactosidase expression by at least 50% compared with a negative mouse serum control (M5905; Sigma-Aldrich).
Results
Design of an artificial miRNA (miR-SOD1) that is highly efficient in vitro
The artificial miRNA miR-SOD1 was designed based on the backbone of cellular miR-155 (Fig. 1a). For the in vitro validation, plasmid DNA was transfected into HEK293 cells. Substantial reduction in SOD1 mRNA (Fig. 1b) and protein levels (Fig. 1c) was verified at 48 and 72 hr posttransfection, respectively. Because it targets a conserved sequence in the SOD1 gene, miR-SOD1 also recognizes the marmoset mRNA sequence (Fig. 1d).
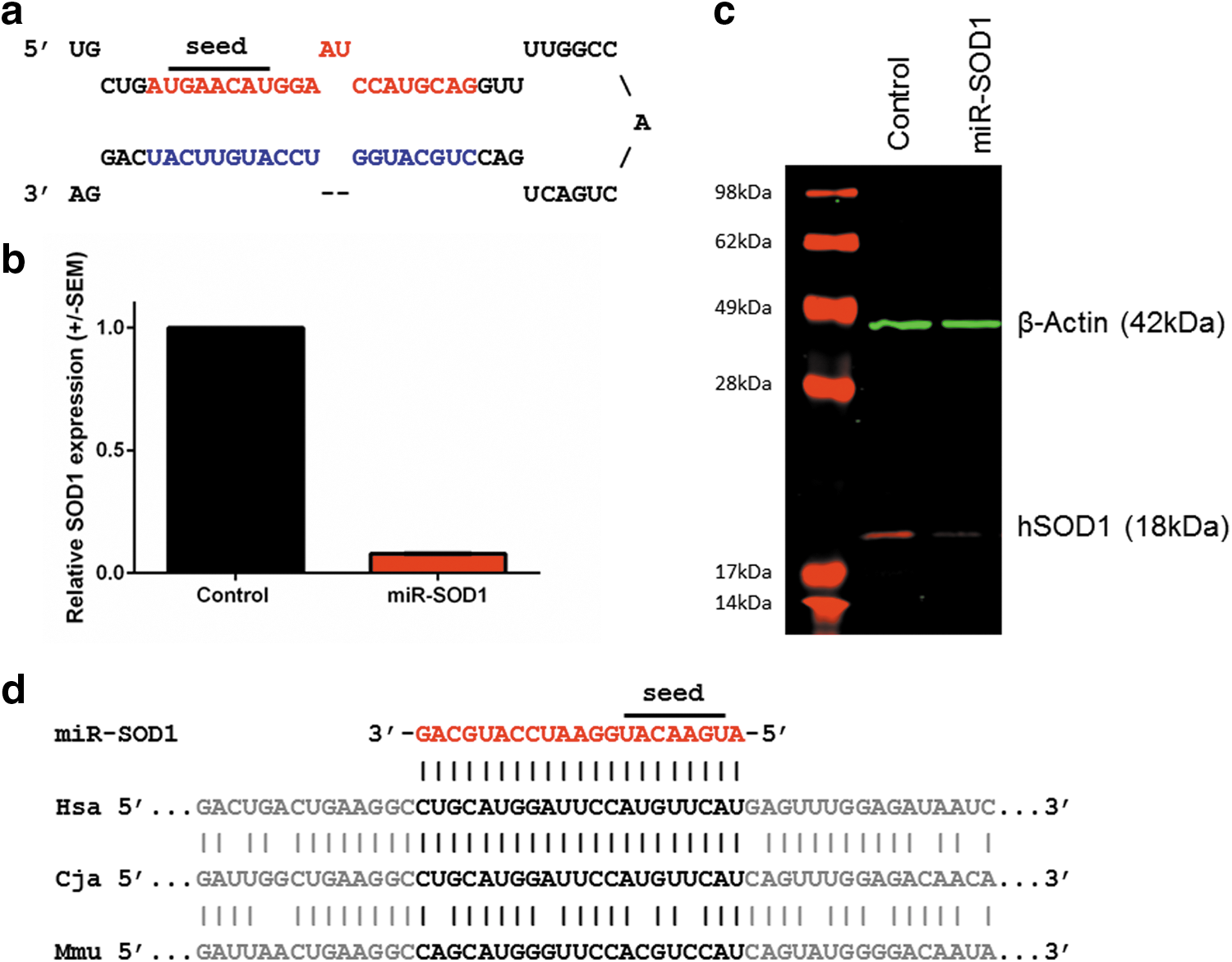
Design and in vitro validation of miR-SOD1, an artificial miRNA targeting human SOD1.
Administration of miR-SOD1 to adult SOD1 G93A mice significantly delays disease onset and extends survival
To determine if miR-SOD1 is effective in vivo after rAAV-mediated delivery, we tested it in SOD1 G93A mice in experiments that compared the efficacy of pol II (chicken beta actin or CB) and pol III (U6) promoters. With either promoter, intravenous rAAVrh10-mediated delivery through the tail vein of miR-SOD1 to adult (P56–68), pre-onset SOD1 G93A mice extends survival (Fig. 2). A significant extension in survival (22 days, or 20%, n = 14–22, p = 0.0485) is observed (Fig. 2a) in the group treated with the CB-driven construct (130 days), compared with an age-matched, gender-matched control group (108 days). Survival is prolonged to a greater extent by the CB-miR-SOD1 treatment in the subset of females (34 days, or 32%, n = 8–10, p = 0.0076; data not shown). A more robust extension of survival (27 days, or 21%, n = 15–17, p < 0.0001) is observed (Fig. 2b) in the group treated with the U6-driven construct (158 days) compared with age-matched, gender-matched littermates (131 days). Such extension in survival is similar to that observed in the only study reporting systemic treatment of adult SOD1 G93A mice, which was 23% using rAAV9-shRNA. 21 No signs of toxicity were observed in either group, based on daily assessment of discomfort as well as spinal cord Hprt levels by RT-qPCR.
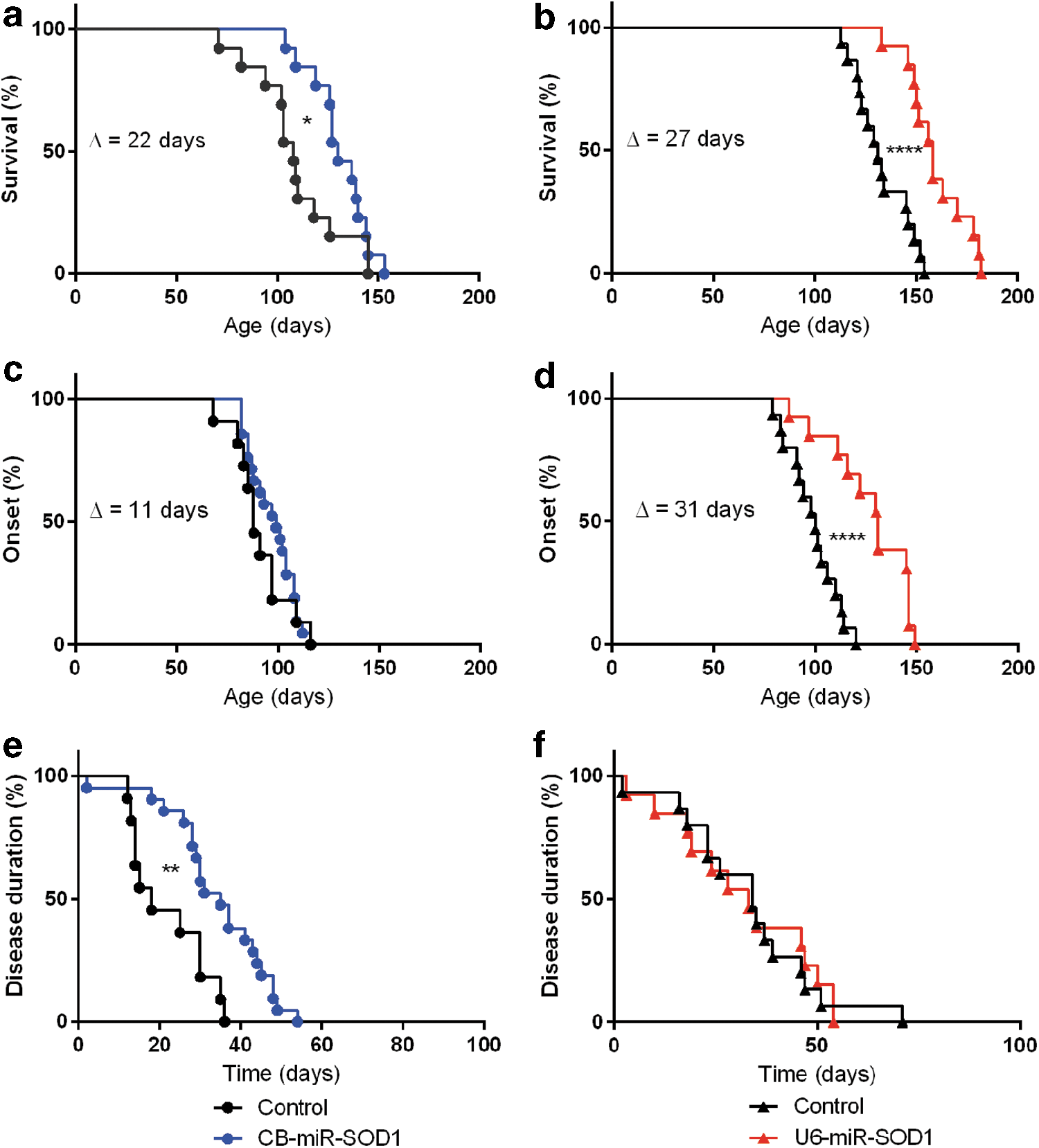
Reducing SOD1 expression prolongs survival in SOD1
G93A mice. SOD1
G93A mice were injected intravenously through the tail vein with 2E12 gc vector and were monitored daily by an experienced, blinded animal caretaker until they were euthanized. The mice were treated with either
For the CB-driven construct, the delay in disease onset of 11 days (12.5%) was not statistically significant (88 days for the control group vs. 99 days with treatment; Fig. 2c). By contrast, the U6-driven construct did achieve a significant 31-day (31%) delay in onset (from 100 to 131 days, n = 15–17, p < 0.0001) for the treated group (Fig. 2d). This exceeds all studies reporting treatment of early adult 23 and adult SOD1 G93A mice, 21,24 where no delay in disease onset was reported. This delay in onset is documented in mice at 144–145 days of age, which is passed median survival of untreated SOD1 G93A mice (Supplementary Videos S1 and S2).
Disease duration was extended by treatment with the CB-miR-SOD1 construct from 18 to 35 days (94%, p = 0.0015; Fig. 2e). A similar disease duration was observed for the U6-miR-SOD1-treated (34 days) and the untreated (33 days) group (Fig. 2f), indicating that the increased survival observed in this group can be attributed primarily to a delay in onset.
Administration of miR-SOD1 to adult SOD1 G93A mice significantly preserves limb strength and motor skills, and stabilizes respiratory physiology
A significant preservation in two and four limbs' strength was observed (Fig. 3a and b), indicating that the treatment efficiently delays muscle wasting (p < 0.0001 for both parameters). Moreover, the treatment significantly preserved motor skills as assessed by rotarod performance test (p = 0.0028), where there is a remarkable difference in latency to fall in mice 120 days of age and older (Fig. 3c).
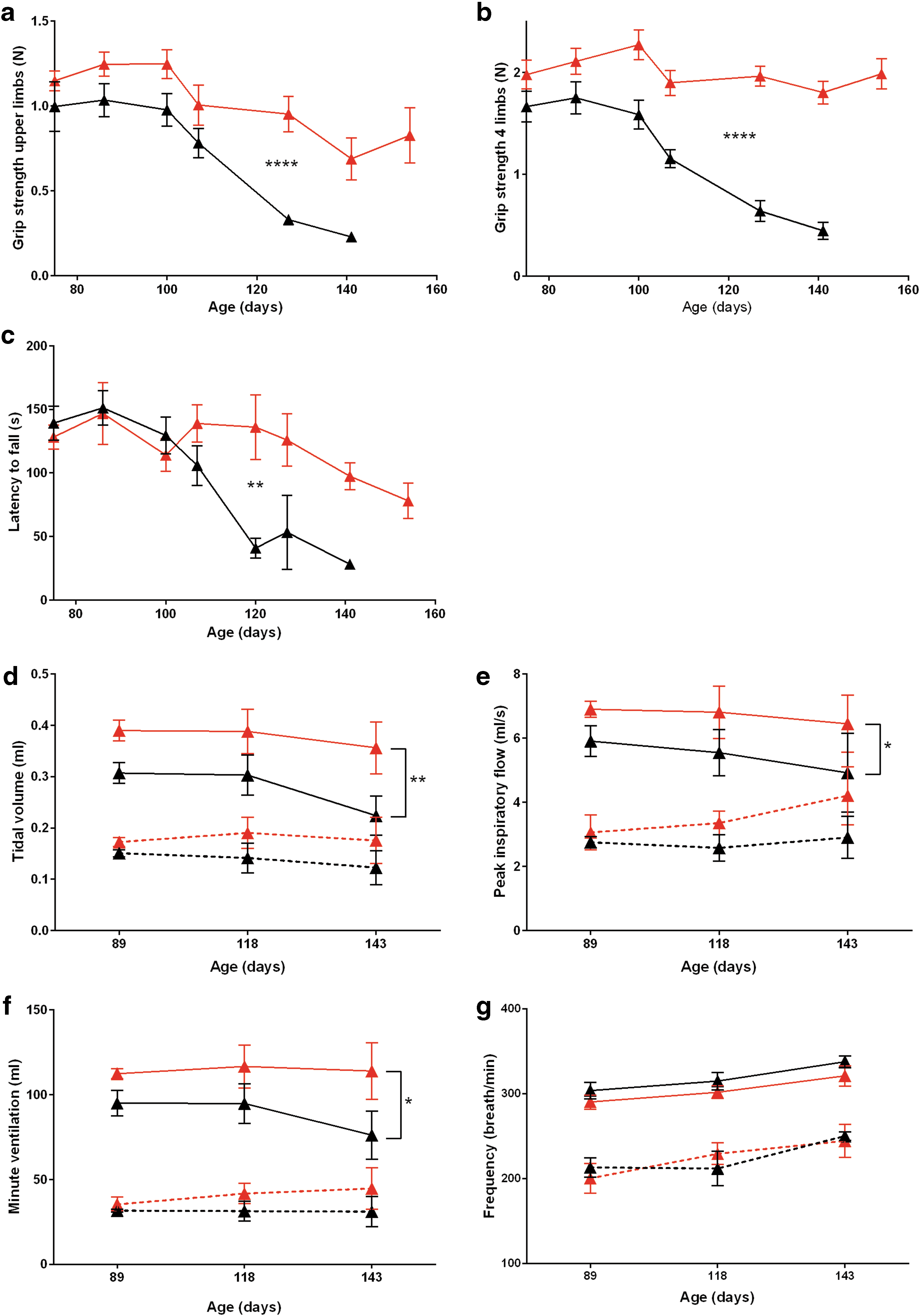
Reducing SOD1 expression preserves limb strength and motor skills, preserves motor neurons, and improves respiratory physiology. Limb strength was quantified by grip strength test for two
During the normoxic baseline conditions, the U6-miR-SOD1-treated and the untreated SOD1 G93A mice showed similar tidal volume, peak inspiratory flow, minute ventilation, and breathing frequency (Fig. 3d–g). However, during a 10 min hypercapnic respiratory challenge, the breathing pattern of the treated SOD1 G93A mice demonstrated a larger tidal volume and minute ventilation (Fig. 3d; p = 0.0043 and Fig. 3f; p = 0.0137) as compared with the control group. In addition, treated mice had a more robust peak inspiratory flow indicative of inspiratory muscle strength (Fig. 3e; p = 0.0455) as compared with controls. There was no significant difference in frequency between the two groups (Fig. 3g; p = 0.1208).
Lumbar intrathecal rAAVrh10-mediated delivery of miR-SOD1 silences SOD1 along the entire spinal cord in C. jacchus
The marmoset (C. jacchus) is a New World primate that is commonly used in neuroscience research because of its small size, high birth rate, and easy handling 32 ; other advantages include efforts toward mapping of the marmoset brain, 33 as well as the successful generation of transgenic marmosets. 34 Marmosets have been used in studies for Huntington's, 35 Parkinson's, 36,37 and Alzheimer's diseases. 38,39 We elected to use marmosets for studies of intrathecal delivery of rAAVrh10-miR-SOD1. Nine marmosets of comparable age and body weight, and prescreened for low levels of NAb against AAVrh.10, received an intrathecal injection at lumbar level of 6E12 gc/kg body weight in 300 μl total volume of rAAVrh10 encoding one of the three constructs previously described: CB-GFP, CB-miR-SOD1, and U6-miR-SOD1. Tail-flick response was monitored as a sign of proper needle placement, indicative of piercing of the dura and contact with intradural nerve roots. The animals were split into three groups of three, each gender-matched (Supplementary Table S3). They were euthanized at 18–23 days postinjection to avoid possible immune response to the GFP. Tissues from one animal from each group were fixed upon euthanasia and used for immunostaining; tissues from the other two animals from each group were frozen upon euthanasia and used for all other end points. The females were subsequently excluded from all further analyses, based on absence of tail-flick response upon needle placement questioning the efficacy of the delivery, which was later confirmed by low GFP levels as determined by RT-qPCR to be significantly lower than in the males.
Biodistribution studies demonstrated that, as expected, the highest number of genome copies per diploid genome (gc/dg) was found at the site of injection in the lumbar spinal cord, varying from 25.07 to 227.45 gc/dg (Supplementary Fig. S2). In the thoracic spinal cord, 16.46–122.93 gc/dg were detected, and 0.63–29.19 gc/dg in the cervical spinal cord (Supplementary Fig. S2). The second highest tissue was the liver with a range of 44.42–138.18 gc/dg (Supplementary Fig. S2). Vector genomes were also detected in the brain with values ranging from 0.08 to 7.73 gc/dg, although a limited number of samples was available for biodistribution (Supplementary Fig. S2).
In addition, CNS transduction was evaluated by immunostaining for GFP. Robust GFP staining was seen at the injection site at LSC level all the way to CSC level (Fig. 4a–c), confirming that this delivery route allows vector delivery to the entire spinal cord. Motor neurons (as distinguished based on morphology) stained positive for GFP at all levels of the spinal cord (Fig. 4d–f). As shown in Fig. 6, GFP expression was detected in motor neurons that were positive for both SOD1 and ChAT as determined by RNA expression.
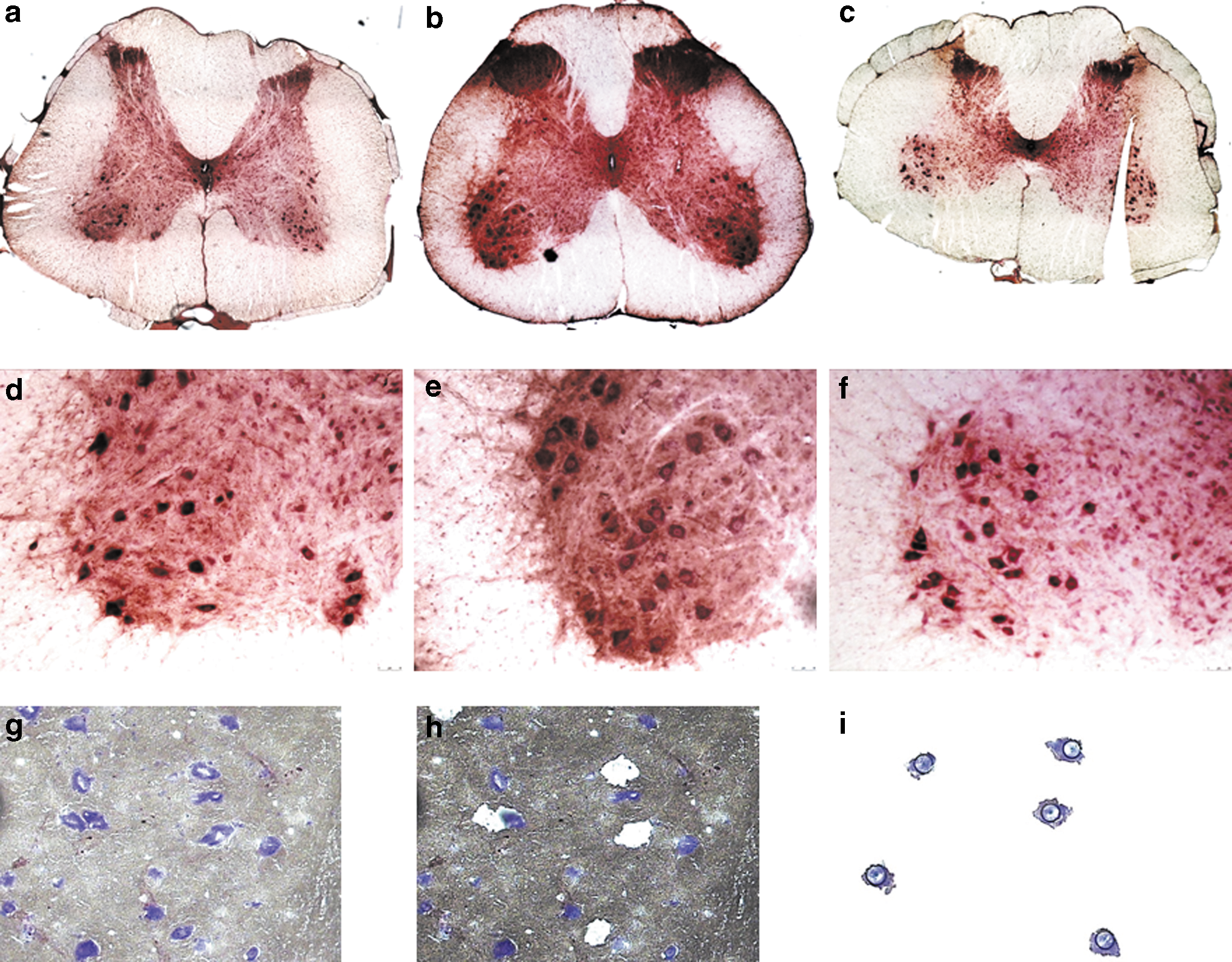
NHP motor neurons are transduced by rAAVrh10.
Next, SOD1 silencing was evaluated in the spinal cord. Motor neurons (n ≈ 2,000) were isolated from 20 μm sections (Fig. 4g) by laser-capture microdissection (Fig. 4g–i); total RNA was isolated from motor neurons (Fig. 4i) and nonmotor neuron tissue (Fig. 4h). SOD1 mRNA levels were quantified by RT-qPCR (Fig. 5a–f). In motor neurons (Fig. 5a–c), SOD1 levels are decreased with the CB-miR-SOD1 by 21% in the lumbar region and 8% in the cervical region (thoracic region data not available). With the U6-miR-SOD1 construct, SOD1 levels are decreased by 93% in the lumbar, 65% in the thoracic, and 92% in the cervical cord region (Fig. 5a). Similar levels of silencing were observed in the nonmotor neuron tissue (Fig. 5d–f). To confirm that the observed reduction in SOD1 mRNA was miRNA-mediated, mature miR-SOD1 levels were quantified by RT-qPCR (Fig. 5g). Levels in the LSC motor neurons of the U6-miR-SOD1-injected animal were four times higher than in the motor neurons of the CB-miR-SOD1-injected animal. Background levels of mature miR-SOD1 were detected in the CB-GFP animal, possibly attributable to contamination during tissue harvesting (Fig. 5g). The levels of SOD1 silencing positively correlated with the levels of mature miR-SOD1 expression (R 2 = 0.99). This analysis was only done in the LSC as limited material was available. SOD1 levels were next evaluated in the pons and medulla by RT-qPCR. In total tissue homogenates, SOD1 levels are decreased by 20% with the CB-miR-SOD1 construct, and by 35% with the U6-miR-SOD1 construct (Fig. 5h).

SOD1 silencing in NHP motor neurons and pons/medulla correlates positively with the levels of mature miR-SOD1. SOD1 and HPRT transcripts were quantified by RT-qPCR in spinal cord motor neuron
Subsequently, RNA multiplex fluorescent in situ hybridization was performed, using a GFP probe in C1 to mark transduced cells, and a SOD1 probe in C2 and a ChAT probe in C3 to mark cholinergic neurons, including motor neurons (Fig. 6). Positive ChAT staining as well as cell morphology were used here for identification of motor neurons in ventral horns. The CB-GFP animal had low levels of GFP and high levels of SOD1 in motor neurons (Fig. 6a and b). The CB-miR-SOD1 animal presented similar fluorescence (data not shown). Consistent with efficient SOD1 silencing, the spinal cord sections in the U6-miR-SOD1 animal had high levels of GFP and low or background levels of SOD1 in motor neurons (Fig. 6c and d). The selected images are representative. The marked difference in fluorescence between the treated and nontreated animals is consistent with the RT-qPCR data and confirms that SOD1 expression was substantially attenuated in the motor neuron population.

SOD1 mRNA is undetectable in GFP-positive, ChAT-positive motor neurons of animal treated with U6-miR-SOD1. Twenty-micrometer sections of frozen spinal cord tissue, from either the control animal
Preexisting NAb response to AAV has a critical impact on transduction efficiency after vector injection. Therefore, NHPs were screened for NAb to rAAVrh10 before injection and only NHP presenting NAb titers lower than 1:5 were selected to prevent any rAAV vector neutralization. Serum samples were then collected 1 week and 3 weeks after vector administration. Consistent with results already published after IC or IT rAAV injection, 40 NAb to injected rAAVrh10 vector were detected in all NHPs (Supplementary Table S4). The NAb titers detected at week 1 ranged from 1:80 to 1:640 and then increased between week 1 and week 3 (range 640–1280). We also measured NAb titers in CSF collected at 3 weeks after AAV administration. In contrast to the results obtained in the serum at the same time point, the titers were significantly lower (range 1:5–1:40) with the exception of NHP No. 18, which had a titer of 1:160.
Discussion
Previous studies have shown that viral vectors permit CNS delivery of constructs that reduce expression of the SOD1 gene and thereby extend survival of transgenic SOD1 G93A ALS mice. In particular, this was shown with lentivirus 20 and rAAV6 and rAAV921,23 in SOD1 G93A mice and with rAAV9 in SOD1 G93A rats. 22 In the only study in which adult mice (defined as >50 days) were treated with rAAV expressing an artificial miRNA, the treated animals survived 11% longer, and disease onset was not delayed. 24 Here we report extending survival by 20% (22 days) with pol II-miR-SOD1 and 21% (29 days) with pol III-miR-SOD1, and we also report delaying disease onset by 31% (31 days) with the pol III, but not with the pol II. Finally, whereas disease duration is not affected by the U6-miR-SOD1 treatment, the CB-miR-SOD1 significantly slows down disease progression. Although the two studies were done at different times, the experimental protocol was identical, and it is not clear why a change of promoter would lead to such difference, and will be further investigated. The male/female ratio is also different in each study and may account for the difference. The strength of the murine study lies in the robust preservation of two and four limbs' strength, rotarod performance, and ventilation, which support our survival data. Interestingly, although the respiratory phenotype of the SOD1 G93A mice has been characterized as early as 2006, 41 to our knowledge no therapy has ever reported to slow down or stabilize the respiratory phenotype in these mice. Because the mice recapitulate the respiratory phenotype of ALS patients, these results highlight the potential of the proposed therapy for clinical use.
Another difference in this study is that we used an artificial miRNA to reduce SOD1 expression, whereas most of the previous studies used shRNA. This allowed us to do a promoter comparison that had never been done in this context. Pol II- and pol III-driven artificial miRNAs present a number of advantages over pol III-driven shRNAs, including improved safety. This is thought to be related to the accuracy of the processing of the miRNA over the shRNA hairpin molecule. 42 As mentioned previously, no toxicity was observed in the current study; in addition to the use of an artificial miRNA, this may also reflect the fact that we used lower vector doses than in prior studies reporting toxicity. 43,44 Nevertheless, the lack of toxicity in this study is noteworthy given that CNS-related toxicity has been reported on various occasions. 42,45 –49 Although in the NHP robust silencing was observed only with the U6-driven construct, optimization of the expression cassette and delivery technique may improve knockdown with the CB-driven construct.
Our study is also distinguished by the use of serotype rh.10 instead of rAAV9 and rAAV6. Although it was shown that rAAV9 transduces motor neurons after systemic injection of a newborn NHP, 50 intrathecal delivery of rAAV9 in NHPs lead primarily to transduction of astrocytes. 51 The use of rh.10 is a point that may be favorable in this case where motor neurons are the primary target. Indeed, we demonstrate here excellent transduction of motor neurons. From an epidemiological perspective, the use of rh.10 over 9 would not be disadvantageous; the prevalence of NAb is equivalent for both serotypes. 52 In addition, several clinical trials with rAAVrh10 are currently ongoing (NCT01414985, NCT01801709, NCT01161576, and NCT02168686); this should generate further safety data for this AAV capsid.
In this proof-of-concept, translational study, another critical issue is the method of delivery of the viral vector; this is particularly important in considering the transition from rodents to a large-animal model. We report here that lumbar intrathecal delivery of an rAAVrh10 vector allows excellent transduction of motor neurons all along the entire spinal cord, extending rostrally into the distal brainstem. It is clear that this approach, when coupled with an appropriately active promoter, results in substantial reduction of SOD1 expression. A major obstacle encountered in this study was the technical challenge of the intrathecal delivery, leading to a high rate of failure (3/9 animals); this was largely a consequence of the relatively small size of the marmosets. Our forthcoming studies will continue to focus on optimizing delivery to the cerebrospinal fluid, examining several variables: (1) using injections into the cisterna magna in addition to the lumbosacral regions; (2) maintaining the subjects in the Tredelenburg position postadministration, a method that improves transduction of the thoracic and cervical segments of the spinal cord 53 ; (3) implanting the catheter into the intrathecal space several days in advance of the infusion, a technique that will allow healing around the catheter entry zone and thereby reduce backflow leakage during infusion; and (4) using larger NHPs. Although more can be done to refine our delivery system, this study nonetheless demonstrates the considerable potential of using rAAVrh10 for intrathecal delivery of an miRNA to silence expression of the SOD1 gene as well as the feasibility of moving to animals larger than ALS mice or rats. In our view, this improved intrathecal delivery protocol will allow us to generate robust and reproducible data that will support clinical development of this therapy for SOD1-mediated ALS.
Footnotes
Acknowledgments
We acknowledge Prize4Life for providing some of the mice used in this study. The laser-capture microdissection was done by Charles R. Vanderburg (Harvard NeuroDiscovery Center). We are grateful to Nicholas Wightman (University of Massachusetts Medical School [UMMS]) for developing the SOD1 Western blot assay and to Alexandra Weiss (UMMS) for help with the mouse colony. This work was funded by ALS Alliance Therapy (R.H.B.), the Rizzuto fund (R.H.B.), and NINDS NS088689 (C.M. and R.H.B.). R.H.B. also receives support from the NINDS (NS079836), the ALS Association, the Angel Fund, the Al-Athel Foundation, the Pierre L. de Bourgknecht ALS Research Foundation, Project ALS, and P2ALS.
Author Disclosure
No competing financial interests exist for F.B., G.G., B.C., J.P.M., G.C.T.C., L.S., Q.S., and M.K.E. C.M. and R.H.B. are both inventors on a patent application that covers SOD1 silencing with the vectors described in this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.